Continuous microcarrier-based cell culture in a benchtop microfluidic bioreactor†
F.
Abeille
abcd,
F.
Mittler
abcd,
P.
Obeid
acd,
M.
Huet
ab,
F.
Kermarrec
acd,
M. E.
Dolega
acd,
F.
Navarro
ab,
P.
Pouteau
ab,
B.
Icard
ab,
X.
Gidrol
acd,
V.
Agache‡
*ab and
N.
Picollet-D'hahan‡
acd
aUniv. Grenoble Alpes, F-38000 Grenoble, France
bCEA, LETI, MINATEC Campus, 17 rue des Martyrs, F-38054 Grenoble Cedex 9, France. E-mail: Vincent.agache@cea.fr
cCEA, iRTSV-BGE, F-38000 Grenoble, France
dINSERM, BGE, F-38000 Grenoble, France
First published on 27th June 2014
Abstract
Microfluidic bioreactors are expected to impact cell therapy and biopharmaceutical production due to their ability to control cellular microenvironments. This work presents a novel approach for continuous cell culture in a microfluidic system. Microcarriers (i.e., microbeads) are used as growth support for anchorage-dependent mammalian cells. This approach eases the manipulation of cells within the system and enables harmless extraction of cells. Moreover, the microbioreactor uses a perfusion function based on the biocompatible integration of a porous membrane to continuously feed the cells. The perfusion rate is optimized through simulations to provide a stable biochemical environment. Thermal management is also addressed to ensure a homogeneous bioreactor temperature. Eventually, incubator-free cell cultures of Drosophila S2 and PC3 cells are achieved over the course of a week using this bioreactor. In future applications, a more efficient alternative to harvesting cells from microcarriers is also anticipated as suggested by our positive results from the microcarrier digestion experiments.
Introduction
Cell culture is performed in incubators to maintain sterility, proper temperature, pH, and osmolality. However, observations and manipulations (media replenishment, sub-culture) of cells are performed by operators. This stage is time-consuming, often leads to variable and poorly reproducible results and increases the risks of contamination.With the development of microfabrication and microfluidic technologies, a large panel of cell culture devices has emerged,1 and potential systems capable of performing integrated characterizations of cell growth2 based on MEMS resonant sensors have been investigated.2–5 Small-scale culture devices offer advantages over standard cell culture and large-scale bioreactors. They are able to better address the cell microenvironment, can be automated and can be integrated with measuring elements that can provide quantitative data.6 These advantages have driven interests in bioprocessing for biopharmaceutical industries (e.g., recombinant protein, vaccine production).7,8 Several successive steps are involved in bioproduction.9 First, a clone selection is performed to discard the cells that would not produce the bioproduct of interest. The remaining clones are separately expanded using increasingly larger reactors: from shake flasks to bench-scale reactors to pilot-plant installations. However, some issues arise when scaling-up production as the clones cultivated in a shake flask may behave differently from those grown in production-scale reactors. Therefore, as the size of the reactor increases, some clones cease being productive and the cultivation has to be stopped. The use of small-scale bioreactors that better mimic the conditions in which clones are cultivated in the reactors used at larger scale would address these issues and optimize the ramping up of bioproduction processes.7,10
Within that framework, Lee et al.11 developed an interesting microfluidic system for semi-continuous bacterial cell culture. A plug of fresh medium is injected into the culture area, while the same amount of medium with cells is simultaneously delivered out of the system. Then, inlets and outlets are closed and mixing to homogenize the culture is achieved through a peristaltic pump. The system also affords control over the temperature, dissolved oxygen level, and cell density. Although bacteria are the most commonly used type of organism for bioproduction, a non-negligible amount of products can only be synthesized by mammalian cells due to their higher level of expression.12,13 To address this issue, Wheeler et al.14 miniaturized a standard mammalian cell culture based on electrowetting on dielectrics (EWOD) technology. This system offers high screening possibilities and an advantage in harvesting cells without interrupting the culture. Only a few other examples of mammalian cell culture systems have implemented the latter function using traditional proteolytic enzymes (e.g., trypsin).15–17 However, the use of such treatments for cell harvest remains questionable because these treatments may damage the cell membranes.18 Other strategies based on hydrodynamic shear stress and thermosensitive polymers19 or on only hydrodynamic shear stress20,21 have been combined with microfluidics to detach mammalian cells. However, these methods increase the complexity of the process fabrication and system handling. One final way to provide cell growth while enabling cell collection is to use solid or porous microcarriers22 or microencapsulation.23 Solid microcarriers are preferable because the cells can be recovered more easily. Similarly, in large-scale cultures, microcarriers are extensively used because in addition to providing higher culture surfaces, they allow for easy cell collection and sub-cultures free of proteolytic agents, thus limiting the need for quality control and handling steps.24 However, to the best of our knowledge, the combination of microcarriers and microfluidics in the continuous culture of cells has not yet been described.
Here, we combine lab-on-a-chip and microcarrier technologies to overcome the aforementioned drawbacks. Therefore, a novel microfluidic cell culture system that combines a perfusion function for continuous media renewal and the use of microcarriers for cell growth support was developed. Perfusion is performed through a porous membrane. Finite element method (FEM) simulations are performed to optimize this perfusion to replenish the entire culture volume. Microcarriers easily enable cells to be harvested from the system because they can be manipulated through fluid transport, which prevents the use of proteolytic enzymes or other harmful techniques. Eventually, this cell culture system has the ability to perform continuous benchtop cell culture outside an incubator, which is not achievable by most of the current cell culture systems. Indeed, most of these systems are made from PDMS, a material that is porous to water vapor and gas, which subsequently results in rapid media drying and media pH drift if not used within a CO2-regulated incubator.
