Microfluidics for single-cell genetic analysis
A. M.
Thompson
a,
A. L.
Paguirigan
b,
J. E.
Kreutz
a,
J. P.
Radich
b and
D. T.
Chiu
*a
aDepartment of Chemistry, University of Washington, Seattle, WA, USA. E-mail: chiu@chem.washington.edu; Fax: +1 206 685 8665; Tel: +1 206 543 1665
bClinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA, USA
First published on 31st March 2014
Abstract
The ability to correlate single-cell genetic information to cellular phenotypes will provide the kind of detailed insight into human physiology and disease pathways that is not possible to infer from bulk cell analysis. Microfluidic technologies are attractive for single-cell manipulation due to precise handling and low risk of contamination. Additionally, microfluidic single-cell techniques can allow for high-throughput and detailed genetic analyses that increase accuracy and decrease reagent cost compared to bulk techniques. Incorporating these microfluidic platforms into research and clinical laboratory workflows can fill an unmet need in biology, delivering the highly accurate, highly informative data necessary to develop new therapies and monitor patient outcomes. In this perspective, we describe the current and potential future uses of microfluidics at all stages of single-cell genetic analysis, including cell enrichment and capture, single-cell compartmentalization and manipulation, and detection and analyses.
Introduction
The sequencing of the human genome through the Human Genome Project (HGP) is a seminal moment in biology. But like many great discoveries, it has created even more questions and spurred research into areas of biology that were previously unknown. Work in proteomics, epigenetics, and posttranscriptional regulation, while significantly aided by the knowledge of the underlying genetic information, has demonstrated that the sequence of human genes alone is a basic framework onto which many layers of genetic regulation are applied. The disease-focused sequencing projects following the HGP, some of which capture multiple levels of genomic data such as The Cancer Genome Atlas, have enabled linking certain consistent genetic changes to specific diseases. However, it has also demonstrated that there is tremendous variation between individuals with similar diseases. Further research into the impact of this genetic information on disease has identified variation between cell populations within individuals. The ability to study this variation in depth will have significant implications for personalized medicine. Our knowledge of the extent to which intercellular variation plays a role in disease evolution and therapy outcome is currently limited by our inability to study small amounts of biological materials, down to the level of an individual cell.Intra-sample heterogeneity likely holds valuable clues for understanding human disease and the variability between the responses of patients with the same disease to a given therapy.1 A clearer picture of how heterogeneity within individuals affects their disease progression and treatment can be a valuable tool for designing therapeutic regimens and defining treatments for different conditions. Perhaps turning an acute condition into a manageable, but chronic, one would be less risky than attempting to cure the individual entirely, especially in the case of therapies that involve alkylating agents or other potential mutation-inducing treatments. Or perhaps, we might improve our ability to choose effective therapies for a given patient by adding heterogeneity information to risk-stratification criteria.
Over the last few decades, research methods for molecular analyses have improved in sensitivity and accuracy because of the technology developed in a wide range of fields, from enzymology to microfluidics. This has resulted in the possibility of studying smaller quantities of starting materials than those traditionally used, along with huge increases in the density and types of data produced. Basic and clinical molecular research laboratories now have the ability to study a range of genetic materials, from uncovering the identity and abundance of small RNAs via RNA sequencing to characterizing large chromosomal alterations via comparative hybridization arrays. The sensitivity increases in molecular techniques have also allowed us to identify the presence of low-frequency features that were not detectable previously. One issue hindering our ability to explore the biology of heterogeneous populations is that the amount of DNA or RNA required for most of the readily available in-depth genetic analysis methods is designed for bulk assays. These assays require nanogram or microgram quantities of genetic material, which is a considerable amount given the minute content of a single cell for which the total available material is in the order of picograms. Beyond the total input issues, the question of isolating and handling single-cell materials without contamination or sample loss poses yet another hurdle for molecular analyses of heterogeneity at the single-cell level.
Probing genetic materials at the level of a single cell will require new technologies to enhance capabilities and deliver accurate, actionable data for the wide range of questions being asked. Although new adaptations of macroscale methods are emerging to address these needs, the field of single-cell genetics requires a variety of fundamentally different strategies. Microfluidic technologies are in a unique position to address the limitations of current methods because they offer the benefits of both fluidic handling and thermal capabilities as well as flexibility in design, throughput and automation. In this perspective, we will discuss the scope and direction of scientific interests in single-cell genetics, highlight some of the ways microfluidics has proven useful in single-cell genetic analysis, and define areas where further improvement is needed.
One cell, many questions
Intra-sample genetic diversity, also known as clonal diversity, has diagnostic value in several diseases, such as predicting progression to malignancy in Barrett's esophagus.2 Clonal diversity has been demonstrated in breast cancer3 and occurs in acute myeloid leukemia (AML) from diagnosis to relapse.4–6 Current strategies for estimating and tracking clonal diversity at the macroscale have used next-generation sequencing (NGS) of bulk tumor samples to determine the frequencies of mutant alleles. Changes in mutant allele frequencies can be observed over the course of therapy (comparing diagnosis to relapse) and between primary tumor sites and metastases. While these mutant alleles can be quantitatively tracked over time and over the course of therapy in the bulk samples, the information about the specific disease-causing clone is lost in the background of all of the other cell types present in any patient sample. This is particularly challenging in samples where there is some ambiguity, such as in a biopsy of a tumor where the boundary of tumor versus normal tissue is not completely clear or in a peripheral blood sample where the amount of leukemic cells varies between patients.Rather than attempting to infer concurrence of different genetic characteristics seen in the averaged data from a bulk sample, being able to assess the genomic or expression characteristics of individual cells themselves can directly link the genotype and expression data that occur simultaneously in a cell. If a cell with a specific set of mutations doesn't actually express those alleles, or has other downstream regulatory changes that cause a different set of targets to be expressed or inhibited, targeting that pathway would incorrectly destroy the wrong cell types and potentially allow the rogue cell to continue to proliferate and cause relapse of disease. RNA and DNA extracted from bulk samples do provide a general description of the population average in the original sample, but it is impossible to reconstruct how the different populations may have contributed to that average. One can find correlations, for example, between mutational allele frequencies and the level of expression of RNAs downstream of that gene, but even this information does not inform whether these events occur concurrently in the same cells. When a population average is measured, the technique used requires a relatively large amount of starting materials to ensure there is enough to avoid sampling issues and stochastic variability in the results. These methods are often not validated in small amounts that would make integration with single-cell assays accurate or reasonable.
Additionally, the separation of measurement or technical variability from biological variability in each measurement platform can be challenging but is crucial for the validation of any single-cell assay where analyte amounts are near the limit of detection. Normalization strategies typically used in bulk measurements are not appropriate for single cells (i.e., technical variability in control genes during a qPCR experiment would cause normalization of the target gene measurement to be erroneous). For this reason, having suitable controls becomes an issue for the validation of single-cell data. Another challenge unique to single-cell molecular analyses is the issue of total sample size (or the total cell number analyzed per tissue sample). As for the degree of heterogeneity in a sample, when the technical variability inherent to the assay and the number of parameters analyzed increases, the number of cells that must be analyzed to describe the overall heterogeneity of a sample with statistical significance must rapidly increase. Available methods for the physical isolation and handling of individual cells for emerging and sensitive genetic analysis techniques limit the sample size because of their low throughput, high cost per cell, or high failure rates.
Currently, there are few powerful tools readily available to identify heterogeneity at the single-cell level. For decades we have been able to process very large populations (millions of cells analyzed per sample) and quickly identify frequencies of different cell types within a single sample by using a wide range of cell surface markers or intracellular stains. For example, flow cytometry is by far the most rapid, complex (multi-parameter) and immediate (protein-based) data producing tool available for single-cell analysis. However, molecular genetic analyses of single cells (both genomic and gene expression) have not had the benefit of decades of tools developed to analyze multiple features simultaneously in individual cells with high enough throughput or data complexity (multiple parameter data). Translating the discovery tools that are effective for bulk samples, such as large scale sequencing and other genetic analysis methods, to the single-cell level will be invaluable to further elucidate mechanisms of disease and how individual cells make choices and regulate their various processes.
Microfluidics as a solution
New methods for single-cell assays must provide the means to link genetic data to an individual cell's characteristics as well as address the major limitations for effective analysis. Specifically, they must: 1) provide the handling precision necessary to isolate and manipulate minute quantities of biological materials, 2) approach single-molecule sensitivity to eliminate bias due to amplification, 3) provide high accuracy as the same cell cannot be measured multiple times, 4) provide throughput high enough to efficiently generate statistically meaningful data, and 5) eliminate contamination from the environment and components within the sample. For a method to be successfully adopted into research and clinical settings, ease of use, integration with existing infrastructure, and cost are critical factors. Microfluidics has shown strong performance in these areas outside of the genetics arena. Research incorporating microfluidics and single-cell genetic analysis, including cell capture and enrichment, cell compartmentalization, and detection, can be used to create simple and more informative tools for single-cell study. Specific advantages to applying a microfluidic approach to this complex field are outlined in Fig. 1 and highlighted in the remainder of this paper. | ||
| Fig. 1 Advantages of microfluidics for single-cell genetic analysis. Microfluidic technologies offer advantages at various stages of single-cell genetic analysis. In this paper, the current and future applications of microfluidics to provide simple and informative analyses in this field are discussed. | ||
Capture and enrichment of single cells
Correlation of genetic data with its single cell of origin requires a method to isolate single cells from a tissue. Currently, methods for the selection and transfer of single cells into wells or tubes include laser capture microdissection, optical tweezer manipulation, micromanipulation, flow cytometry, or microfluidic methods. These methods differ in their equipment requirements, cost, degree of user skill, tissue compatibility, and throughput. Flow cytometry is attractive because of its multiparameter sorting and high throughput, but depositing cells into microliter volume wells results in dilution of analytes and does not allow the user to easily confirm that cells were deposited successfully into the analysis volume. Laser capture microdissection can provide certainty of cell isolation but at very low throughput.7 Enrichment and compartmentalization within a microfluidic platform can be designed for high throughput while minimizing dilution and contamination risk. A number of microfluidic techniques have been developed to address these needs.A variety of techniques have been used to indiscriminately trap single cells from a cell suspension in microfluidic systems. These methods include hydrodynamic mechanisms or use electrical, optical, magnetic or acoustic fields to control trapping. An extensive review of cell trapping methods is presented by Nilsson et al.8 To be integrated with downstream genetic analysis, these devices must be compatible with isolation, manipulation, and analysis or retrieval mechanisms. The commercial microfluidic C1™ Single-Cell Auto Prep System from Fluidigm uses hydrodynamic capture and isolation of single cells from suspension before cell lysis and processes the single-cell genetic material before retrieval and use with multiplex PCR, RT-PCR, or NGS methods. A disadvantage of this and other hydrodynamic trapping devices is that an excess of cells are needed for high trapping density, resulting in a loss of the majority of the single cells from the incoming sample. These methods are also unable to select specific types of cells in a sample, even those types that are fairly common. For many single-cell applications, a simple, straightforward device for trapping thousands of single cells, rather than hundreds, would expand the studies that could be executed with statistically significant data.
Some questions regarding single cells are focused on assessing genetic heterogeneity in only a small sub-population of single cells in a biological sample. Rare cells, usually thought of as having cellular abundance less than 0.1 percent, are relevant markers in cancer, prenatal diagnosis, and infectious disease. In order to gather sufficient data from patient samples, single-cell enrichment techniques typically must offer high sample throughput and yield high recoveries of target cells. If these enrichment devices are to be used upstream of cell trapping, isolation, manipulation and genetic analysis methods, the techniques should allow for high purity retrieval of viable cells. Circulating tumor cells (CTCs), an extraordinarily rare cell type, are present in quantities near one cell per 1 billion blood cells in patients with advanced stage cancer. Methods such as fluorescence activated cell sorting (FACS), magnetic activated cell sorting (MACS), and cell affinity separations are high-throughput and very effective for some applications, but they typically have low-recovery efficiency for CTCs and will deposit 0.1% of background cells with the target population. This background noise limits their applicability in cases such as genetic analysis of single CTCs.9 In contrast, microfluidic mechanisms for isolation of CTCs have been demonstrated that enrich these rare cells with high recovery. These techniques utilize laminar flow and/or high surface-to-volume ratios to operate. For example, antibodies can be used to immobilize specific cell types in static10 (Fig. 2A) or flow11 (Fig. 2B) systems, optical methods can be combined with valves to create CTC-containing aliquots of sample12 (Fig. 2C), and fabricated filters and flow focusing can be integrated upstream of labelled CTC separation and collection by magnetophoresis13 (Fig. 2D).
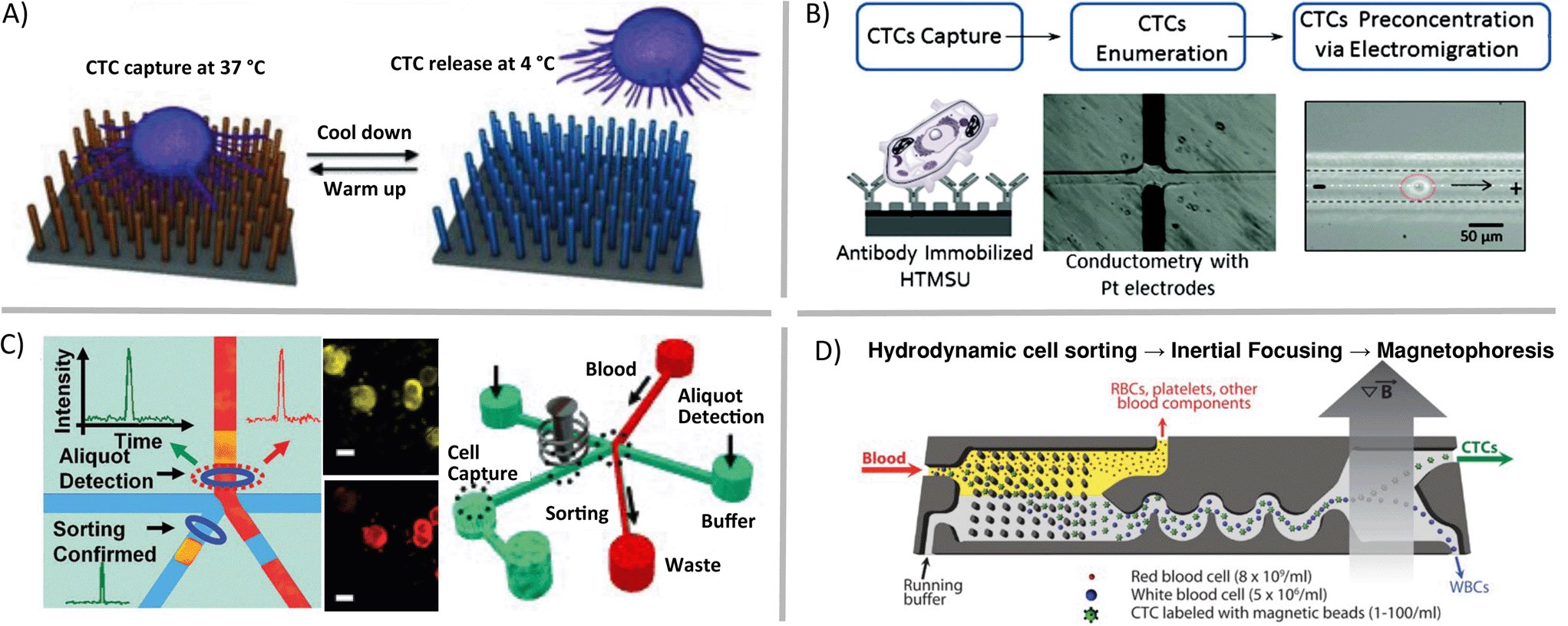 | ||
| Fig. 2 Microfluidic enrichment of rare cells. Circulating tumor cells (CTCs) can be enriched from whole blood using a variety of techniques including A) nanostructure- and antibody-based reversible surface capture [reprinted (adapted) with permission from ref. 10. Copyright 2013 Nature Publishing Group], B) affinity capture combined with electrokinetic enrichment [reprinted (adapted) with permission from ref. 11. Copyright 2011 American Chemical Society], C) microfluidic aliquot sorting of target cells combined with on-chip filtration [reprinted (adapted) with permission from ref. 12. Copyright 2012 Angew. Chem., Int. Ed.], and D) combined hydrodynamic–magnetic methods [reprinted (adapted) with permission from ref. 13. Copyright 2013 AAAS]. | ||
Compartmentalization
On its own, trapping cells is insufficient for single-cell analysis because the contents of those cells, once lysed, aren't necessarily isolated from each other. The cells also have to be compartmentalized in such a way as to maintain all biological materials from a single cell in an isolated fluidic space that is separate from other cells. There are a variety of methods that have been developed for the compartmentalization of cells for downstream analysis (Fig. 3). Valve-based systems can pneumatically isolate cells and often utilize traps upstream of the sample handling (Fig. 3A).14 Droplet generation systems can also be used to isolate cells, without the need for traps or valves, which can drastically simplify the device design/fabrication. These droplet systems also have the potential for much higher throughput than valve-based systems. However, droplet platforms typically rely on the partitioning of cells into droplets following a Poisson distribution, which can limit throughput due to a large number of droplets being empty and a few with two or more cells. If throughput is high enough, a sufficient rate of single-cell measurements can be obtained despite these uninformative droplets. Techniques also exist to selectively encapsulate single cells into individual droplets (Fig. 3B)15 or to “beat” Poisson statistics (Fig. 3C).16 | ||
| Fig. 3 Compartmentalization of single cells. Numerous microfluidic methods have been developed to form discrete aqueous volumes to spatially confine aqueous volumes on-chip or to trap single cells. A) A hydrodynamic single-cell trap creates spatial separation of single cells before compartmentalization using valves [reprinted (adapted) with permission from ref. 14. Copyright 2011 National Academy of Sciences, USA]. B) Example of selective encapsulation of single particles/cells in aqueous droplets [reprinted (adapted) with permission from ref. 15. Copyright 2005 American Chemical Society]. C) Single cells in suspension are manipulated in-flow before droplet generation, resulting in the majority of discrete volumes that contain a single-cell [adapted with permission from ref. 16]. | ||
Another approach is to isolate cells in chambers but without using valves. Typically systems that isolate chambers (containing cells, beads, or some other components) have the sample fully fill the device and then cap the chambers with oil or a physical barrier.17,18 Other systems are essentially extensions of droplet platforms but keep the volumes isolated using physical barriers rather than emulsion stabilizing surfactants. This maintains some of the advantages of droplet systems (reduced risk of fouling of the device surface and crosstalk) while facilitating the tracking of individual samples over an extended time period. Examples of systems that enable additional sample processing include the SlipChip,19 SD chip,20 some valve-based chips,21 and some hybrid emulsion/physical isolation systems.22
It is relevant to note that by isolating single cells into compartments, intercellular interactions and any effects of the native cell matrix on the genome are removed. Methods to probe such interactions in a controlled environment, through arrays that incorporate cell culture or media exchange prior to isolation and lysis, for instance, would both enhance our understanding of cellular processes and might validate the results of techniques studying cells in isolation. A recent review of microfluidic devices to probe cell–cell communication is given by Guo et al.23
Once isolated, performing cell lysis in these individual, microfluidic compartments minimizes exposure to contamination from other cells within the sample or from materials in the laboratory. Lysis methods should preserve the integrity of the genetic material, and chemical lysis methods should be compatible with downstream enzymatic reactions such as PCR. Also, stress induced cell-signalling that might alter transcript levels should be avoided. Methods for cell lysis include physical, chemical, thermal, and electrical techniques with varying lengths of time to lyse the cell and different design requirements for the microfluidic device. Each of these techniques has been utilized in microfluidic nucleic acid analysis devices reviewed by Kim et al.24
Analysis of single-cell genetic materials
Analyzing materials from a single cell is challenging in many regards. In the single cell, RNA is present in picogram quantities; some low abundance RNA transcripts are present in 1–10 copies.7 While qPCR and qRT-PCR theoretically are able to amplify and detect single-molecule quantities of nucleic acids, it is challenging to quantify low quantities in microliter-volume reactions. For one, amplification bias causes copy number uncertainty.15 Additionally, the qPCR signal is analog, requiring “real time” monitoring and signal calibration25 or internal reference standards for relative gene expression that are complicated by the stochastic nature of gene expression at the single-cell level.26 Whole-genome or whole-transcriptome amplification is a requisite for single-cell analysis using NGS platforms. But in the process of amplification, information about the spatial arrangement of sequences, copy number variation, or relative gene expression is not fully conserved due to variations in amplification efficiency and transcript length limitations. Additionally, while NGS provides a huge amount of data per cell, it is currently cost-prohibitive to perform NGS on sufficient numbers of cells to describe a population or to describe the contribution of measurement/technical error in any statistically relevant way.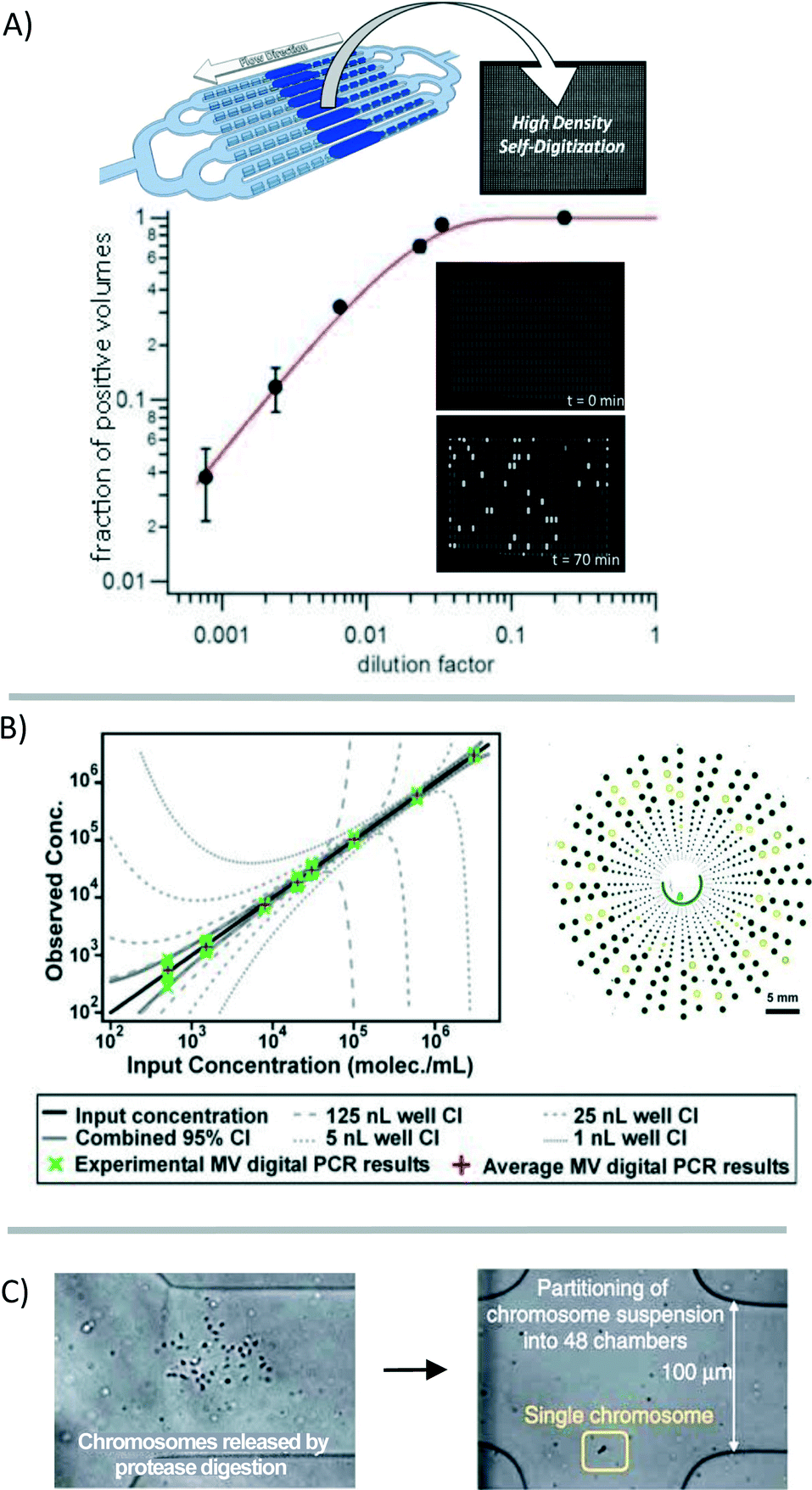 | ||
| Fig. 4 Digitization of the genetic material for analysis. (A) The genetic material in a sample can be broken into many small volumes before gene-specific amplification [reprinted (adapted) with permission from ref. 27. Copyright 2013 American Chemical Society]. Signal accumulation allows for the counting of positive reactions. The fraction of positive volumes correlates to the absolute copy number without the need for a reference standard [adapted from ref. 28]. (B) Multi-volume (MV) digital PCR can reduce the number of volumes necessary to achieve a high dynamic range [reprinted (adapted) with permission from ref. 29. Copyright 2011 American Chemical Society]. (C) Single chromosomes are partitioned to preserve haplotype information through multiple strand displacement and sequencing [reprinted (adapted) with permission from ref. 17. Copyright 2011 Nature Publishing Group]. | ||
Despite improvements in the accuracy, sensitivity, and reproducibility of digital PCR for the quantification of DNA, gene expression analysis has not achieved the same performance standards. Digital RT-PCR requires a reverse-transcription step for the construction of a cDNA library before preamplification and digitization into microfluidic volumes. This reverse transcription step is known to suffer from variations in efficiency between transcripts.25,26,30 Variations in sample preparation steps have vastly different results in digital RT-PCR;25 although with consistent sample preparation, results may be highly reproducible between measurements.30 Performing reverse-transcription and preamplification steps in smaller volumes may offer some reduction in amplification bias. In one study comparing various methods of single-cell preparation, performing these enzymatic reactions in nanoliter, rather than in microliter, volumes resulted in the best correlation between quantitative sequencing and PCR results.30
Digitization of the sample volume derived from a single cell can also be useful for preserving haplotype information. Typically, information about co-localization of mutations on a single chromosome is lost because of limitations in product length that result from enzymatic nucleic-acid amplification. Fan et al. showed that by using a microfluidic device to compartmentalize the individual chromosomes of a single cell, this information is preserved during downstream analysis (Fig. 4C).17
Future outlook
Despite improvements in single-cell genetic analysis capabilities, further improvements in single-cell handling, enrichment, and analysis techniques are necessary for these methods to make an impact on our understanding of biology. There are a number of needs that are priorities for generating a strong set of single-cell data for human biology research. Microfluidic device designs that address these issues early on in their development will be far more likely to allow researchers to access a broader range of single-cell characteristics in a statistically meaningful way.Strategies for manipulation and interrogation of single cells should aim to improve upon information accuracy, amount of information obtained per cell, and single-cell throughput. Performing whole genome amplification in small volumes has already been shown to better preserve relative gene abundance for more accurate gene quantification, and future analysis systems requiring whole genome amplification should continue to use microfluidic volumes for these operations. Accuracy of single-cell genetic analysis systems will also be improved if whole genome amplification is limited or avoided, which may be possible using innovative single-molecule detection strategies that take advantage of minimal dilution offered by microfluidic systems. Future methods to increase the amount of information per cell might come in the form of incorporating increasingly accurate whole genome amplification with highly informative NGS. The ability to integrate multiple manipulation operations and analytical detection strategies on a single microfluidic device could also lead to complex systems generating data on multiple gene targets or multiple macromolecule types. Currently, the throughput of microfluidic single-cell genetic analysis systems has been limited to hundreds of single-cells. Future microfluidic designs should explore avenues to decrease the number of cells wasted during trapping and compartmentalization, increase the density of single cell arrays, and ultimately increase the number of single cells analysed per device. As always, ease of use, cost, and analysis time should be considered for any technology moving towards commercialization.
Beyond the platforms employed for isolating and analyzing single cells, additional computational methods will be crucial for researchers to address technical variation and identify the degree of significance of any biological variation detected. Large data sets (as from single-cell sequencing data sets),35 and smaller, more focused data sets (as from digital PCR and RT-PCR assays),36 will need slightly different validation strategies. Ideally, the integration of multiple data types originating from the same single cells will be possible (for example, cell surface markers with gene expression with genotyping). The generation and curation of single-cell data sets from both normal and diseased human tissues would provide a valuable understanding of the types of variation that are normal in human development and those that are hallmarks of disease evolution and progression.
Acknowledgements
We gratefully acknowledge support from the National Institutes of Health (R01CA175215).Notes and references
- Q. F. Wills, K. J. Livak, A. J. Tipping, T. Enver, A. J. Goldson, D. W. Sexton and C. Holmes, Nat. Biotechnol., 2013, 31, 748 CrossRef CAS PubMed.
- L. M. Merlo, N. A. Shah, X. Li, P. L. Blount, T. L. Vaughan, B. J. Reid and C. C. Maley, Cancer Prev. Res., 2010, 3, 1388–1397 CrossRef PubMed.
- S. Y. Park, M. Gonen, H. J. Kim, F. Michor and K. Polyak, J. Clin. Invest., 2010, 120, 636–644 CAS.
- L. Ding, T. J. Ley, D. E. Larson, C. A. Miller, D. C. Koboldt, J. S. Welch, J. K. Ritchey, M. A. Young, T. Lamprecht, M. D. McLellan, J. F. McMichael, J. W. Wallis, C. Lu, D. Shen, C. C. Harris, D. J. Dooling, R. S. Fulton, L. L. Fulton, K. Chen, H. Schmidt, J. Kalicki-Veizer, V. J. Magrini, L. Cook, S. D. McGrath, T. L. Vickery, M. C. Wendl, S. Heath, M. A. Watson, D. C. Link, M. H. Tomasson, W. D. Shannon, J. E. Payton, S. Kulkarni, P. Westervelt, M. J. Walter, T. A. Graubert, E. R. Mardis, R. K. Wilson and J. F. DiPersio, Nature, 2012, 481, 506–510 CrossRef CAS PubMed.
- M. Jan, T. M. Snyder, M. R. Corces-Zimmerman, P. Vyas, I. L. Weissman, S. R. Quake and R. Majeti, Sci. Transl. Med., 2012, 4, 149ra118 Search PubMed.
- J. S. Welch, T. J. Ley, D. C. Link, C. A. Miller, D. E. Larson, D. C. Koboldt, L. D. Wartman, T. L. Lamprecht, F. Liu, J. Xia, C. Kandoth, R. S. Fulton, M. D. McLellan, D. J. Dooling, J. W. Wallis, K. Chen, C. C. Harris, H. K. Schmidt, J. M. Kalicki-Veizer, C. Lu, Q. Zhang, L. Lin, M. D. O'Laughlin, J. F. McMichael, K. D. Delehaunty, L. A. Fulton, V. J. Magrini, S. D. McGrath, R. T. Demeter, T. L. Vickery, J. Hundal, L. L. Cook, G. W. Swift, J. P. Reed, P. A. Alldredge, T. N. Wylie, J. R. Walker, M. A. Watson, S. E. Heath, W. D. Shannon, N. Varghese, R. Nagarajan, J. E. Payton, J. D. Baty, S. Kulkarni, J. M. Klco, M. H. Tomasson, P. Westervelt, M. J. Walter, T. A. Graubert, J. F. DiPersio, L. Ding, E. R. Mardis and R. K. Wilson, Cell, 2012, 150, 264–278 CrossRef CAS PubMed.
- V. Sanchez-Freire, A. D. Ebert, T. Kalisky, S. R. Quake and J. C. Wu, Nat. Protoc., 2012, 7, 829–838 CrossRef CAS PubMed.
- J. Nilsson, M. Evander, B. Hammarstrom and T. Laurell, Anal. Chim. Acta, 2009, 649, 141–157 CrossRef CAS PubMed.
- Y. Gao, W. Li and D. Pappas, Analyst, 2013, 138, 4714–4721 RSC.
- S. Hou, H. Zhao, L. Zhao, Q. Shen, K. S. Wei, D. Y. Suh, A. Nakao, M. A. Garcia, M. Song, T. Lee, B. Xiong, S. C. Luo, H. R. Tseng and H.-h. Yu, Adv. Mater., 2013, 25, 1547–1551 CrossRef CAS PubMed.
- U. Dharmasiri, S. K. Njoroge, M. A. Witek, M. G. Adebiyi, J. W. Kamande, M. L. Hupert, F. Barany and S. A. Soper, Anal. Chem., 2011, 83, 2301–2309 CrossRef CAS PubMed.
- P. G. Schiro, M. Zhao, J. S. Kuo, T. Schneider, K. M. Koehler, D. E. Sabath and D. T. Chiu, Angew. Chem., Int. Ed., 2012, 51, 4618–4622 CrossRef CAS PubMed.
- E. Ozkumur, A. M. Shah, J. C. Ciciliano, B. L. Emmink, D. T. Miyamoto, E. Brachtel, M. Yu, P. Chen, B. Morgan, J. Trautwein, A. Kimura, S. Sengupta, S. L. Stott, N. M. Karabacak, T. A. Barber, J. R. Walsh, K. Smith, P. S. Spuhler, J. P. Sullivan, R. J. Lee, D. T. Ting, X. Luo, A. T. Shaw, A. Bardia, L. V. Sequist, D. N. Louis, S. Maheswaran, R. Kapur, D. A. Haber and M. Toner, Sci. Transl. Med., 2013, 5, 179ra47 CAS.
- A. K. White, M. Vanlnsberghe, O. I. Petriv, M. Hamidi, D. Sikorski, M. A. Marra, J. Piret, S. Aparicio and C. L. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13999–14004 CrossRef CAS PubMed.
- M. He, J. S. Edgar, G. D. M. Jeffries, R. M. Lorenz, J. P. Shelby and D. T. Chiu, Anal. Chem., 2005, 77, 1539–1544 CrossRef CAS PubMed.
- J. F. Edd, D. D. Carlo, K. J. Humphry, S. Köster, D. Irimia, D. A. Weitz and M. Toner, Lab Chip, 2008, 8, 1262–1264 RSC.
- C. H. Fan, J. Wang, A. Potanina and S. R. Quake, Nat. Biotechnol., 2011, 29, 51–56 CrossRef PubMed.
- D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang, A. J. Rivnak, E. P. Ferrell, J. D. Randall, G. K. Provuncher, D. R. Walt and D. C. Duffy, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed.
- W. B. Du, L. Li, K. P. Nichols and R. F. Ismagilov, Lab Chip, 2009, 9, 2286–92 RSC.
- D. E. Cohen, T. Schneider, M. Wang and D. T. Chiu, Anal. Chem., 2010, 82, 5707–5717 CrossRef CAS PubMed.
- K. Leung, H. Zahn, T. Leaver, K. M. Konwar, N. W. Hanson, A. P. Pagé, C. C. Lo, P. S. Chain, S. J. Hallam and C. L. Hansen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7665–7670 CrossRef CAS PubMed.
- C. H. J. Schmitz, A. C. Rowat, S. Köster and D. A. Weitz, Lab Chip, 2009, 9, 44–49 RSC.
- F. Guo, J. B. French, P. Li, H. Zhao, C. Y. Chan, J. R. Fick, S. J. Benkovic and T. J. Huang, Lab Chip, 2013, 13, 3152–3162 RSC.
- J. Kim, M. Johnson, P. Hill and B. K. Gale, Integr. Biol., 2009, 1, 574–586 RSC.
- R. Sanders, D. J. Mason, C. A. Foy and J. F. Huggett, PLoS One, 2013, 8, e75296 CAS.
- A. Raj and A. van Oudenaarden, Annu. Rev. Biophys., 2009, 38, 255–70 CrossRef CAS PubMed.
- T. Schneider, G. S. Yen, A. M. Thompson, D. R. Burnham and D. T. Chiu, Anal. Chem., 2013, 85, 10417–10423 CrossRef CAS PubMed.
- A. Gansen, A. M. Herrick, I. K. Dimov, L. P. Lee and D. T. Chiu, Lab Chip, 2012, 12, 2247–2254 RSC.
- J. E. Kreutz, T. Munson, T. Huynh, F. Shen, W. Du and R. F. Ismagilov, Anal. Chem., 2011, 83, 8158–8168 CrossRef CAS PubMed.
- A. R. Wu, N. F. Neff, T. Kalisky, P. Dalerba, B. Treutlein, M. E. Rothenberg, F. M. Mburu, G. L. Mantalas, S. Sim, M. F. Clarke and S. R. Quake, Nat. Methods, 2014, 11, 41–46 CrossRef CAS PubMed.
- Y. Marcy, T. Ishoey, R. S. Lasken, T. B. Stockwell, B. P. Walenz, A. L. Halpern, K. Y. Beeson, S. M. Goldberg and S. R. Quake, PLoS Genet., 2007, 3, 1702–1708 CAS.
- S. Picelli, A. K. Bjorklund, O. R. Faridani, S. Sagasser, G. Winberg and R. Sandberg, Nat. Methods, 2013, 10, 1096–1098 CrossRef CAS PubMed.
- G. K. Geiss, R. E. Bumgarner, B. Birditt, T. Dahl, N. Dowidar, D. L. Dunaway, H. P. Fell, S. Ferree, R. D. George, T. Grogan, J. J. James, M. Maysuria, J. D. Mitton, P. Oliveri, J. L. Osborn, T. Peng, A. L. Ratcliffe, P. J. Webster, E. H. Davidson, L. Hood and K. Dimitrov, Nat. Biotechnol., 2008, 26, 317–325 CrossRef CAS PubMed.
- T. Matsunaga, M. Hosokawa, A. Arakaki, T. Taguchi, T. Mori, T. Tanaka and H. Takeyama, Anal. Chem., 2008, 80, 5139–5145 CrossRef CAS PubMed.
- S. Yilmaz and A. K. Singh, Curr. Opin. Biotechnol., 2012, 23, 437–443 CrossRef CAS PubMed.
- A. McDavid, G. Finak, P. K. Chattopadyay, M. Dominguez, L. Lamoreaux, S. S. Ma, M. Roederer and R. Gottardo, BMC Bioinf., 2013, 29, 461–467 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |