Isolation of breast cancer and gastric cancer circulating tumor cells by use of an anti HER2-based microfluidic device†
Giuseppe
Galletti
a,
Matthew S.
Sung
a,
Linda T.
Vahdat
a,
Manish A.
Shah
a,
Steven M.
Santana
b,
Giuseppe
Altavilla
c,
Brian J.
Kirby
d and
Paraskevi
Giannakakou
*a
aWeill Medical College of Cornell University, New York, NY, USA. E-mail: pag2015@med.cornell.edu
bBiomedical Engineering, Cornell University, Ithaca, NY, USA
cDepartment of Human Pathology, University of Messina, Messina, Italy
dSibley School of Mechanical and Aerospace Engineering, Cornell University, Ithaca, NY, USA
First published on 7th October 2013
Abstract
Circulating tumor cells (CTCs) have emerged as a reliable source of tumor cells, and their concentration has prognostic implications. CTC capture offers real-time access to cancer tissue without the need of an invasive biopsy, while their phenotypic and molecular interrogation can provide insight into the biological changes of the tumor that occur during treatment. The majority of the CTC capture methods are based on EpCAM expression as a surface marker of tumor-derived cells. However, EpCAM protein expression levels can be significantly down regulated during cancer progression as a consequence of the process of epithelial to mesenchymal transition. In this paper, we describe a novel HER2 (Human Epidermal Receptor 2)-based microfluidic device for the isolation of CTCs from peripheral blood of patients with HER2-expressing solid tumors. We selected HER2 as an alternative to EpCAM as the receptor is biologically and therapeutically relevant in several solid tumors, like breast cancer (BC), where it is overexpressed in 30% of the patients and expressed in 90%, and gastric cancer (GC), in which HER2 presence is identified in more than 60% of the cases. We tested the performance of various anti HER2 antibodies in a panel of nine different BC cell lines with varying HER2 protein expression levels, using immunoblotting, confocal microscopy, live cells imaging and flow cytometry analyses. The antibody associated with the highest capture efficiency and sensitivity for HER2 expressing cells on the microfluidic device was the one that performed best in live cells imaging and flow cytometry assays as opposed to the fixed cell analyses, suggesting that recognition of the native conformation of the HER2 extracellular epitope on living cells was essential for specificity and sensitivity of CTC capture. Next, we tested the performance of the HER2 microfluidic device using blood from metastatic breast and gastric cancer patients. The HER2 microfluidic device exhibited CTC capture in 9/9 blood samples. Thus, the described HER2-based microfluidic device can be considered as a valid clinically relevant method for CTC capture in HER2 expressing solid cancers.
Introduction
Circulating tumor cells (CTCs) have emerged during the last decade as a viable and readily accessible alternative source of tumor cells in the form of a “liquid biopsy”, with numerous studies that report how CTCs can be successfully isolated from the peripheral blood of patients with advanced solid tumors using a variety of techniques.1–3 The clinical relevance of CTC isolation lies in a real-time access to tissue putatively closely related to the disease state without subjecting the patient to a more invasive biopsy; furthermore, analyzing CTCs in real time can potentially elucidate the molecular and biological changes of the tumor that occur during treatment, perhaps providing insight into the onset of drug resistance.4Although enormous efforts have been applied to improve the efficiency and the purity of CTC capture and identification, isolation of this rare population of tumor cells remains challenging. Existing technologies rely primarily on the use of EpCAM-based immunocapture, such as the FDA-approved CellSearch system (Veridex, Raritan, NJ, USA). Although this technique is able to detect and enumerate fixed CTCs from metastatic cancer patients,5–7 viable CTCs are required for molecular and functional characterization of tumor cells. More importantly, tumor cells that gain access to the vascular system could undergo drastic molecular changes as a consequence of the process of epithelial to mesenchymal transition (EMT), causing the down regulation of several epithelial markers.8,9 Thus, EpCAM protein levels can be significantly reduced during the EMT process, limiting the effectiveness of the EpCAM-dependent approach for CTC capture. Several non-EpCAM based alternative strategies have been developed and proven to be effective in isolation and molecular characterization of CTCs from the peripheral blood of metastatic cancer patients.10,11 We have recently developed a prostate cancer specific microfluidic device for CTC isolation that operates on the principle of geometrically enhanced differential immunocapture (GEDI), using anti prostate-specific-membrane antigen (PSMA) antibody-coated microposts in a geometry that generates cell-size-dependent collision and adhesion and shown that this innovative design achieved capture of viable CTCs using only 1 ml of blood with minimal leukocyte contamination.12,13 In addition, we showed that the PSMA-GEDI microdevice achieved capture of 10–400 higher CTC numbers compared to CellSearch, in a study of 30 patients using same-patient and same-day blood draw design. The higher CTC recovery of the PSMA-GEDI was attributed to both the enhanced geometry and microfluidic technology and to the very low levels of EpCAM staining of the captured CTCs.
Despite the success of the PSMA-GEDI device, the use of PSMA was applicable to prostate cancer patients only. To expand this technology to other solid tumor types and to avoid problems with low EpCAM expression on CTCs, we chose HER2 as a surface antigen to capture CTCs from additional tumor types.
HER2 is one of the most studied membrane markers in solid tumor cancer biology. HER2 is a ligand-less transmembrane receptor that belongs to the human epidermal growth factor receptor (HER) family.14 HER2 heterodimerizes with other receptors of the HER family and amplifies the triggered intracellular signal networks that drive cell proliferation and tumor progression.15 HER2 is expressed at high levels (3+ based on immunohistochemical assessment) in 25–30% of breast cancer patients while it is expressed to some degree (1+ to 3+) in more than 90% of breast cancer patients.16 HER2-targeted clinical therapies, such as the anti-HER2 monoclonal antibody trastuzumab (Herceptin™) and the small molecule tyrosine kinase inhibitor lapatinib, show activity in HER2-overexpressing patients.17,18 The receptor is also biologically and therapeutically relevant in other tumor types such as gastric cancer, in which HER2 is expressed in 67% of the cases and overexpressed in 13%–30% of patients, depending on disease subtype.9,19,20 Importantly, cell line and animal model studies have shown that HER2 protein expression is not affected by EMT in breast cancer cells.21 Taken together, the broad expression of HER2 receptor in these two cancer types together with EpCAM's susceptibility to EMT prompted its use as the membrane target for CTCs selection and capture from peripheral blood in this study.
Here we report the development and the characterization of the anti-HER2 GEDI microfluidic device, and its performance and clinical application in breast and gastric cancer patients.
Materials and methods
Reagents
Breast cancer cell lines MCF-7, T-47D, SK-BR-3, BT-474, MDA-MB-231, MDA-MB-468, HCC-1937, Hs 578T and BT-549 were purchased from the American Type Culture Collection (ATCC, Manassas, VA). These cell lines were selected as representative of the five clinically relevant intrinsic subtypes of breast cancer.22 MCF-7, SK-BR-3, MDA-MB-231, MDA-MB-468 and Hs 578T cells were cultured using Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin antibiotic solution. BT-549 cells were cultured using RPMI 1640 media supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin antibiotic solution. BT-474 cells were cultured in DMEM HG supplemented with non-essential amino acids (NEAA), 10% fetal bovine serum and 1% penicillin–streptomycin antibiotic solution. Cells were grown at 37 °C and 5% CO2. Primary antibodies for HER2 (clone 42 BD Biosciences, San Jose, CA, USA; clone SP3 and clone 9G6.10 Thermo Fisher Scientific, Lafayette, CO, USA), pan-cytokeratin (clone C-11 Biolegend, San Diego, CA, USA), tyrosinated tubulin (clone YL 1/2 Millipore, Billerica, MA, USA), CD-45-QD800 (clone H130 Invitrogen, Carlsbad, CA, USA) were purchased. Alexa Fluor secondary antibodies for western blot, immunofluorescence and flow cytometry were obtained from Invitrogen, CF594 was obtained from Biotium. Pan-CK antibody was conjugated to secondary CF594 using the Mix-n-Stain kit (Biotium) according to the manufacturer's instructions. Calcein green and calcein red (Invitrogen, Carlsbad, CA, USA) were used.Immunoblotting
Cells were collected and lysed in lysis buffer (50 mM Tris pH 7.5, 100 mM NaCl, 2 mM EDTA, 1% NP-40) supplemented with 7× protease inhibitors cocktail (Roche Diagnostics, Pleasanton, CA, USA). Cell lysates were resuspended in Laemmli buffer and 50 μg of protein were separated via electrophoresis; proteins were transferred onto polyvinylidene difluoride and blocked in 5% milk. Data were acquired using Odyssey Imaging System from LICOR Biosciences (Lincoln, NE, USA) and densitometry was performed with ImageJ software (National Institute of Health, Bethesda, MD, USA).Immunofluorescence
For fixed-sample immunofluorescence, cells were plated on 1.5 mm coverslips (Electron Microscopy Services, Hatfields, PA, USA); cells were fixed with PHEMO buffer (68 mM PIPES, 25 mM HEPES, 15 mM EGTA, 3 mM MgCl2, 10% DMSO) supplemented with 3.7% formaldehyde, 0.05% glutaraldehyde and 0.5% Triton X-100 and blocked with 10% normal goat serum (Jackson ImmunoResearch, PA, USA).For live cell immunofluorescence, cells were plated on Lab-Tek II chambered coverglass (Lab-Tek, Rochester, NY, USA) and incubated with primary and secondary antibodies without prior fixation or permeabilization; this preserved the native tertiary conformation of the antigen as it is present on the surface of living cells. For CTC identification on the GEDI chip, captured cells were fixed with 2% formaldehyde + 50% PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2), permeabilized with 0.25% Triton X-100 solution and blocked with 6% bovine serum albumin (BSA) + 10% normal goat serum (NGS). Cells were then stained with the following antibodies: anti-pan-CK directly conjugated with CF594, anti-tyrosinated-α-tubulin followed by anti-rat Alexa 488, anti-CD45 directly conjugated with QD800 and anti-HER2 (clone 9G6.10) followed by anti mouse 647. DNA was counterstained with DAPI. Images were acquired with Zeiss LSM 700 confocal microscope under a 63×/1.4 NA objective (Zeiss, Germany). For live cell immunofluorescence, cells were imaged with an AxioVision 200 epifluorescent microscope (Zeiss, Germany).
Flow cytometry
Live breast cancer cells were collected and incubated with anti-HER2 clone 9G6.10 primary antibody for 1 h at 4 °C. Data were acquired with LSR II system (BD Biosciences, San Jose, CA, USA) and results were analyzed with FlowJo flow cytometry analysis software (TreeStar, Ashland, OR, USA).Adsorption assay
To quantify biotinylated antibody adhesion to and saturation on the surface an immunofluorescence assay was performed. A series of solutions with different 9G6.10 antibody concentrations (0.3–100 μg mL−1) were prepared via serial dilution. 100 microliters of each dilution was incubated on wells of a NeutrAvidin-coated 96-well plate (Thermo Scientific, Rockford, IL, USA) for 1 h. Following incubation, all wells were washed with PBS and subsequently incubated with a 1% (m/v) BSA in PBS solution as a blocking buffer. The blocking buffer was removed and the wells were washed with PBS. Finally, a fluorophore-conjugated murine secondary antibody in PBS was incubated in the antibody-conjugated wells for 1 h. After incubation, all wells were washed with PBS and read by a plate reader.Microfluidic device and cell capture
All device fabrication, preparation and functionalization were carried out as previously described.13 The 9G6.10 monoclonal antibody was biotinylated (EZ-Link NHS–LC–LC–Biotin, Thermo Fisher Scientific, Lafayette, CO, USA) and immobilized on the device surfaces using MPTMS/GMBS/NeutrAvidin–biotin chemistry. Briefly, polydimethylsiloxane (PDMS) sheet was clamped to the top of the device with a custom jig to create closed channels populated with post arrays. Inlet and outlet holes were created with a biopsy punch, and 23-gauge metal tubes were inserted into the PDMS to connect inlet and outlets to external tubing. Devices were primed with a 50/50 ethanol–water mixture, and then flushed with DI water and 1% bovine serum albumin before experiments; 1 ml of blood from each sample was flowed through the microfluidic device at 1 ml h−1 rate. For capture efficiency tests, between 200 and 500 breast cancer cells were stained with calcein, according to manufacturer direction and were spiked in 1 ml of healthy donor blood. For CTC capture, 1 ml of peripheral blood was processed through each GEDI microfluidic device; the isolated cells were fixed and stained for cytokeratin, CD-45, HER2, tyrosinated tubulin and DAPI as described above. The device was subsequently mounted on a coverglass (VWR, Radnor, PA, USA) and analyzed under the microscope. Each post within the entire functional area of the microfluidic device was manually visualized throughout the z-axes and CTCs were defined as DAPI+/cytokeratin+/CD-45− events.Blood sample collection
Peripheral blood samples were collected in sodium citrate tubes (Greiner Bio-One, Monroe, NC, USA) from healthy donors or patients with metastatic breast cancer or metastatic gastric cancer under clinical trials approved by the Internal Review Board (IRB) of Weill Cornell Medical College (Analysis of Circulating Tumor Cells in Breast Cancer in Predicting Response to Microtubular Targeting Agents, protocol number: 1204012297; An Open-Labeled, Multicenter Phase II Study of Cabazitaxel in Refractory Metastatic Gastric or Gastroesophageal Adenocarcinoma, protocol number: 1208012946).Results and discussion
Our first effort focused on the identification of a suitable monoclonal antibody to use to functionalize our microfluidic device; fundamental features of a CTC-capturing candidate antibody are mainly represented by high binding affinity and specificity for the extracellular domain of the selected plasma membrane protein, so that CTC capture can be achieved even in low-expressing cells while avoiding capture of non-HER2-expressing leukocytes.Initial screening of several commercially available anti-HER2 antibodies allowed us to narrow down to three different anti-HER2 clones that exhibited high sensitivity and specificity. We tested the performance of these antibodies in a panel of nine breast cancer cell lines representative of the five clinically relevant molecular breast cancer subtypes (luminal A and B, HER2-amplified, basal and normal like; Table 1).
| Cell lines | Intrinsic subtypes | HER2 protein expression | HER2 gene amplification |
|---|---|---|---|
| MCF-7 | Luminal | + | No |
| T-47D | Luminal | + | No |
| BT-474 | Luminal | +++ | Yes |
| SK-BR-3 | HER2 ampl | +++ | Yes |
| MDA-MB-468 | Basal | − | No |
| HCC 1937 | Basal | + | N/A |
| MDA-MB-231 | Normal | + | No |
| Hs 578T | Normal | + | No |
| BT-549 | Normal | − | N/A |
Antibody performance was first tested by immunoblotting (Fig. 1A). Clone 42 recognized HER2 protein expression in six of the nine total cell lines in our panel including not only HER2 overexpressing cell lines (SK-BR-3), but also luminal-like cells (BT-474), basal-like and normal like breast cancer cells (HCC 1937, MDA-MB-231, Hs 578T, BT-549). Clones SP3 and 9G6.10 proved less proficient, with clone SP3 able to recognize the antigen only in high HER2-expressing cells (SK-BR-3 and BT-474) while clone 9G6.10 detected only one of the two HER2-overexpressing cell lines (BT-474) with dim staining in a few of the others. Based on this initial assessment, clone 42 appeared to provide the best signal in the majority of cell lines, which was corroborated by subsequent immunofluorescence evaluation in a subset of cell lines (Fig. 1B).
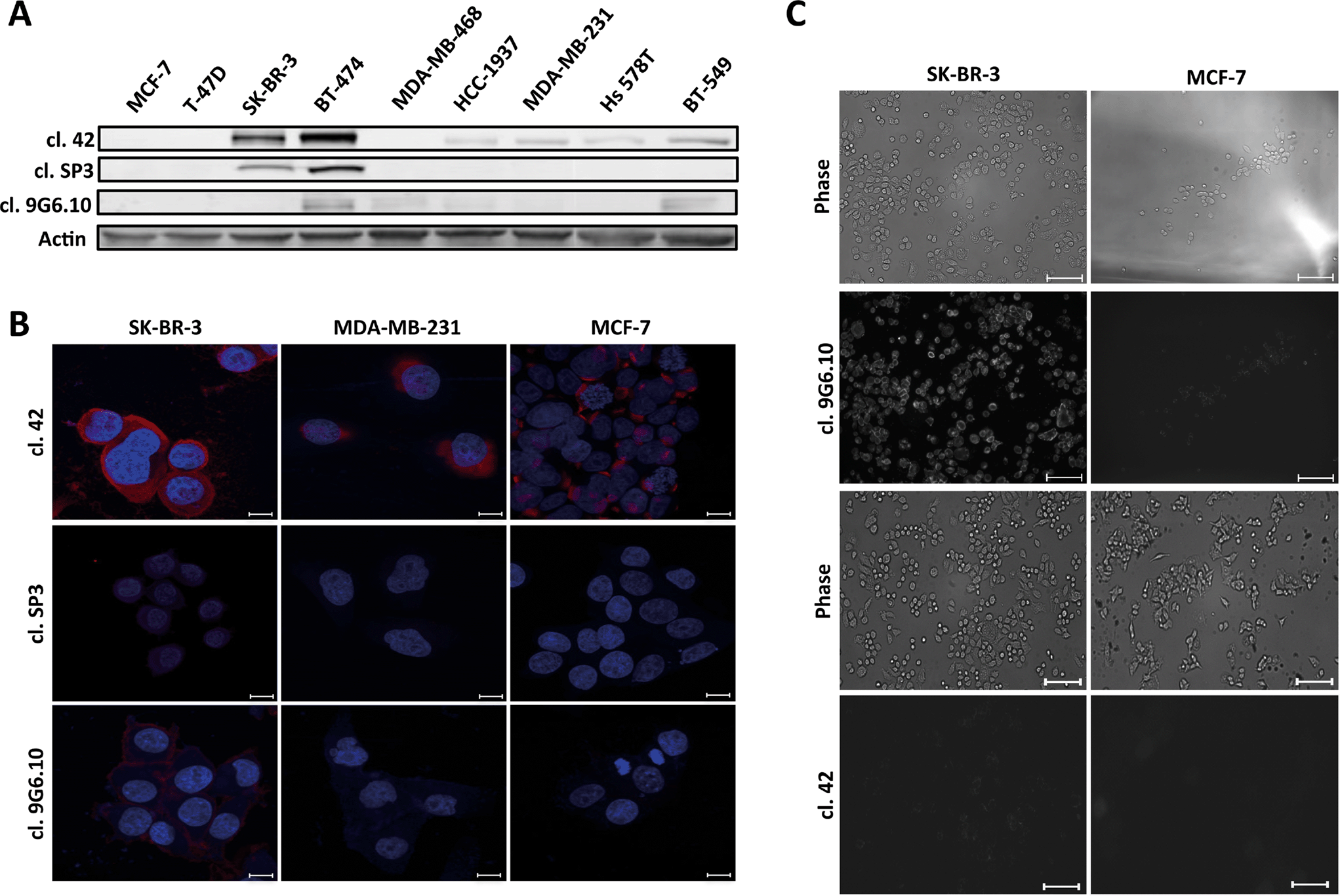 | ||
| Fig. 1 Anti-HER-2 antibodies tested by immunoblot (A) and immunofluorescence (B) or live cell microscopy (C) analyses. A. Breast cancer cell lines representative of the five different breast cancer molecular subtypes were used (MCF-7, T-47D and BT-474, luminal like; SK-BR-3, HER2 overexpressing; MDA-MB-468 and HCC 1937, basal like; MDA-MB-231, Hs 578T and BT-549, normal like). Fifty μg of total cell lysate was loaded per lane. Actin was used as a loading control. B. Three breast cancer cell lines with different HER2 expression levels based on immunoblot (SK-BR-3 high; MDA-MB-231, intermediate; MCF-7, low) were subjected to indirect immunofluorescence followed by confocal microscopy. HER2 primary antibodies as indicated, secondary antibodies Alexa 568, DNA, DAPI. Scale bar: 20 μm. C. Anti-HER2 antibodies tested by live cell immunofluorescence microscopy. SK-BR-3 and MCF-7 breast cancer cell lines were used as positive and negative controls for HER2 protein expression. Cells were plated on Lab-Tek dishes and incubated with the indicated anti-HER2 antibody for 1 h followed by 30 min incubation with secondary Alexa 488 antibody. Live cells were imaged by phase contrast (top panels) or fluorescence (bottom panels). Scale bar 10 μm. | ||
To test the ability of clone 42 to bind to the native conformation of HER2 antigen present on the surface of live breast cancer cells we performed a live-cell incubation experiment using glass cover slips functionalized with biotinylated clone 42 antibody and assessed retention of HER2-expressing cells following incubation and washout. Our results showed poor cell retention (data not shown) which led us to hypothesize that likely clone 42 does not recognize the native conformation of HER2 in living cells.
Since recognition of the native antigen conformation is a critical requirement for CTC capture we next tested the performance of anti-HER2 antibodies using live cell microscopy (Fig. 1C). We used breast cancer cell lines with high and low levels of HER2 protein (SK-BR-3 and MCF-7, respectively) and incubated them with the primary antibodies without any prior process of fixation to preserve the structure of the receptor. Surprisingly, only clone 9G6.10 was able to recognize efficiently HER2 antigen in its native conformation; conversely, clone 42, which performed well with western blot and fixed cell immunofluorescence, lost its efficiency to bind HER2.
To further quantitate the sensitivity and specificity of clone 9G6.10 to recognize HER2 in live cells we used flow cytometry in the panel of the nine breast cancer cell lines as well as peripheral blood mononucleated cells (PBMCs) isolated from healthy donors. Clone 9G6.10 proved to recognize a wide range of HER2 expression across the five different breast cancer subtypes (Fig. 2A and B); it detected HER2 protein not only in HER2 overexpressing breast cancer cell lines (SK-BR-3, BT-474), but also in breast cancer cells characterized by intermediate or low protein levels (MCF-7, T-47D, MDA-MB-231 and HCC 1937). The purity of the cell population captured on a microfluidic device is critical when the target population is as rare as CTCs in the peripheral blood; to keep a high purity of the isolated cells, a key feature is represented by the specificity of the surface marker chosen to identify the CTCs and of the corresponding antibody. Clone 9G6.10 showed high specificity for epithelial cells as it failed to detect any HER2 expression on PBMCs. Quantitation of the mean fluorescence intensity normalized to matching IgG showed that the signal detected in PBMCs was lower than that of the HER2-negative cell line MDA-MB-468 (Fig. 2B).
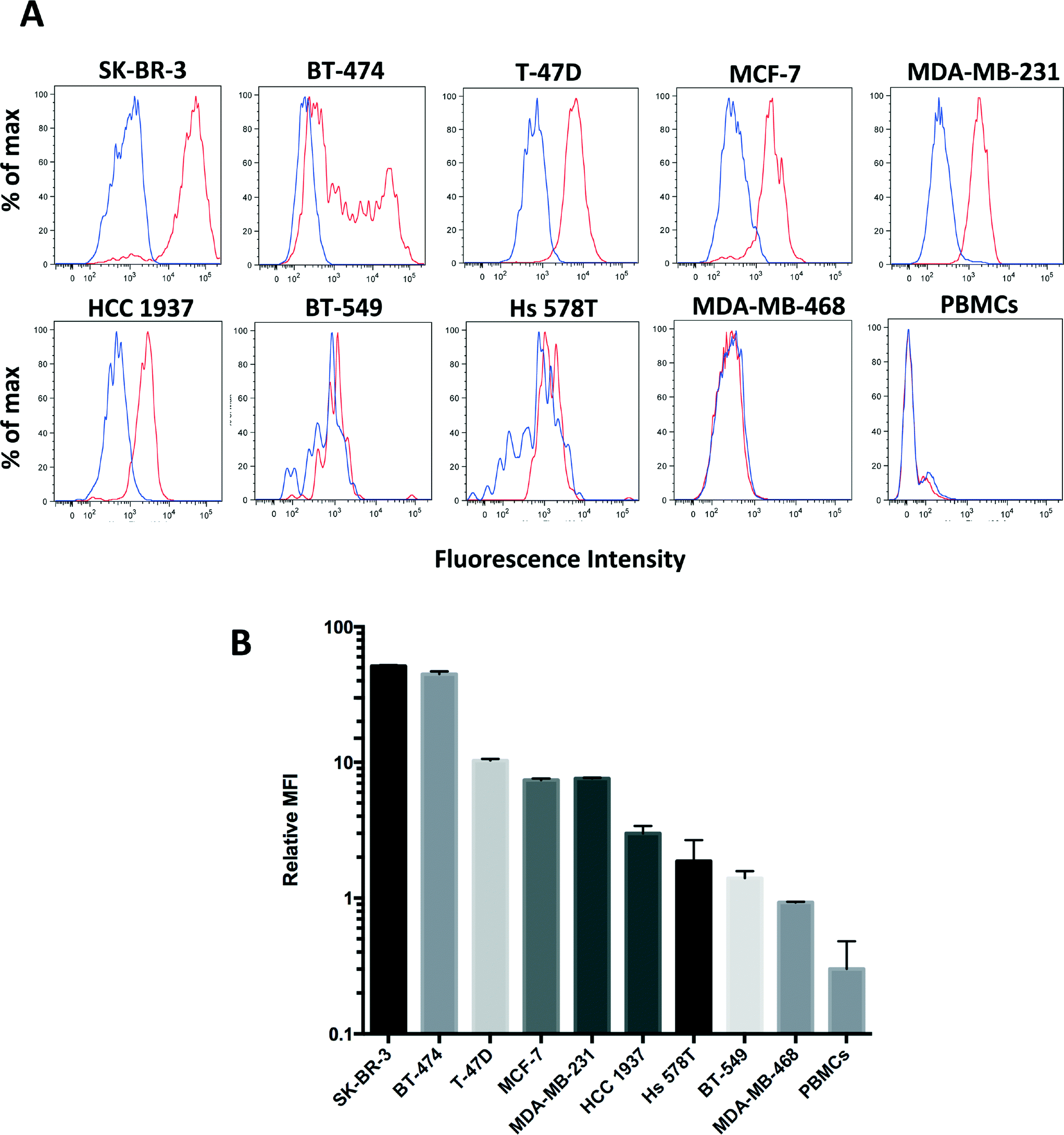 | ||
| Fig. 2 A. HER2 protein expression levels in breast cancer cell lines and healthy donor PBMCs tested by flow cytometry using the anti-HER2 antibody clone 9G6.10 (red lines) or IgG control (blue lines). Live cells were incubated with anti-HER2 antibody or matching IgG followed by secondary Alexa 488. Ten thousand events were acquired for each condition. The x-axis shows logarithmic fluorescence intensity; the y-axis shows percent of max. B. Relative mean fluorescence intensity (MFI) of HER2 protein assayed by flow cytometry; HER2 MFI was normalized using the IgG control MFI in each cell line. (SEM shown as error bar). | ||
These results suggest a minimal nonspecific binding of HER2-negative cells from clone 9G6.10 and underline the specificity of clone 9G6.10 in identifying a HER2-positive cell population in the blood, reducing the likelihood of potential leukocyte contamination on an anti-HER2 9G6.10-coated microfluidic device.
In order to establish the concentration of antibody solution to optimally functionalize the surface of our microfluidic device, we performed an immunofluorescence-based adsorption assay using solutions of biotinylated 9G6.10 antibody with concentrations ranging from 0.3 μg ml−1 to 100 μg ml−1. Antibody-binding levels were measured by quantifying fluorescence from a secondary antibody incubated with the functionalized surface. As shown in Fig. 3, results indicated that the saturation on the surface occurred at a biotinylated antibody concentration of 10 μg ml−1, suggesting that this concentration is optimal for GEDI chip surface functionalization for cell capture experiments. The concentration of 10 μg ml−1 of biotinylated antibody is similar to what was shown in our previous work, which described the optimization of anti-PSMA antibody concentration for the functionalization of the active area of the prostate cancer specific GEDI device.23 The antibody concentration is also comparable to what was used to functionalize other microfluidic devices described in the literature characterized by similar design and biochemical properties.2
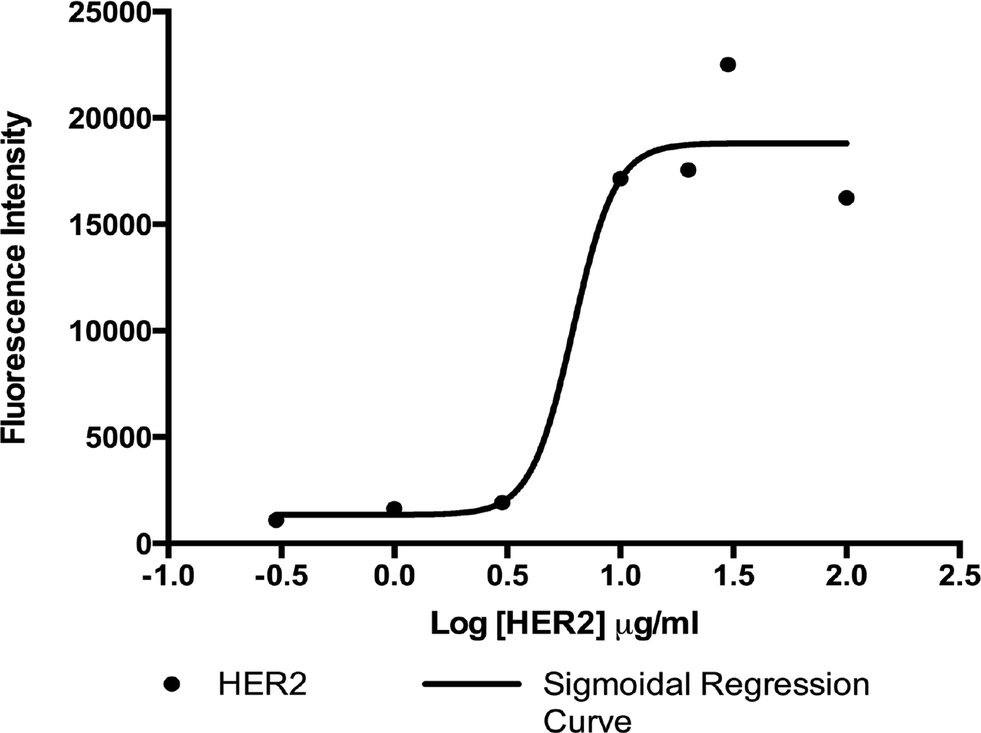 | ||
| Fig. 3 Adsorption test for biotinylated anti-HER2 clone 9G6.10. Increasing concentrations of biotinylated mouse anti-HER2 antibody clone 9G6.10 were incubated on a NeutrAvidin-coated plate. A fluorescently labeled secondary anti mouse antibody was used to detect the amount of primary antibody. | ||
In order to define HER2-based GEDI capture performance, we conducted cell capture efficiency tests with breast cancer cells expressing different levels of HER2 protein (SK-BR-3, high; MCF-7, intermediate; and MDA-MB-468, negative). Cells were fluorescently prelabeled with calcein green or red, counted and spiked in 1 ml of peripheral blood of healthy donor at a concentration of ~200–500 cells ml−1. Cell suspensions were then processed through the HER2-based GEDI microfluidic device at 1 ml h−1 flow rate. Captured cells were fixed on the microfluidic device and were subjected to microscopy enumeration based on calcein and DAPI (nuclear stain) positivity. HER2-based GEDI chip capture efficiency ranged from 78% for high HER2-expressing breast cancer cells to 26% for low HER2-expressing breast cancer cells (Fig. 4). In particular, capture efficiency was significantly higher for both HER2 high- and low-expressing cells than for HER2-negative cells. These results support our initial hypothesis that HER2 can be used to capture CTCs from patients that not only overexpress the receptor but also from patients that express moderate to low levels of HER2.
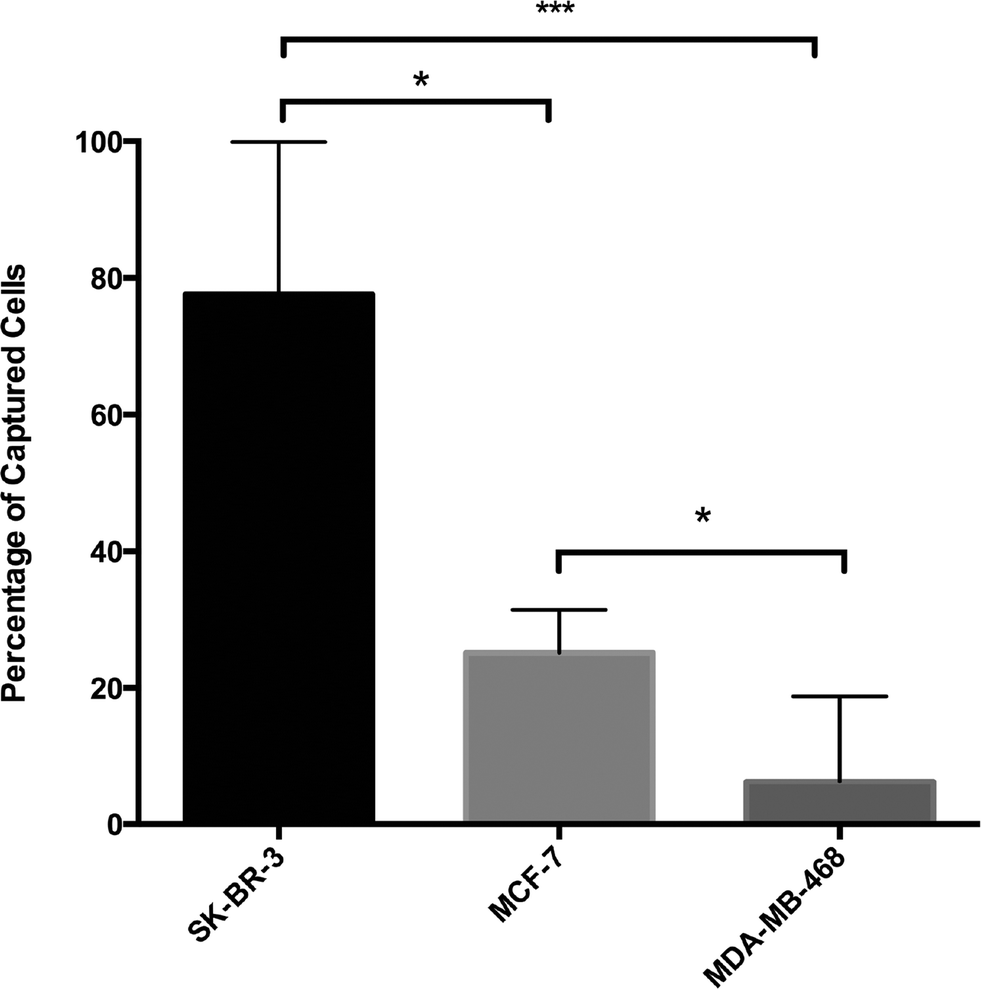 | ||
| Fig. 4 HER2-GEDI chip capture efficiency. Breast cancer cell lines with different HER2 protein expression levels were spiked into healthy donor blood, processed through the device and enumerated. Capture efficiency was calculated as the ratio of the number of captured cells over the number of spiked cells and displayed as a percentage (SD shown as error bar. *** p < 0.001; * p < 0.05). | ||
Following capture efficiency validation in cell lines, we tested the performance of our HER2-GEDI chip using peripheral blood from metastatic breast cancer or metastatic gastric cancer patients. We analyzed a total of nine blood samples obtained from five breast cancer patients, and from two gastric cancer patients with samples collected before treatment and on chemotherapy treatment for a total of four samples (for patients characteristics see Table 2). Blood samples from healthy donors (n = 3) were also collected as negative control. One ml of each blood specimen was processed through the HER2-functionalized microfluidic device and, following capture, cells were fixed and stained (see materials and methods). The GEDI chips were analyzed using a confocal microscope and high-resolution images were acquired using a 63×/1.4 NA objective. CTCs were identified as DAPI+/cytokeratin+/CD-45− cells. CTCs were found in 9/9 (100%) of the cancer samples (Fig. 5). Of the seven patients, five of them had confirmed HER2-positive status as assessed in the primary tumor by means of immunohistochemistry and pathological evaluation and/or HER2 gene amplification (Table 2). The number of CTCs captured for each blood sample ranged from 37 to 224 (mean 94 CTCs ml−1). In particular, CTCs number ranged from 31 to 115 (mean 74 CTCs ml−1) in breast cancer patients and from 33 to 224 (mean 120 CTCs ml−1) in gastric cancer patients. DAPI+/cytokeratin+/CD-45− cells were also found in healthy donors but at a lower extent (Fig. 5A; p < 0.01). Importantly, CTCs captured from HER2-positive and HER2-negative patients showed a wide range of HER2 expression (Fig. 5C), suggesting that this device can be used clinically to quantify HER2 protein expression on patient CTCs and correlate with clinical response to HER2-targeted therapies. The sensitivity of our assay in quantifying HER2 expression on GEDI captured cells was further confirmed by a cell spiking experiment using established cell lines with high to low HER2 expression (ESI† Fig. S1). These results show that the HER2-based GEDI microfluidic device is able to selectively capture CTCs from breast and gastric cancer patients using just one ml of peripheral blood and that this device can be used in the context of a larger clinical study of patients with HER2-expressing cancer.
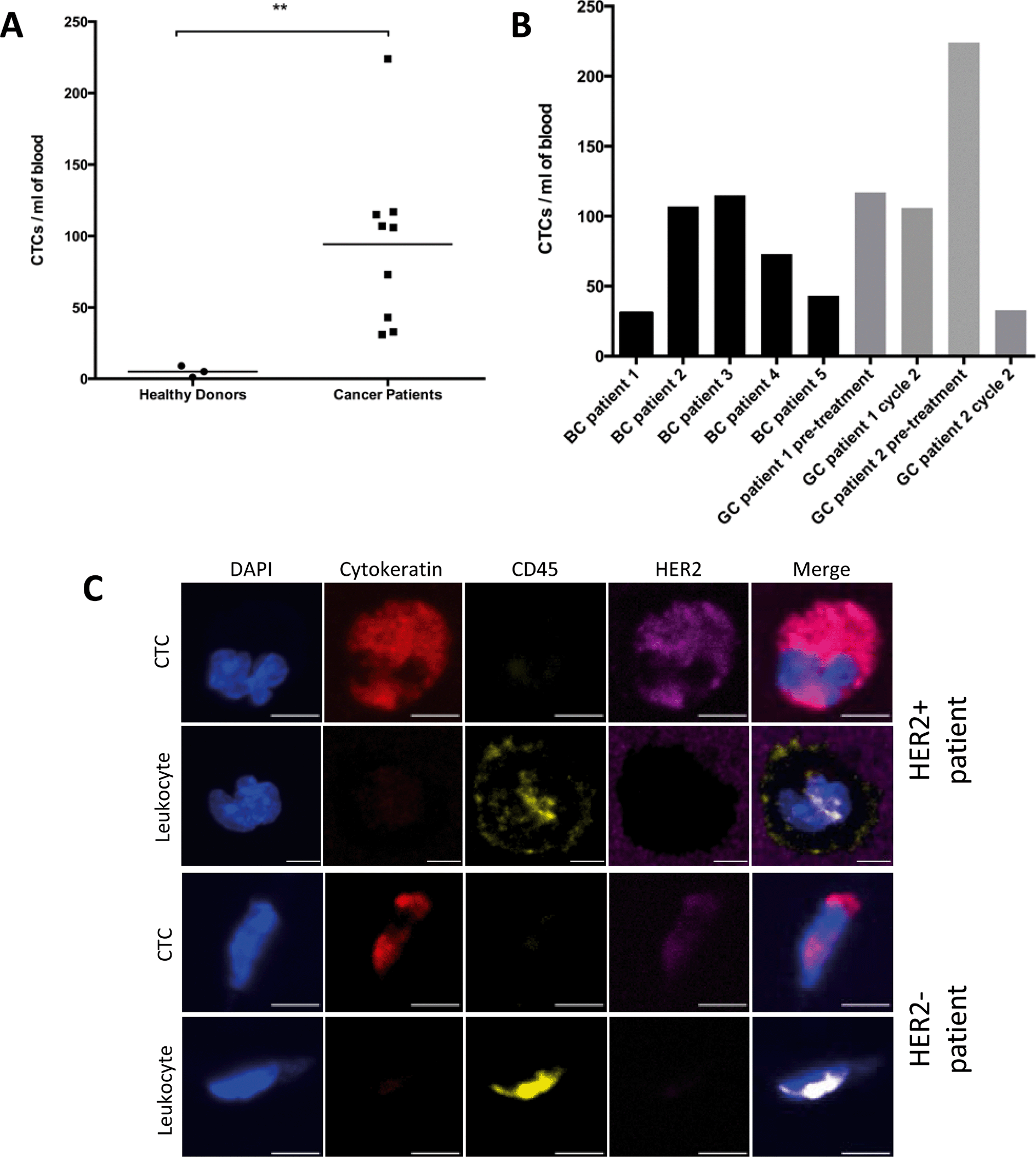 | ||
| Fig. 5 HER2-GEDI CTC capture from metastatic breast cancer and gastric cancer patients. A. Disease-specific HER2-GEDI capture of CTCs. CTC enumeration (CTCs ml−1) was performed using blood from healthy donors (median = 5) and breast or gastric cancer patients (median = 94). ** p < 0.01. B. CTC enumeration (CTCs ml−1) was performed using blood from breast and gastric cancer patients BC: breast cancer; GC: gastric cancer. C. Representative images of gastric cancer CTCs captured on the HER2-GEDI chip and stained for the selected panel of molecular markers. CTCs (DAPI+, cytokeratin+, CD45−); leukocytes (DAPI+, cytokeratin−, CD45+). HER2 staining reflects a wide range of HER2 expression on the captured CTCs. Leukocytes are also shown for comparison. Scale bar: 5 μm. | ||
| Patient ID | HER2 IHC | HER2 FISH | ER | PgR | Treatment |
|---|---|---|---|---|---|
| BC patient 1 | 1+/2+ | 2.2 | + | − | Anti HER2 therapy (trastuzumab) |
| BC patient 2 | 0 | N/A | − | − | Chemotherapy (ixabepilone) |
| BC patient 3 | 2+ | 1.6 | + | + | Chemotherapy (paclitaxel) |
| BC patient 4 | 1+ | N/A | + | + | Biological/hormonal therapy (denosumab/letrozole) |
| BC patient 5 | 2+ | 1.1 | − | − | Chemotherapy (anthracycline) |
| GC patient 1 | 0 | N/A | N/A | N/A | Chemotherapy (cabazitaxel) |
| GC patient 2 | 3+ | 3.4 | N/A | N/A | Chemotherapy (cabazitaxel) |
Conclusions
CTCs have emerged as a readily accessible source of tumor cells and the downstream molecular analysis of this source of tumor cells has opened an exciting new field of investigation in cancer research. The currently FDA-approved methodology for the isolation of CTCs (CellSearch) relies on an EpCAM-dependent capture, as this antigen is expressed primarily on the surface of cells of epithelial origin. Using this technique investigators were able to show CTC capture in 41–70% of metastatic breast cancer patients and CTC counts assessed by CellSearch were shown to correlate with disease progression in metastatic breast cancer patients receiving chemotherapy or endocrine therapy.24However, in order to get access to the bloodstream and potentially migrate to new sites of metastasis, cancer cells undergo several molecular changes, known as epithelial-to-mesenchymal transition (EMT), characterized by the down regulation of most of their epithelial markers and the acquisition of a less differentiated phenotype compared to the original primary tumor. It has been shown how EpCAM protein expression is down regulated as a consequence of the EMT process,8,25 making alternative markers important for capturing CTCs from metastatic cancer patients.
Here, we describe a HER2-based microfluidic device to capture circulating tumor cells from the peripheral blood of breast cancer and gastric cancer patients. We proposed HER2 as an antigen alternative to EpCAM-based capture, as HER2 is expressed in ~90% of breast tumors and more than 65% of gastric cancer patients. Using the HER2-GEDI device we were able to capture CTCs from 100% of the metastatic breast (n = 5) and gastric patient samples (n = 4), with a mean of 94 CTCs ml−1 of blood, and there was minimal CTC false negative capture in healthy donor blood (mean = 5).
In this study we did not directly compare CTC capture between the HER2-GEDI and the FDA-cleared CellSearch, as we have previously shown significantly higher CTC capture in prostate cancer patients, partially attributed to low EpCAM expression, and based on the results of the CellSearch clinical study showing CTC-detection (>5 CTC per 7.5 ml of blood) in only 49% of the breast cancer patients analyzed.1
The mean CTC detection rate per ml of our microfluidic device in cancer patients is comparable or even superior to what has been already reported in the literature by other authors using microfluidic devices. Nagrath et al. reported a mean CTC detection rate of 79 CTC ml−1 in breast cancer patients using their EpCAM-based chip;2 however, the authors did not describe extensively the clinical characteristics of the analyzed patients population and, consequently, it is not possible to evaluate the performance across the different clinical breast cancer subtypes. Yu et al. tested their herringbone CTC-chip coated with an anti-EpCAM/HER2/EGFR antibody cocktail and CTCs were isolated only from 17 out of 41 (41%) patients analyzed with a mean of 38 captured CTCs per 3 ml using their innovative RNA in situ hybridization (RNA-ISH) based staining.4 This value is lower compared to what we demonstrated for our HER2-based GEDI chip and a possible explanation could rely on a potential interference of the three antibodies against each other in CTC capture.
Circulating tumor cells were detected not only in HER2 high-expressing breast cancer patients (BC patient 1, 3 and 5 and GC patient 2) but also in HER2 low-expressing breast cancer patients (BC patient 4) (Fig. 5 and Table 2). The small number of patients analyzed here precludes firm conclusions regarding the correlation between the number of captured CTCs and HER2 positivity of the patients' primary tumors. Interestingly, CTC capture was observed in the triple negative breast cancer patient (BC patient 2) and the HER2 negative gastric cancer (GC patient 1). Importantly, in both gastric cancer patients, CTC counts before and during taxane chemotherapy (cabazitaxel 25 mg m−2) revealed a trend with the patients' clinical response to therapy. Specifically, GC patient 2 showed radiographic response to therapy, assessed by CT scan to quantify tumor burden, which was accompanied by a sharp decrease in CTC counts (from 224 to 37). In contrast, GC patient 1, who did not benefit from cabazitaxel therapy, experienced radiographic disease progression with no significant change in CTC counts (Fig. 5B).
In the case of GC patient 1, HER2 status was assessed on the tumor biopsy within 6 months of our CTC evaluation. The tumor was deemed HER2-negative, and the patient was ineligible for HER-targeted therapies. Our CTC results contradict the tumor evaluation and suggest that perhaps CTCs can be a better descriptor of the overall tumor burden as opposed to the tissue biopsy, which reflects only one of the metastatic sites and cannot capture tumor heterogeneity. In addition, our analyses revealed variable HER2 expression on CTCs suggesting that the HER2-GEDI chip and the accompanying CTC characterization can be used in the context of future clinical trials correlating HER2 expression on CTCs with clinical response to targeted therapies. Moreover, several groups have reported discordance between HER2 status of the primary site and CTCs in metastatic breast cancer.26 Our results are thus, in line with data reported in the literature and highlight the potential importance of HER2 analysis on CTCs as prognostic and predictive markers, in particular in those patients undergoing anti-HER2 treatment.
Other HER2-based devices have been described in the literature with the intent to isolate CTCs from peripheral blood of breast cancer patients. Several groups have reported the potential application of the anti-HER2 monoclonal antibody trastuzumab (Herceptin™) to isolate rare populations of breast cancer cells. Thierry et al. tested a PDMS microfluidic device coated with trastuzumab and the device proved effective when tested with HER2 overexpressing cell line (SK-BR3); however, this device was not tested with breast cancer patient samples therefore, its clinical application for CTC capture was not directly exploited.27 Similarly, Mi et al. have developed trastuzumab-coated nanoparticles able to bind and recognize HER2 overexpressing cells and proposed this strategy as suitable for CTCs isolation; however, even in this case, there were no data regarding CTC capture in breast cancer patients.28
A recent report showed CTC capture from the blood of metastatic breast cancer patients using a combination of EpCAM, HER2 and EGFR antibodies.4,29 However, the performance of each antibody individually in patient samples has not been assessed, therefore the benefit of having three versus one antibody for capture was not clinically tested. Moreover, the method reported by Pecot et al. requires extensive peripheral blood manipulation before CTC isolation leading potentially to a significant loss of rare cell populations like CTCs.
Our HER2-based GEDI microfluidic device does not require any blood processing as peripheral blood is directly analyzed through the device, thus minimizing potential CTC loss. In addition, our device utilizes only 1 ml of patient blood for CTC isolation – this volume is significantly lower than the blood volume required for other methods, such as 7.5 ml of blood for CellSearch or 10 ml of blood for MagSweeper – and as such it is much easier to incorporate into clinical trials.
We are currently testing the potential clinical application and usefulness of the HER2-GEDI microfluidic device in a large cohort of patients in the context of two clinical trials of patients with metastatic breast cancer or metastatic gastric cancer receiving chemotherapy containing a microtubule-targeting drug. We plan to monitor patient response to therapy in real-time and develop CTC-based biomarkers predictive of response.
Acknowledgements
This work was in part supported by the National Institute of Health (NCI U54 CA143876), by the Manhasset Women’s Coalition Against Breast Cancer (MWCABC) and by the Ann Moore Breast Cancer Fund.References
- M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, J. Matera, M. C. Miller, J. M. Reuben, G. V. Doyle, W. J. Allard, L. W. Terstappen and D. F. Hayes, N. Engl. J. Med., 2004, 351, 781–791 CrossRef CAS PubMed.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- P. Pinzani, B. Salvadori, L. Simi, S. Bianchi, V. Distante, L. Cataliotti, M. Pazzagli and C. Orlando, Hum. Pathol., 2006, 37, 711–718 CrossRef CAS PubMed.
- M. Yu, A. Bardia, B. S. Wittner, S. L. Stott, M. E. Smas, D. T. Ting, S. J. Isakoff, J. C. Ciciliano, M. N. Wells, A. M. Shah, K. F. Concannon, M. C. Donaldson, L. V. Sequist, E. Brachtel, D. Sgroi, J. Baselga, S. Ramaswamy, M. Toner, D. A. Haber and S. Maheswaran, Science, 2013, 339, 580–584 CrossRef CAS PubMed.
- S. Riethdorf, H. Fritsche, V. Muller, T. Rau, C. Schindlbeck, B. Rack, W. Janni, C. Coith, K. Beck, F. Janicke, S. Jackson, T. Gornet, M. Cristofanilli and K. Pantel, Clin. Cancer Res., 2007, 13, 920–928 CrossRef CAS PubMed.
- J. S. de Bono, H. I. Scher, R. B. Montgomery, C. Parker, M. C. Miller, H. Tissing, G. V. Doyle, L. W. Terstappen, K. J. Pienta and D. Raghavan, Clin. Cancer Res., 2008, 14, 6302–6309 CrossRef CAS PubMed.
- S. J. Cohen, C. J. Punt, N. Iannotti, B. H. Saidman, K. D. Sabbath, N. Y. Gabrail, J. Picus, M. Morse, E. Mitchell, M. C. Miller, G. V. Doyle, H. Tissing, L. W. Terstappen and N. J. Meropol, J. Clin. Oncol., 2008, 26, 3213–3221 CrossRef PubMed.
- A. M. Sieuwerts, J. Kraan, J. Bolt, P. van der Spoel, F. Elstrodt, M. Schutte, J. W. Martens, J. W. Gratama, S. Sleijfer and J. A. Foekens, J. Natl. Cancer Inst., 2009, 101, 61–66 CrossRef CAS PubMed.
- J. W. Kornfeld, S. Meder, M. Wohlberg, R. E. Friedrich, T. Rau, L. Riethdorf, T. Loning, K. Pantel and S. Riethdorf, Br. J. Cancer, 2011, 104, 138–145 CrossRef CAS PubMed.
- H. C. Lin, H. C. Hsu, C. H. Hsieh, H. M. Wang, C. Y. Huang, M. H. Wu and C. P. Tseng, Clin. Chim. Acta, 2013, 419, 77–84 CrossRef CAS PubMed.
- E. Ozkumur, A. M. Shah, J. C. Ciciliano, B. L. Emmink, D. T. Miyamoto, E. Brachtel, M. Yu, P. I. Chen, B. Morgan, J. Trautwein, A. Kimura, S. Sengupta, S. L. Stott, N. M. Karabacak, T. A. Barber, J. R. Walsh, K. Smith, P. S. Spuhler, J. P. Sullivan, R. J. Lee, D. T. Ting, X. Luo, A. T. Shaw, A. Bardia, L. V. Sequist, D. N. Louis, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Sci. Transl. Med., 2013, 5, 179ra147 CrossRef PubMed.
- J. P. Gleghorn, E. D. Pratt, D. Denning, H. Liu, N. H. Bander, S. T. Tagawa, D. M. Nanus, P. A. Giannakakou and B. J. Kirby, Lab Chip, 2010, 10, 27–29 RSC.
- B. J. Kirby, M. Jodari, M. S. Loftus, G. Gakhar, E. D. Pratt, C. Chanel-Vos, J. P. Gleghorn, S. M. Santana, H. Liu, J. P. Smith, V. N. Navarro, S. T. Tagawa, N. H. Bander, D. M. Nanus and P. Giannakakou, PLoS One, 2012, 7, e35976 CAS.
- D. J. Slamon, G. M. Clark, S. G. Wong, W. J. Levin, A. Ullrich and W. L. McGuire, Science, 1987, 235, 177–182 CAS.
- L. N. Klapper, M. H. Kirschbaum, M. Sela and Y. Yarden, Adv. Cancer Res., 2000, 77, 25–79 CrossRef CAS.
- C. J. Witton, J. R. Reeves, J. J. Going, T. G. Cooke and J. M. Bartlett, J. Pathol., 2003, 200, 290–297 CrossRef CAS PubMed.
- D. J. Slamon, B. Leyland-Jones, S. Shak, H. Fuchs, V. Paton, A. Bajamonde, T. Fleming, W. Eiermann, J. Wolter, M. Pegram, J. Baselga and L. Norton, N. Engl. J. Med., 2001, 344, 783–792 CrossRef CAS PubMed.
- C. E. Geyer, J. Forster, D. Lindquist, S. Chan, C. G. Romieu, T. Pienkowski, A. Jagiello-Gruszfeld, J. Crown, A. Chan, B. Kaufman, D. Skarlos, M. Campone, N. Davidson, M. Berger, C. Oliva, S. D. Rubin, S. Stein and D. Cameron, N. Engl. J. Med., 2006, 355, 2733–2743 CrossRef CAS PubMed.
- M. Hofmann, O. Stoss, D. Shi, R. Buttner, M. van de Vijver, W. Kim, A. Ochiai, J. Ruschoff and T. Henkel, Histopathology, 2008, 52, 797–805 CrossRef CAS PubMed.
- Y. Y. Janjigian, D. Werner, C. Pauligk, K. Steinmetz, D. P. Kelsen, E. Jager, H. M. Altmannsberger, E. Robinson, L. J. Tafe, L. H. Tang, M. A. Shah and S. E. Al-Batran, Ann. Oncol., 2012, 23, 2656–2662 CrossRef CAS PubMed.
- M. Ai, K. Liang, Y. Lu, S. Qiu and Z. Fan, Cancer Biol. Ther., 2013, 14, 237–245 CrossRef CAS PubMed.
- C. M. Perou, T. Sorlie, M. B. Eisen, M. van de Rijn, S. S. Jeffrey, C. A. Rees, J. R. Pollack, D. T. Ross, H. Johnsen, L. A. Akslen, O. Fluge, A. Pergamenschikov, C. Williams, S. X. Zhu, P. E. Lonning, A. L. Borresen-Dale, P. O. Brown and D. Botstein, Nature, 2000, 406, 747–752 CrossRef CAS PubMed.
- S. M. Santana, H. Liu, N. H. Bander, J. P. Gleghorn and B. J. Kirby, Biomed. Microdevices, 2012, 14, 401–407 CrossRef CAS PubMed.
- D. F. Hayes, M. Cristofanilli, G. T. Budd, M. J. Ellis, A. Stopeck, M. C. Miller, J. Matera, W. J. Allard, G. V. Doyle and L. W. Terstappen, Clin. Cancer Res., 2006, 12, 4218–4224 CrossRef CAS PubMed.
- M. Santisteban, J. M. Reiman, M. K. Asiedu, M. D. Behrens, A. Nassar, K. R. Kalli, P. Haluska, J. N. Ingle, L. C. Hartmann, M. H. Manjili, D. C. Radisky, S. Ferrone and K. L. Knutson, Cancer Res., 2009, 69, 2887–2895 CrossRef CAS PubMed.
- M. Pestrin, S. Bessi, F. Galardi, M. Truglia, A. Biggeri, C. Biagioni, S. Cappadona, L. Biganzoli, A. Giannini and A. Di Leo, Breast Cancer Res. Treat., 2009, 118, 523–530 CrossRef CAS PubMed.
- B. Thierry, M. Kurkuri, J. Y. Shi, L. E. Lwin and D. Palms, Biomicrofluidics, 2010, 4, 32205 CrossRef PubMed.
- Y. Mi, K. Li, Y. Liu, K. Y. Pu, B. Liu and S. S. Feng, Biomaterials, 2011, 32, 8226–8233 CrossRef CAS PubMed.
- C. V. Pecot, F. Z. Bischoff, J. A. Mayer, K. L. Wong, T. Pham, J. Bottsford-Miller, R. L. Stone, Y. G. Lin, P. Jaladurgam, J. W. Roh, B. W. Goodman, W. M. Merritt, T. J. Pircher, S. D. Mikolajczyk, A. M. Nick, J. Celestino, C. Eng, L. M. Ellis, M. T. Deavers and A. K. Sood, Cancer Discovery, 2011, 1, 580–586 CrossRef CAS PubMed.
- K. Subik, J. F. Lee, L. Baxter, T. Strzepek, D. Costello, P. Crowley, L. Xing, M. C. Hung, T. Bonfiglio, D. G. Hicks and P. Tang, Breast Cancer: Basic Clin. Res., 2010, 4, 35–41 Search PubMed.
- A. Mackay, N. Tamber, K. Fenwick, M. Iravani, A. Grigoriadis, T. Dexter, C. J. Lord, J. S. Reis-Filho and A. Ashworth, Breast Cancer Res. Treat., 2009, 118, 481–498 CrossRef CAS PubMed.
- S. Riethdorf, V. Muller, L. Zhang, T. Rau, S. Loibl, M. Komor, M. Roller, J. Huober, T. Fehm, I. Schrader, J. Hilfrich, F. Holms, H. Tesch, H. Eidtmann, M. Untch, G. von Minckwitz and K. Pantel, Clin. Cancer Res., 2010, 16, 2634–2645 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc51039e |
| This journal is © The Royal Society of Chemistry 2014 |