Materials and methods
System design and fabrication
The bioreactor is composed of a cell culture card (CCC) mounted on a homemade aluminum holder (Fig. 1A).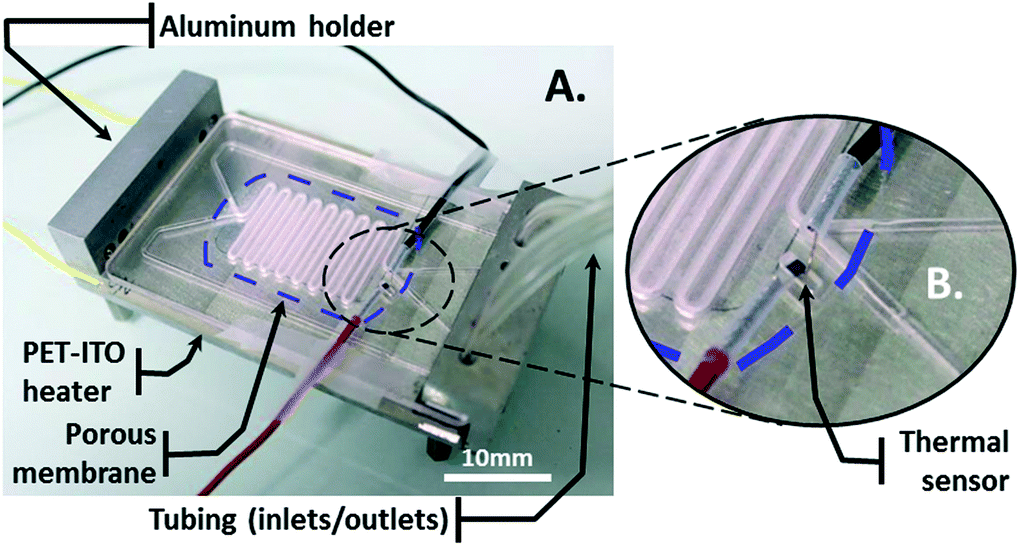 | ||
| Fig. 1 Device design and fabrication. A) Image of the PMMA fabricated device. The culture card is mounted onto an aluminum holder. The heating element is inserted between the holder and the card. A thermal sensor is integrated close to the culture area at the same height that the cells will be cultured. B) Enlarged view of thermal sensor integration. A trench is milled to glue the sensor inside. | ||
The holder is composed of a clamping system to hermetically connect the tubing to the CCC. A PET-ITO heater, which consists of a 200-μm-thick polyethylene terephthalate (PET) sheet coated with 400 nm of indium tin oxide (ITO), is inserted between the CCC and the holder. A silver-filled epoxy glue (EPO-TEK®, H20E, Epoxy Technology, Bussy-Saint-Georges, France) is deposited sideways along the length of the sheet onto the ITO layer to create the electrodes. A thermal sensor is integrated on top of the CCC in a small trench, which is covered with glue, for thermal read-out (Fig. 1B).
The CCC is composed of two channels at different levels. These channels meet and face each other in the area where they have a snake-like shape. There, a porous membrane with 0.4-μm-diameter pores (BGCM00010, Millipore) is embedded in glue and separates the channels (Fig. 2A and B).
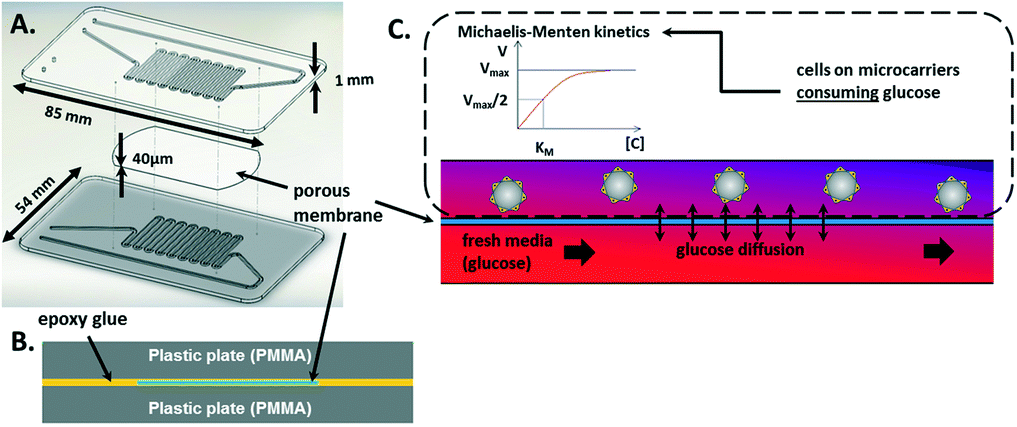 | ||
| Fig. 2 Device design and fabrication. A) Exploded view of the CCC illustrating the assembly of the device: a porous membrane is inserted between two PMMA plates, where the channels are milled. B) Schematic of the embedded membrane. C) Working principle of the culture area and the model considered for the media-feeding simulation. The upper channel contains cells that consume glucose in the entire volume, while fresh medium flows into the lower channel to supply glucose by diffusion through the porous membrane. | ||
This configuration guarantees that cells in one channel are confined while nutrients can be provided from the other channel by diffusion through the membrane pores (Fig. 2C). Fresh medium is dispensed using a syringe-pump set-up.
The fabrication process of the CCC consists of the following steps: the two PMMA plates are milled on one face by a milling machine (Charly4U, Mecanumeric) to create 1 mm-wide channels with a depth of 500 μm. Then, epoxy glue (EPO-TEK® OG116-31, Epoxy Technology, Bussy-Saint-Georges, France) is deposited on one PMMA plate face presenting the channel structures using a screen-printing machine (Ekra M2H, ASYS). The porous membrane is placed on the “snake-like” channel area. Glue is deposited on the other PMMA plate on the milled face. It must be noted that no glue is deposited on the channel walls due to the configuration of the screen-printing process and the small width of the channel structures (Fig. S1†). Both glued areas are faced and then pressed together by inserting the plates in a plastic bag in a vacuum pack (Boxer 35, HENKELMAN Vacuum Systems). The bag is exposed to UV light (100 mW cm−2, 365 nm) for 2 min to cross-link the glue, which completes the sealing of the device.
Cell culture
Mouse embryonic fibroblast NIH 3T3 cells (ref. CRL-1658, ATCC) were cultured in a 5% CO2 incubator (Incusafe, Sanyo) in 75 cm2 tissue culture flasks (Falcon, BD Biosciences) containing supplemented culture medium: red phenol DMEM (Dulbecco's Modified Eagle Medium) with 10% (v/v) newborn calf serum and 1% penicillin/streptomycin (P/S). The medium was changed every other day. Once cells reached 70% confluence, they were rinsed with Ca2+- and Mg2+-free phosphate-buffered saline (PBS) solution and sub-cultured using trypsin-EDTA solution for 4 min at 37 °C. All of the cell culture reagents were purchased from Life Technologies™ unless stated otherwise.
Human prostate cancer PC3 cells (ref. CRL-1658, ATCC) were cultured in a 5% CO2 incubator (Incusafe, Sanyo) in 75 cm2 tissue culture flasks (Falcon, BD Biosciences) containing supplemented culture medium: red phenol F-12K Medium (Kaighn's Modification of Ham's F-12 Medium) with 10% (v/v) fetal bovine serum and 1% P/S. The medium was changed every other day. Once cells reached 70% confluence, they were rinsed with Ca2+- and Mg2+-free PBS solution and sub-cultured using trypsin-EDTA solution for 10 min at 37 °C.
Microcarrier preparation
Cytodex 3 microbeads (ref. C3275, Sigma) with a final mean diameter of 175 μm later served as cell (PC3 or NIH 3T3) supports for adhesion. The microbeads were hydrated with a solution of 0.2% Tween-80 (Sigma) in PBS, and 24 h later, the beads were autoclaved (Systec VE-55, Fisher Scientific) at 120 °C for 1 h. After sterilization, the microcarriers were washed twice and stored in PBS.Cell proliferation on microcarriers
The influence that cell culture on microcarriers has on cell proliferation was studied. PC3 cells were seeded at a density of 40 cells mm−2 either on the surface of standard Petri dishes treated for cell culture or on microcarriers in non-cell culture-treated Petri dishes to force the cells to adhere only to the microcarriers. A microcarrier density of 5 mg mL−1 was used. Triplicate samples were performed for each condition. The cell proliferation rate was determined by counting the cells daily for approximately 4 days.Bioreactor set-up
The entire system was sanitized prior to performing bioreactor cell culture. The CCC was incubated at 100 °C for 4 h. The outer sides of the tubing (Tygon®, Cole-Parmer) and bioreactor were thoroughly cleaned with 70% (v/v) ethanol. The inner side of the tubing was flushed with the same solution for 10 min. The tubing and CCC were rinsed with 1% (v/v) P/S in PBS for 10 min.Mammalian cell culture requires a temperature of 37 °C. Heating is achieved by Joule dissipation of an electrical current flowing through the 400 nm resistive ITO layer. Its regulation is achieved by adjusting the voltage from the power supply connected to the PET-ITO heater according to the temperature recorded by the integrated thermal sensor. The homogeneity and temperature levels were characterized using an infrared camera (FLIR A20).
PC3 prostate cancer cells were first seeded at 2.4 × 104 cells mL−1 and incubated on the microcarriers (Cytodex 3) at 2 mg mL−1 for 24 h in 2 mL of culture medium inside a non-cell-culture-treated Petri dish to force the cells to adhere only to the microcarriers. Drosophila S2 cells were prepared at 2.4 × 104 cells mL−1. Approximately 250 μL of either culture was withdrawn to fill the upper channel of the CCC with cells. The inlet and outlet of this channel were closed to begin the perfusion of culture media into the lower channel. The perfusion rate was adapted from studies that simulated feeding for NIH 3T3 cells. Images were captured daily with Motic Image Plus software using a camera (Moticam 1000) mounted on a stereomicroscope (Lumar V12, Zeiss).
Media feeding simulation
Finite element simulations were performed (COMSOL, Multiphysics 4.3a) to determine the perfusion flow rate to maintain the cell culture. The stationary 2D model that was built is representative of the cross section along the snake-like channels where media renewal occurs (Fig. 2C). The “Porous media” and “Diluted species” modules from COMSOL were used as the governing physics in this model, where the Supplementary Table (ESI†) summarizes the set parameter values.In both channels, the initial concentration was fixed at 25 mM, as referenced by the culture medium formulation.
In the upper channel, the glucose consumption due to the presence of cells was based on a Michaelis–Menten kinetic law (see eqn (1)):
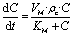 | (1) |
A no-flow condition was imposed on the upper channel inlet and outlet. The flow rate of the lower channel inlet was varied to simulate fresh media feeding, where a zero pressure condition was applied to the outlet. Computing at each flow rate allowed the glucose concentration distribution over the channel that contained the cells to be studied. Additional details about the model and the physics used to construct the simulations are provided in Fig. S2.†
Transmembrane diffusion
After fabricating the CCC, the ability of the membrane to enable transmembrane transport was evaluated. A red solution was prepared by diluting 300 μL of a red food dye (azorubin E122, McCormick) in 50 mL of PBS. The red solution was perfused into the lower channel of the device at the same rate as the color-free PBS solution, which was only dispensed into the upper channel. The solution flow rates were 10, 30, 100, 300, and 1000 μL min−1. For each flow rate, the liquids were collected at the outlets of the device, and 200 μL of the collected solutions were dispensed in a 96-well plate and analyzed using a microplate reader (Infinite M1000, Tecan) to measure their absorbance level.Cell counting
Dextranase (ref. D0443, Sigma) at 1% (v/v) was added and incubated in the culture medium that contained the cells on the microcarriers for 20 min to completely dissolve the beads. Dissolution was evaluated by microscopic observation. Cell clusters were centrifuged (300g, 5 min, 20 °C) and washed twice with PBS and then immersed in a trypsin-EDTA solution for 5 min to obtain single floating cells. Eventually, the cells were centrifuged and immersed in culture medium before being counted. The number of cells was determined using an automated cell counter (Millipore).Immunostaining
Cells grown on microcarriers were characterized by immunostaining of actin filaments after 4 days of culture. Cells that adhered to the microcarriers were fixed for 20 min using 4% (v/v) paraformaldehyde (PFA) in Tris-buffered saline (TBS) solution followed by 2 washes with phosphate-buffered saline solution supplemented with Mg2+ and Ca2+ (PBS+). Then, the cells were permeabilized for 10 min with 0.5% Triton X-100 in TBS followed by two 10-min washes with PBS+. Actin filaments were stained for 40 min with phalloidin tetramethylrhodamine B isothiocyanate (Jackson ImmunoResearch) diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :300 in 0.1% (v/v) Tween-20 (Sigma) in TBS, followed by a 10-min wash with PBS+. Nuclei were counterstained for 5 min with Hoechst (Jackson ImmunoResearch) diluted 1:7000 in PBS+. A final 10-min wash was performed with PBS+. A Gene Frame® (ThermoFisher Scientific) was used to create a thin-height well (250 μm) on a standard microscope glass slide. Stained cells that adhered to the microcarriers were transferred into this well, and a coverslip was mounted using the Gene Frame® with a fluorescence mounting medium kit (Dako) and stored at 4 °C before imaging. All reagents were purchased from Life Technologies™ unless stated otherwise.
:300 in 0.1% (v/v) Tween-20 (Sigma) in TBS, followed by a 10-min wash with PBS+. Nuclei were counterstained for 5 min with Hoechst (Jackson ImmunoResearch) diluted 1:7000 in PBS+. A final 10-min wash was performed with PBS+. A Gene Frame® (ThermoFisher Scientific) was used to create a thin-height well (250 μm) on a standard microscope glass slide. Stained cells that adhered to the microcarriers were transferred into this well, and a coverslip was mounted using the Gene Frame® with a fluorescence mounting medium kit (Dako) and stored at 4 °C before imaging. All reagents were purchased from Life Technologies™ unless stated otherwise.
Confocal observations
The PC3 cell behavior during their growth on the microcarriers was investigated using a Leica TCS SP2 laser scanning confocal microscope with a 20×/0.5 NA objective. The PC3 cells were cultured as described in the cell culture section and imaged after 2 days of culture.Stained PC3 cells that adhered to the microcarriers were imaged using a Nikon Eclipse Ti spinning disk confocal microscope with a 40×/1.3 NA objective.
Cytotoxic assays
Two types of cytotoxic assays were conducted: one concerned the glues used to seal the device and the other concerned the enzyme used to dissolve the microcarriers.The cytotoxicity of the glues was evaluated in two steps. First, culture experiments were performed with different glues to identify potential biocompatible candidates. Single drops of UV-cured acrylic 6108-T and epoxies KB4597, KB4594, and OG116-31 were deposited at the center of separate Petri dishes. NIH 3T3 cells were then seeded at 4 × 104 cells mL−1 in each dish and cultured for 24 h before image acquisition.
Then, a quantitative assay based on the International Organization for Standardization (ISO) 10993-5 was performed on the biocompatible candidates previously identified. The assay comprised two types of tests: a contact test in which cells were cultured with the materials to test and a conditioning test in which cells were cultured with medium previously incubated with the materials.
More precisely, NIH 3T3 cells were inoculated in 6-well plates at a density of 4 × 104 × cells mL−1 in 2 mL of solution. After 24 h, an 8 × 8 mm2 square of the material was added to each well for 24 h for the contact experiment. Silicon dioxide and zinc were used as negative25 (no death induced) and positive26 (death induced) control materials, respectively. UV-cured glue OG116-31 was the material that was tested. Triplicate samples were performed for each type of material and for cells not exposed to any material. Additionally, for the conditioning experiment, the medium was changed for each well using 2 mL of the medium exposed to one material over 24 h. Triplicate samples were also performed for the contact experiment and were incubated for 24 h. Then, the materials were removed, and each well was washed twice with 2 mL of PBS before adding 1 mL of the culture medium.
Cytotoxicity was assessed 24 h after using the WST-1 assay (cell proliferation reagent, Roche), which is similar to the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide reagent) assay. WST-1 reagent was added (10% (v/v)) to the culture medium and remained in the incubator for 3 h. The absorbance was then recorded at 450 nm (soluble formazan titration) and 690 nm (background subtraction) using a microplate reader (Infinite M1000, Tecan). The absorbance difference (450–690 nm) was directly proportional to the number of viable cells. The percentage cell viability was determined using the following equation:
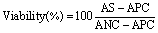 | (2) |
Regarding the cytotoxicity of dextranase, cells were seeded at 104 cells mL−1 in 200 μL of solution in a 96-well plate and cultured for 24 h. Media were replaced with 200 μL of media containing a dextranase concentration of either 10−3, 10−2, 10−1, 1%, 3%, 5%, 7%, or 10% (v/v). Media without dextranase or with 10 mM H2O2 were used for the negative and positive controls, respectively. Six samples were performed for each condition. The cells were incubated for 24 h and then rinsed twice with 200 μL of PBS. Afterward, the cells were incubated for 24 h with 100 μL of culture medium. Then, 10 μL of WST-1 was added to each well. After 3 h, the absorbance was recorded at 450 nm (soluble formazan titration) and 690 nm (background subtraction) using a microplate reader (Infinite M1000, Tecan). The absorbance difference (450–690 nm) was directly proportional to the number of viable cells. The percentage cell viability was determined using the previous equation (eqn (2)).
Results and discussion
Fluidic characterization
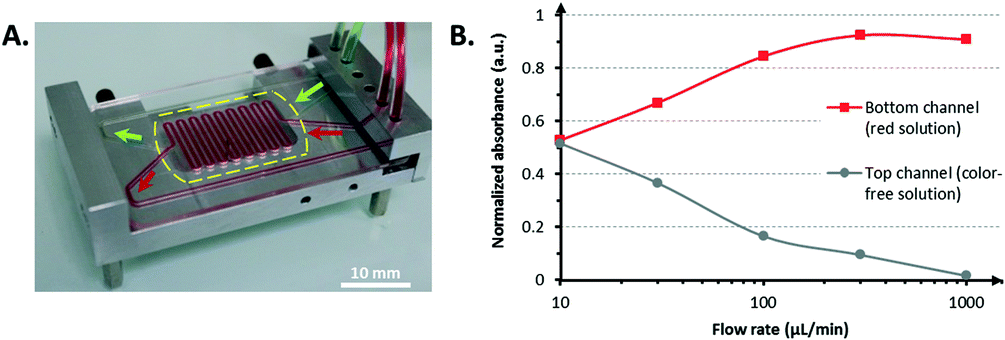 | ||
| Fig. 3 Fluidic characterization. A) Leakage test. Image of the bioreactor composed of the CCC mounted on an aluminum holder. The dashed line represents the edge of the porous membrane. B) Transmembrane diffusion assay. Validation of the transmembrane diffusion after CCC fabrication. The flow rates in both the upper and the lower channels were equal at each measurement point. The absorbance values have been normalized so that the absorbance of the red stock solution is equal to 1 and the absorbance of the PBS (color-free) solution is equal to 0. | ||
The membrane, which is situated inside the CCC, appears transparent almost everywhere due to the glue wetting. Only the part of the membrane in the snake-like area maintains its white color, which indicates that the glue did not wet this area of the membrane (i.e., by capillarity action, for instance). It was assumed that the pore integrity was not compromised for future mass transport. The described transmembrane transport assay further confirmed this hypothesis.
In addition to the good liquid confinement within the bioreactor that was demonstrated, the perfusion of green and red stained fluids through the bioreactor at 1 mL min−1 (33 mm s−1) in the lower and upper channels, respectively, did not display any leakage. This result validated the successful bonding between the device and the porous membrane. The same observation held for the tight interconnections between the CCC and the tubing to ensure that sterility would remain during cell culture.
A lower flow rate resulted in closer absorbance values. For flow rates less than or equal to 10 μL min−1, the absorbance values were equal to half of the maximum absorbance (i.e., the absorbance of the stock red solution), which indicates that a complete exchange of the red dye occurred from one channel to the other through a diffusion process. The red food dye has a molecular size comparable to that of the cell culture medium constituents. Therefore, the possibility of transmembrane transport across the porous membrane with this molecule confirmed that nutrients could reach the cells.
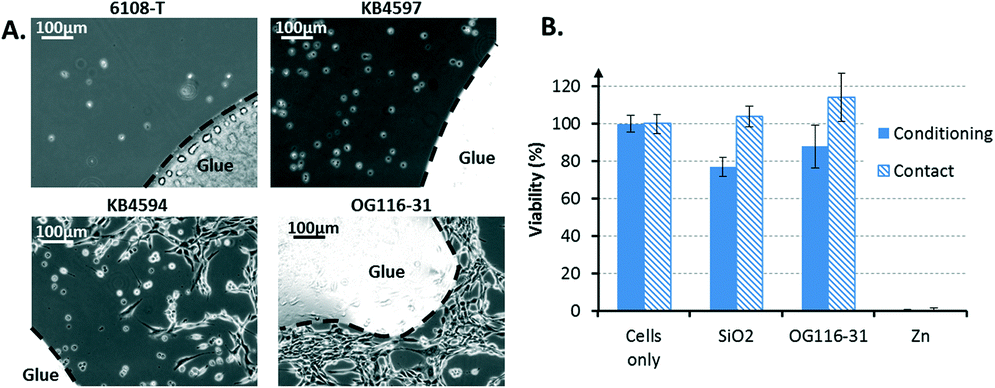 | ||
| Fig. 4 System biocompatibility. A) A test of the effects of different glues (used for porous membrane embedding) on NIH 3T3 cells grown in the presence of the glue for 24 h. One acrylic (6108-T) and three epoxy (KB4597, KB4594, and OG116-31) glues were examined. B) Cytotoxicity assay (N = 6) for OG116-31: NIH 3T3 viability as a function of the materials used in conditioning and in contact mode. In contact mode, the cells were cultured with the different materials, and in conditioning mode, the cells are cultured with medium previously incubated with the different materials. | ||
Dead cells were characterized by a round shape, whereas healthy cells exhibited an elongated morphological shape. The 6108T acrylic glue and the epoxy KB4597 glue were highly toxic because all cells were found dead. The epoxy KB4594 glue had a relatively low toxicity according to the small cell-depleted area that was visible around the glue. However, the epoxy OG116-31 displayed a marginal toxicity because the cells appeared to have even proliferated on it. Thus, the OG116-31 glue was selected to perform a cytotoxic assay to obtain more quantitative results (Fig. 4B). In both the conditioning and contact modes, the glue exhibited a toxicity comparable to that of the SiO2 substrate, which was used as a non-toxic reference material for cells. Consequently, cell proliferation was not expected to be altered by the direct contact of the cells with the OG116-31 glue or by the possible release of the by-products of this glue into the culture medium. Therefore, epoxy OG116-31 was chosen to complete the fabrication of the biocompatible device to host mammalian cell culture. This method is a simple tool to integrate a membrane within a microfluidic system in a biocompatible manner. However, other methods, such as thermal bonding29 or mechanical sealing,30 can achieve biocompatible membrane integration. These methods may be considered instead of glue-based bonding in the case where the glue may have more subtle effects on the cells in the long term.
Control of the cell microenvironment
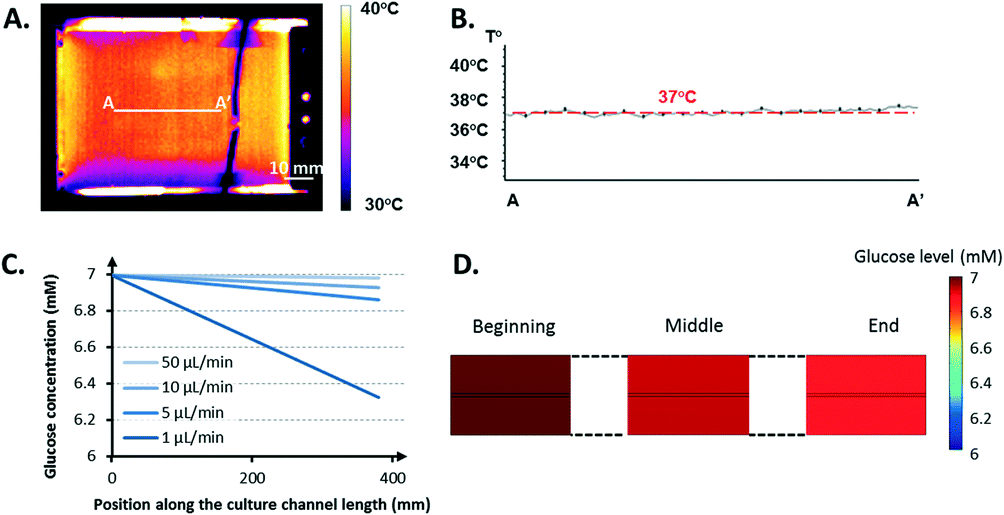 | ||
| Fig. 5 Cell microenvironment control. A) IR thermal characterization. IR imaging of the entire bioreactor. B) Thermal profile along the AA′ line across the culture area. C–D) Glucose levels. Glucose concentration at the middle height of the culture channel (upper channel) versus the fresh medium perfusion rate in the lower channel (C). Glucose distribution within the culture area obtained from the simulation, where the lower channel is perfused with fresh medium at 5 μL min−1 (D). | ||
This difference, approximately 2–3 °C, was due to the poor insulating properties of the glue used to attach the sensor. An underestimation of 2–3 °C can easily jeopardize the cell culture because mammalian cells can rarely survive at temperatures greater 40 °C.31 The thermal images validated that such a limit was not reached over the entire culture area. Moreover, the heating set-up provided a homogeneous, stable thermal distribution across the area of interest (Fig. 5B). The PET-ITO heater coupled to a thermal sensor proved to be an effective, easy-to-integrate solution to satisfy the requirements of the cell culture in terms of thermal management after IR characterization.
Fig. 5D displays the global glucose repartition at a perfusion rate of 5 μL min−1. The gradient observed along the height of the culture channel appears negligible compared to the gradient over its length.
Other simulations were performed by considering oxygen consumption. Data found in the literature regarding the Michaelis–Menten parameters for oxygen used in these simulations led to the conclusion that glucose was consumed considerably faster by the cell (data not shown). This result is partially due to a difference of at least one order of magnitude between the saturated consumption rate of glucose (≈10−16–10−17 mol per cell s−1) and oxygen (≈10−18 mol per cell s−1).32 Because the studies were stationary, a fixed density of 106 cells mL−1 was considered in the model, which corresponds to the maximum cell density expected once the microcarriers are completely covered with cells. Thus, the gradient observed in the simulation of the culture channel from one end to the other corresponds to the maximum spatial variation of the glucose concentration present at the final stage of a culture run. This gradient is null at the beginning of a culture run and slowly increases with an increasing number of cells until it reaches its maximum value, when the critical cell density is reached.
If control over the nutrient distribution can be achieved, it is more likely that the concentration of the biochemical species can be controlled as well. According to the drug perfusion flow rate, gradients or a stable concentration of drugs could be supplied to cells to study their response or trigger specific cell mechanistic responses (e.g., differentiation). Therefore, drug testing could also be supported.
Cell growth
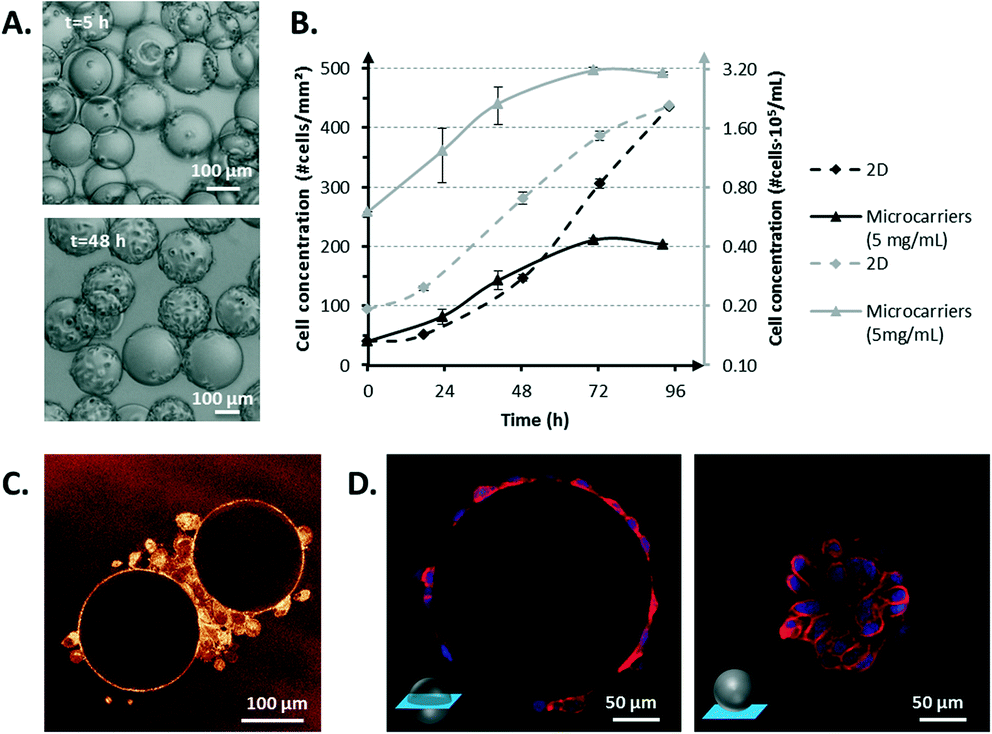 | ||
| Fig. 6 Cell proliferation on microcarriers. A) An example of PC3 cell culture on microcarriers (2 mg mL−1) in a non-cell-culture-treated Petri dish at 5 h (top) and at 48 h (bottom). B) PC3 growth on microcarriers (5 mg mL−1) versus 2D PC3 cell culture (N = 3). The vertical axis uses a base 2 logarithmic scale. The data are plotted either in cells per surface unit (black lines and left vertical axis) or in cells per culture volume (grey lines and right vertical axis). C) Confocal images illustrating the cell transfer between bead carriers during cell culture. D) Immunostaining images of PC3 cells cultured for 4 days on a microcarrier at two different planes (see scheme in the bottom left corner): the middle plane (left) and bottom plane (right) of the microcarrier. The nuclei are stained in blue, and phalloidin (actin filaments) is shown in red. | ||
Initially, the cells aggregated and adhered all around the beads. Then, the cells proliferated and colonized a higher surface area on the beads. Cell proliferation on the microcarriers was studied at different microcarrier densities and compared to a standard 2D cell culture (Fig. 6B). The microcarrier density was chosen based on the maximum density recommended by the manufacturer. The seeding density of cells per surface area and volume was identical from one condition to the other: 40 cells mm−2 in 2 mL. The maximal cell proliferation rates on microcarriers and in 2D culture were comparable. However, the 2D cell culture exhibited a lag phase during the first 24 h. The microcarrier cell culture growth rate started to decrease more significantly and sooner (≈1 day) than that of the 2D cell culture, certainly due to the accumulation of cell waste and/or a lack of nutrients as the cell concentration per milliliter was very high (Fig. 6B). This result reaffirms the need to integrate a mechanism by which the culture medium can be renewed. Even if the cell seeding on microcarriers was not homogeneous (Fig. 6A), the cells were not confined to the beads to which they first adhered. Cells have the ability to transfer from one bead to another by forming a cell bridge if these microcarriers are close enough, as shown in Fig. 6C. Cells can therefore grow in a similar manner as those grown on 2D flat surfaces until complete coverage of all of the surfaces. Once the beads are confluent (i.e., completely covered with cells), contact inhibition occurs, and the cells stop proliferating, as observed in regular 2D cell culture.33
In addition, PC3 cells presented comparable spread morphology when adhered to the microcarriers compared with PC3 cells adhered to flat culture surfaces (Fig. 6D). These cells also developed tight cell–cell junctions (Fig. 6D).
Cell bead transfer is a well-known phenomenon in bioprocess engineering34 and is intentionally used to expand cell culture and increase the productivity of a batch. This cell feature creates opportunities to perform long-term cell culture: empty microcarriers can be added inside the bioreactor once a certain number of cells is reached, and part of the microcarriers already covered with cells can be simultaneously extracted from the system. The remaining cells will be able to colonize the empty beads, and proliferation will continue. Repetitions of this process can significantly increase the culture duration and open routes toward continuous cell culture processes.
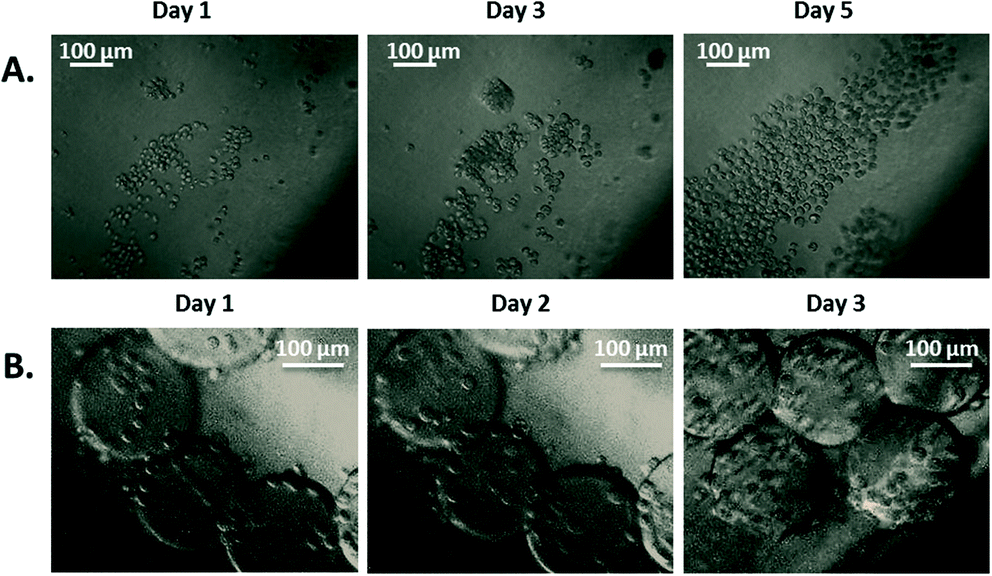 | ||
| Fig. 7 Bioreactor cell culture. Images of cells proliferating in the culture area of the bioreactor. A) Drosophila S2 proliferation. B) PC3 proliferation on microcarriers. Images were captured daily. | ||
Drosophila cells were used as a simple model and without microcarriers to validate the entire system operation. The culture of such cells does not require CO2 regulation when exposed to air; room temperature and ambient oxygen level are sufficient for their normal growth. Additionally, this cell type can be cultured as adherent cells or in suspension due to their weak adherence. PC3 cells were used as the more complex model and were cultured on microcarriers. These cells require a constant temperature of 37 °C, and when exposed to air, their medium requires a partial CO2 pressure of 5% to maintain its pH level. Proliferation of both cell types was observed within the bioreactor over 3 to 5 days. The growth of Drosophila cells (Fig. 7A) validated the global operation of the bioreactor to move toward the culture of PC3 cells on microcarriers (Fig. 7B). The ability to culture different types of cells highlighted the versatility of the bioreactor.
The CCC can be used inside an incubator or coupled to a gas exchanger35 to regulate the CO2 partial pressure in the medium for a longer cultivation period. However, these systems occupy a substantial amount of space or require heavy installation equipment (gas source, control equipment) and safety certifications. To address these issues, we are using a different approach aimed at developing portable culture systems that would rely as little as possible on such regulations. During the culture of PC3 cells, no CO2 regulation was necessary because the materials composing the CCC were chosen to limit the escape of CO2 from the medium. Furthermore, commercial CO2-independent media are currently under investigation to enable long-term cell culture without the use of a CO2 regulation system.
As outlined in the introduction, microcarrier cell culture easily allows for the manipulation of adherent cells through flow transport and avoids using proteolytic enzymes to collect the microcarriers covered with cells from a culture system or to perform enzyme-free subcultures. These criteria are critical for preserving the quality of the cultured cells, particularly in the case of stem cells, as repeated enzymatic treatments have been shown to compromise their genetic integrity.36
Microcarrier technology has additional advantages. Indeed, microcarriers can be made from different materials, such as dextran, glass, or polystyrene,37 which can be functionalized to allow for the grafting of biopolymers, recombinant proteins, extracellular matrix proteins, peptides, or charged molecules.37–39 Therefore, various studies are possible regarding the influence of these surface chemistries on cell behavior and fate.40 Additionally, thermosensitive polymers can be coated onto carriers to realize in situ cell harvest.39
Cell harvesting
NIH 3T3 cells grown on microcarriers were collected after the 4th day of culture inside the bioreactor. A plug of medium was sent into the channel that contained the cells to be extracted from the system. Afterwards, the number of collected cells was determined. Standard protocol for cell harvest using trypsin was not reproducible and was unsuccessful in completely detaching the cells from the microcarriers, as shown in Fig. 8A.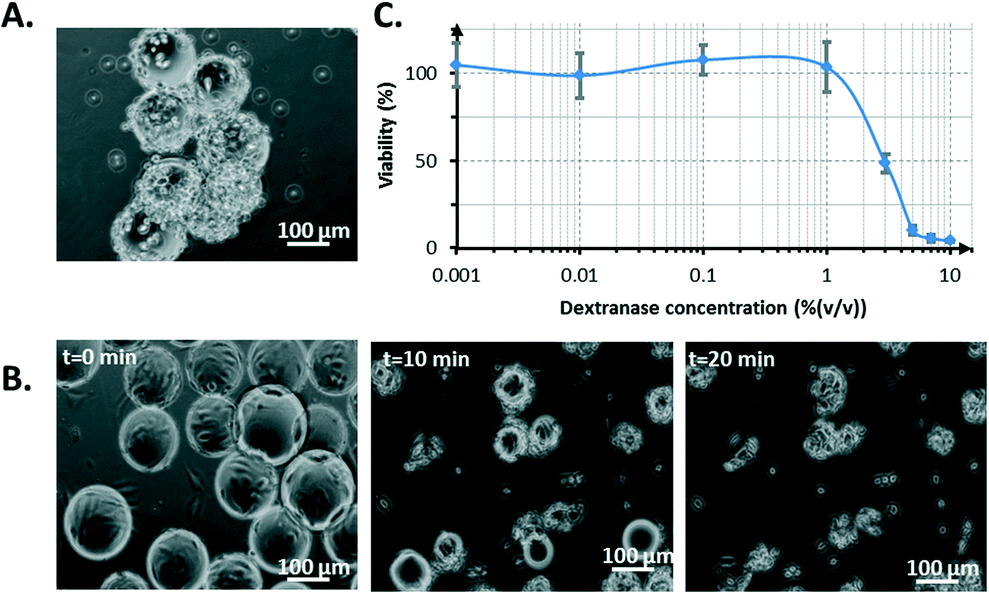 | ||
| Fig. 8 Cell harvest from microcarriers comparing the use of trypsin (A) or dextranase (B–C). A) Cells on microcarriers treated with trypsin for 10 min. B) Microcarrier dissolution using 1% (v/v) dextranase for 10 or 20 min. C) NIH 3T3 viability versus dextranase concentration (N = 6). A 1% (v/v) dextranase solution was chosen for the cell counting protocol. | ||
However, the prior dissolution of microcarriers by dextranase revealed that cells could be efficiently recovered. Dextranase degraded the constitutive dextran matrix of the beads, which left cell clusters (Fig. 8B). This method exhibited a marginal toxicity of up to 1% (v/v) dextranase (after 24 h of incubation with cells). The IC50 (concentration threshold corresponding to 50% viability) was 3% (v/v) dextranase (Fig. 8C). After the dextranase treatment, the cells were exposed to trypsin to obtain a homogeneous suspension of isolated cells, which is important for cell counting. The use of trypsin was the most straightforward way to obtain single-cell populations. However, other methods based on gentle vortexing, gentle pipetting or the use of a dissociation buffer could have been used instead to guarantee a proteolytic-free single-cell recovery. It is also important to note that single-cell recovery represents only a single goal to reach. More extensively, a microcarrier-based cell culture enables proteolytic-free collection for numerous other subsequent cell manipulations such as transfection, immunostaining, cell co-cultures and seeding of other culture systems.
3D cell spheroids have been shown to be a promising tool for drug discovery applications because, to date, they are the in vitro model that most closely mimics an in vivo model.41 Interestingly, our method based on microcarrier dissolution could provide a new way of generating multicellular 3D spheroids in addition to the existing methods.41 When the cells proliferate on a microcarrier, they develop cell–cell junctions that bind the cells together. These junctions, when using dextranase, keep the cells close when the microcarrier is dissolved. A cluster is formed and can evolve into a spheroid.
With further developments, dextranase could be perfused directly into the bioreactor to dissolve the microbeads and produce 3D cell spheroids, which could be cultured under controlled conditions. Subsequent drug testing could even be performed though the perfusion function of the bioreactor, as suggested earlier. In this configuration, our bioreactor could be turned into a 3D-model-based drug testing platform.
Conclusion
A microfluidic system for microcarrier cell culture was developed and validated for different cell types. An original fabrication process using biocompatible glue was shown to facilitate the integration of a porous membrane, the core element of the perfusion function. Diffusion experiments ensured that the integration process did not affect the capacity to support transmembrane mass transfer. The culture of Drosophila S2 cells demonstrated the ability of the device to support benchtop and long-term cell growth. In the case of PC3 cell culture on microcarriers, the perfusion flow rate was optimized to 5 μL min−1 based on simulations to ensure a stable environment, even when a maximum cell density is reached in the system. Moreover, the thermal management solution provided a homogeneous temperature of 37 °C in the culture area. These results resulted in the effective benchtop microcarrier cell culture of PC3 cells without the need for an incubator. This system paves the way toward the development of portable systems for continuous cell culture that provide adequate, controlled environments.Acknowledgements
The authors gratefully acknowledge Didier Grunwald from CEA (France) for the confocal observations and helpful discussions. M.E. Dolega acknowledges the Irtelis PhD program of CEA (France) for financial support.References
- M. Tehranirokh, A. Z. Kouzani, P. S. Francis and J. R. Kanwar, Biomicrofluidics, 2013, 7, 51502 CrossRef PubMed.
- S. Son, A. Tzur, Y. Weng, P. Jorgensen, J. Kim, M. W. Kirschner and S. R. Manalis, Nat. Methods, 2012, 9, 910–912 CrossRef CAS PubMed.
- G. Blanco-Gomez, E. Trioux and V. Agache, IEEE Electron Device Lett., 2012, 33, 609–611 CrossRef.
- G. Blanco-Gomez and V. Agache, J. Microelectromech. Syst., 2012, 21, 224–234 CrossRef CAS.
- V. Agache, G. Blanco-Gomez, F. Baleras and P. Caillat, Lab Chip, 2011, 11, 2598–2603 RSC.
- D. Schäpper, S. M. Stocks, N. Szita, A. E. Lantz and K. V. Gernaey, Chem. Eng. J., 2010, 160, 891–898 CrossRef PubMed.
- M. Micheletti and G. J. Lye, Curr. Opin. Biotechnol., 2006, 17, 611–618 CrossRef CAS PubMed.
- D. Schapper, M. N. Alam, N. Szita, A. Eliasson Lantz and K. V. Gernaey, Anal. Bioanal. Chem., 2009, 395, 679–695 CrossRef PubMed.
- M. Matasci, D. L. Hacker, L. Baldi and F. M. Wurm, Drug Discovery Today: Technol., 2008, 5, e37–e42 CrossRef PubMed.
- F. Li, N. Vijayasankaran, A. Shen, R. Kiss and A. Amanullah, mAbs, 2010, 2, 466–479 CrossRef.
- K. S. Lee, P. Boccazzi, A. J. Sinskey and R. J. Ram, Lab Chip, 2011, 11, 1730–1739 RSC.
- B. Griffiths, Mol. Biotechnol., 2001, 17, 225–238 CrossRef CAS PubMed.
- A. L. Demain and P. Vaishnav, Biotechnol. Adv., 2009, 27, 297–306 CrossRef CAS PubMed.
- I. Barbulovic-Nad, S. H. Au and A. R. Wheeler, Lab Chip, 2010, 10, 1536–1542 RSC.
- B. Y. Zhang, M. C. Kim, T. Thorsen and Z. H. Wang, Biomed. Microdevices, 2009, 11, 1233–1237 CrossRef CAS PubMed.
- X. Y. Zhu, L. Y. Chu, B. H. Chueh, M. W. Shen, B. Hazarika, N. Phadke and S. Takayama, Analyst, 2004, 129, 1026–1031 RSC.
- S. K. Moore and S. J. Kleis, Acta Astronaut., 2008, 62, 632–638 CrossRef CAS PubMed.
- J. Yang, M. Yamato, K. Nishida, T. Ohki, M. Kanzaki, H. Sekine, T. Shimizu and T. Okano, J. Controlled Release, 2006, 116, 193–203 CrossRef CAS PubMed.
- D. Ma, Z.-M. Li, Q.-H. He and H.-W. Chen, 14th International Conference on Miniaturized Systems for Chemistry and Life Sciences, 2010 Search PubMed.
- H. Lu, L. Y. Koo, W. C. M. Wang, D. A. Lauffenburger, L. G. Griffith and K. F. Jensen, Anal. Chem., 2004, 76, 5257–5264 CrossRef CAS PubMed.
- L. Liu, K. Loutherback, D. Liao, D. Yeater, G. Lambert, A. Estevez-Torres, J. C. Sturm, R. H. Getzenberg and R. H. Austin, Lab Chip, 2010, 10, 1807–1813 RSC.
- J. H. Park, R. A. Perez, G. Z. Jin, S. J. Choi, H. W. Kim and I. B. Wall, Tissue Eng., Part B, 2013, 19, 172–190 CrossRef CAS PubMed.
- P. R. Marcoux, M. Dupoy, R. Mathey, A. Novelli-Rousseau, V. Heran, S. Morales, F. Rivera, P. L. Joly, J. P. Moy and F. Mallard, Colloids Surf., A, 2011, 377, 54–62 CrossRef CAS PubMed.
- C. L. Crespi and W. G. Thilly, Biotechnol. Bioeng., 1981, 23, 983–993 CrossRef.
- G. Kotzar, M. Freas, P. Abel, A. Fleischman, S. Roy, C. Zorman, J. M. Moran and J. Melzak, Biomaterials, 2002, 23, 2737–2750 CrossRef CAS.
- J. Borovansky and P. A. Riley, Chem.-Biol. Interact., 1989, 69, 279–291 CrossRef CAS.
- M. Ni, W. H. Tong, D. Choudhury, N. A. A. Rahim, C. Iliescu and H. Yu, Int. J. Mol. Sci., 2009, 10, 5411–5441 CrossRef CAS PubMed.
- J. H. Brauker, V. E. Carrbrendel, L. A. Martinson, J. Crudele, W. D. Johnston and R. C. Johnson, J. Biomed. Mater. Res., 1995, 29, 1517–1524 CrossRef CAS PubMed.
- Z. F. Wang, Y. P. Seah and Z. P. Wang, Microelectron. Eng., 2013, 110, 386–391 CrossRef CAS PubMed.
- A. Ould-Dris, P. Paullier, L. Griscom, C. Legallais and E. Leclerc, J. Membr. Sci., 2010, 352, 116–125 CrossRef CAS PubMed.
- S. Fulda, A. M. Gorman, O. Hori and A. Samali, Int. J. Cell Biol., 2010, 2010, 214074 Search PubMed.
- B. A. Wagner, S. Venkataraman and G. R. Buettner, Free Radical Biol. Med., 2011, 51, 700–712 CrossRef CAS PubMed.
- P. K. Davis, A. Ho and S. F. Dowdy, BioTechniques, 2001, 30, 1322–1331 CAS.
- Y. Wang and F. Ouyang, Bioprocess Eng., 1999, 21, 211–213 CrossRef CAS.
- S. Martewicz, F. Michielin, E. Serena, A. Zambon, M. Mongillo and N. Elvassore, Integr. Biol., 2012, 4, 153–164 RSC.
- M. M. Mitalipova, R. R. Rao, D. M. Hoyer, J. A. Johnson, L. F. Meisner, K. L. Jones, S. Dalton and S. L. Stice, Nat. Biotechnol., 2005, 23, 19–20 CrossRef CAS PubMed.
- J. Malda and C. G. Frondoza, Trends Biotechnol., 2006, 24, 299–304 CrossRef CAS PubMed.
- M. Hervy, J. L. Weber, M. Pecheul, P. Dolley-Sonneville, D. Henry, Y. Zhou and Z. Melkoumian, PLoS One, 2014, 9, e92120 Search PubMed.
- A. Tamura, J. Kobayashi, M. Yamato and T. Okano, Biomaterials, 2012, 33, 3803–3812 CrossRef CAS PubMed.
- J. Varani, M. Dame, J. Rediske, T. F. Beals and W. Hillegas, J. Biol. Stand., 1985, 13, 67–76 CrossRef CAS.
- S. Breslin and L. O'Driscoll, Drug Discovery Today, 2013, 18, 240–249 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI): Supplementary Table, Fig. S1 and S2. See DOI: 10.1039/c4lc00570h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |