 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactive self-assembled monolayers: from surface functionalization to gradient formation
Carlo
Nicosia
and
Jurriaan
Huskens
*
Molecular Nanofabrication group, MESA+ Institute for Nanotechnology, University of Twente, Enschede, The Netherlands. E-mail: j.huskens@utwente.nl; Fax: +31-53-4894645; Tel: +31-53-4892980/2937
First published on 13th September 2013
Abstract
This review describes the progress of the development of surface chemical reactions for the modification of self-assembled monolayers (SAMs) and the fabrication of surface chemical gradients. Various chemical reactions can be carried out on SAMs to introduce new functionalities. “Click” reactions, which are highly efficient and selective, have largely contributed to the development and implementation of surface chemical reactions in the fields of biotechnology, drug discovery, materials science, polymer synthesis, and surface science. Besides full homogeneous functionalization, SAMs can be modified to exhibit a gradual variation of physicochemical properties in space. Surface-confined chemical reactions can be used for the fabrication of surface chemical gradients making the preparation of exceptionally versatile interfaces accessible.
 Carlo Nicosia | Carlo Nicosia (1983) studied industrial chemistry at the University of Parma, Italy. In 2007 he obtained his undergraduate degree (cum laude) under the supervision of Professor Enrico Dalcanale. In 2009 he became a PhD student in the group of Professor Jurriaan Huskens. His work involves the investigation of chemical reactions of reactive monolayers assembled onto planar surfaces for the fabrication of surface gradients and biomolecular patterns. |
 Jurriaan Huskens | Jurriaan Huskens (1968) studied chemical engineering at the Eindhoven University of Technology. He obtained his PhD from the Delft University of Technology in 1994. Following a postdoctoral stay at the University of Texas at Dallas, USA and an EU Marie Curie fellowship at the Max-Planck-Institut für Kohlenforschung, Mülheim an der Ruhr, Germany, he began his academic career at the University of Twente, Enschede, The Netherlands in 1998, where he is now a full professor of “Molecular Nanofabrication”. The research in his group is focused on fundamental and applied aspects of supramolecular surface science and nanotechnology. |
1 Introduction
In 1959 the physicist Richard Feynman delivered a vision of exciting new technological discoveries based on the fabrication of materials and devices at the atomic/molecular scale that we call today nanotechnologies.1 The interesting size range where nanotechnologies operate is typically from 100 nm down to the atomic level. In this range the properties of materials are different compared with the same materials at larger size mainly due to the higher surface area and the prevalence of quantum effects.2 These new properties of nanomaterials are conveniently employed over a wide range of fields ranging from catalysis, optics, electronics and informatics, to bio-nanotechnology and nanomedicine.3 In the 1980s nanoscience discoveries experienced impressive propulsion with the invention of the scanning tunnelling microscope (STM) and the atomic force microscope (AFM) allowing the imaging of surfaces with molecular or even atomic resolution.Self-assembled monolayers (SAMs) are two-dimensional nanomaterials formed spontaneously by the highly ordered assembly of the molecular constituents onto the surface of a variety of solids.4,5 SAMs, formed by adsorption of a one-molecule-thick layer on the surface, are excellent systems to study interfacial reactions. The exponential growth in SAM research is justified by the multi-disciplinarity of the field that gathers chemists, physicists, biologists and engineers.
The two most common families of SAMs are alkylsilanes on oxide surfaces,6–9 and sulfur-containing molecules on gold5,10–12 (Scheme 1). Because of their ease of preparation, the spontaneous formation of a densely packed monolayer, and the conductivity of the substrate, the assembly of ω-functionalized thiols (or disulfide or sulfide) on gold has been extensively studied. However, organosilane monolayers on SiO2 (silicon or glass) can be integrated into silicon technology and their covalent nature results in high chemical and physical stability allowing extensive modification steps.
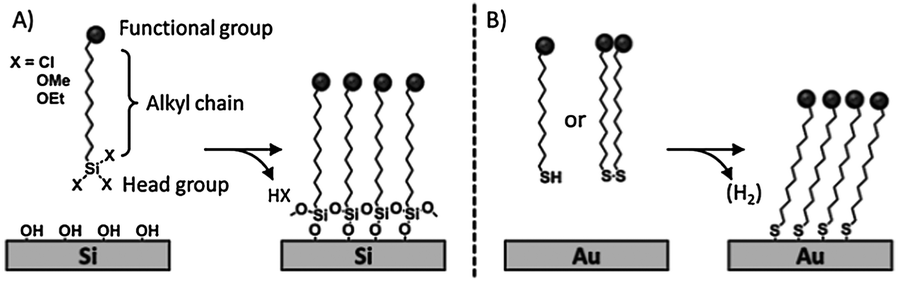 | ||
| Scheme 1 Formation of functional silane/SiO2 (A) and thiol/gold (B) monolayers. | ||
The mechanism of formation of SAMs of sulfur-containing molecules on gold and organosilanes on SiO2 has been extensively described elsewhere.4,5,7,8 Interfacial reactions are versatile and essential modification schemes to control the surface composition and density of monolayers. The terminal groups of the building blocks of SAMs allow the fine-tuning of the interfacial surface properties in terms of chemical reactivity, conductivity, wettability, adhesion, friction, corrosion resistance and (bio)compatibility.13 The introduction of components with different functional end groups in the monolayers can be performed through two different routes: (i) the adsorption of pre-functionalized molecules or (ii) the modification of the monolayer after formation. While the former route requires the complete synthesis of the molecular constituent of the monolayer, the latter method, based on a stepwise process, offers intrinsic advantages: (i) it enables the incorporation of groups that are not compatible with the synthesis of the building block (e.g. silane or thiol groups); (ii) it does not affect the order of the underlying monolayer; (iii) it allows the quick preparation of multiple samples; (iv) it employs ordinary synthetic procedures; (v) it requires low amounts of reagents.5 On the other hand, since purification of the functionalized monolayer is impossible, high-yielding, efficient, selective and clean reactions are essential.
The implementation of reactions for the chemical modification of monolayers on planar surfaces is pivotal to expand the function and to tailor the properties of surfaces and materials. Haensch and coworkers recently described, in a critical review,9 the chemical modification of silane-based monolayers involving nucleophilic substitution and Huisgen 1,3-dipolar cycloaddition of organic azides and acetylenes. Sullivan and Huck illustrated nucleophilic substitution, esterification, acylation, and nucleophilic addition reactions on thiols/Au and silanes/SiO2 functionalized with terminal amines, hydroxyls, carboxylic acids, aldehydes, and halogens.14 Jonkheijm and coworkers outlined, in a comprehensive review,15 strategies for the fabrication of reactive interfaces for the fabrication of biochips. Numerous reactions are available to modify the surface chemistry of SAMs9,14,16 (e.g. nucleophilic substitutions,17–19 esterification,20 amidation,21–23etc.).
In the last decade a lot of effort was focused on the implementation of methods to obtain selective, efficient, robust, quantitative, simple and rapid surface transformations to reduce the formation of by-products, avoiding the need for purification and allowing easy surface analysis. Click chemistry encompasses all these properties. The click reaction paradigm delivered by Sharpless and coworkers in 200124,25 is based on implementing highly efficient and selective reactions that reach quantitative conversion under mild conditions, essential qualities for the development of surface and materials sciences.26–31
Microcontact printing (μCP), scanning probes, UV and e-beam lithographies, the so-called “top-down” methods, are commonly employed to generate patterns of SAMs with sizes ranging from tens of nanometers to millimeters. Microcontact printing, in particular, was introduced by Whitesides and coworkers as a fast, flexible, simple and inexpensive way to replicate patterns generated via photolithography.32–34 In the conventional μCP, a microstructured elastomeric poly(dimethylsiloxane) (PDMS) stamp was employed to transfer molecules of the “ink” (e.g. thiols) to the surface of the substrate (e.g. gold) by conformal contact. Patterns of alkanethiols on gold were conveniently used as an etch-protecting layer for the fabrication of microstructures with potential application in microelectronics.
Soon after its development, μCP evolved as a powerful tool to pattern functional reactive monolayers by means of the local chemical reaction between an ink transferred by the stamp and the functional groups introduced on the substrate. This process is called “reactive μCP” or microcontact chemistry35–37 (Scheme 2).
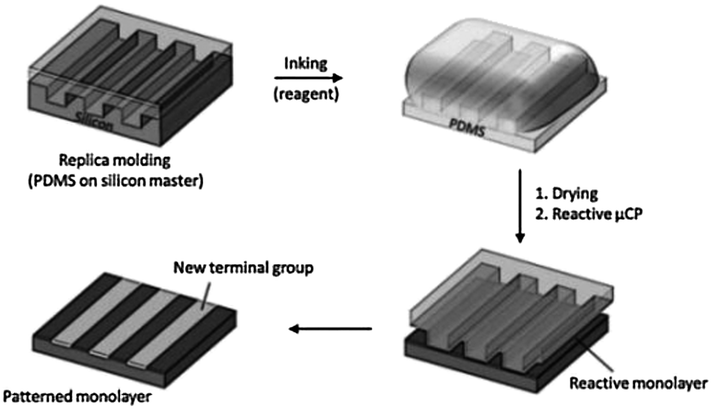 | ||
| Scheme 2 Reactive microcontact printing: stamping of a reagent onto a reactive monolayer yields a patterned monolayer on a substrate. | ||
μCP is an efficient method, and enhances slow and uncatalyzed reactions at the interface.36 Reactions induced by μCP allow for near-quantitative yield, mild reaction conditions, and short reaction times, mainly due to the high degree of preorganization of the surface-immobilized reactant and the high local concentration of reagents (ink) at the stamp/substrate interface.
Surface gradients are surfaces with a gradual variation of at least one physicochemical property in space that may evolve in time. Surface chemical gradients (Scheme 3) have allowed for the gradual modulation of interfacial properties and have been employed to generate smart materials and to investigate surface-driven transport phenomena like the motion of water droplets on a wettability gradient,38 or the study of biological processes, for example the directed migration (haptotaxis) and polarization of cells on biomolecular gradients.39 Moreover, surface gradients integrate a wide range of properties in a single sample thus providing a valuable tool for the fast high-throughput analysis of several parameters avoiding the effect of the distribution of properties in different specimens and tedious analysis of multiple samples. Two general methods are commonly employed for the development of monolayer-based surface chemical gradients: (i) the controlled adsorption/desorption of SAMs on gold or silicon and (ii) the chemical post-modification of reactive SAMs.39–42
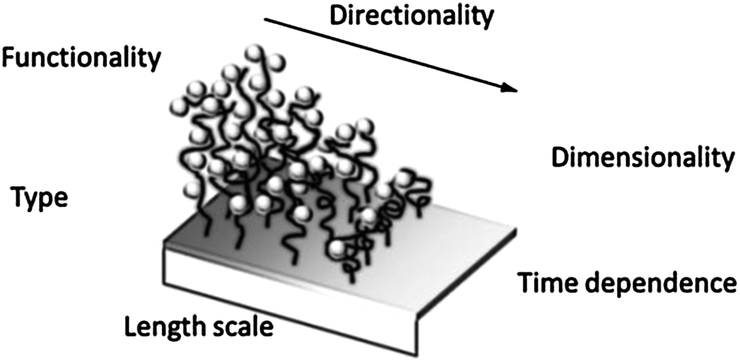 | ||
| Scheme 3 Surface chemical gradient attributes. Adapted from ref. 39. | ||
In the first part of this review the reactivity of SAMs and the covalent modifications that have been carried out on monolayers by means of “click chemistry” from solution and by soft lithography are described. In the second part the fabrication of surface chemical gradients exploiting recent strategies based on “click”-based chemical modifications of terminal functional groups of SAMs is illustrated. Practically all examples deal with monolayers on flat surfaces, not on (nano/micro)particles, although many processes would be compatible with both.
2 Chemical transformation of monolayers
Both thiols on gold and organosilanes on silicon or glass produce robust dense monolayers and have been used for subsequent modification using chemical reactions. A common modification of the surface properties is obtained via modular stepwise selective functionalization of pre-formed reactive SAMs.Below are described the main recent examples in which reactive monolayers are employed in combination with click chemistry, either by reaction in solution or by soft lithography (e.g. μCP). Herein, the most attractive and common click reactions were considered: the azide–alkyne cycloaddition, the thiol-ene reaction, the Michael addition, the imine and oxime formation, and the Diels–Alder cycloaddition (Scheme 4).
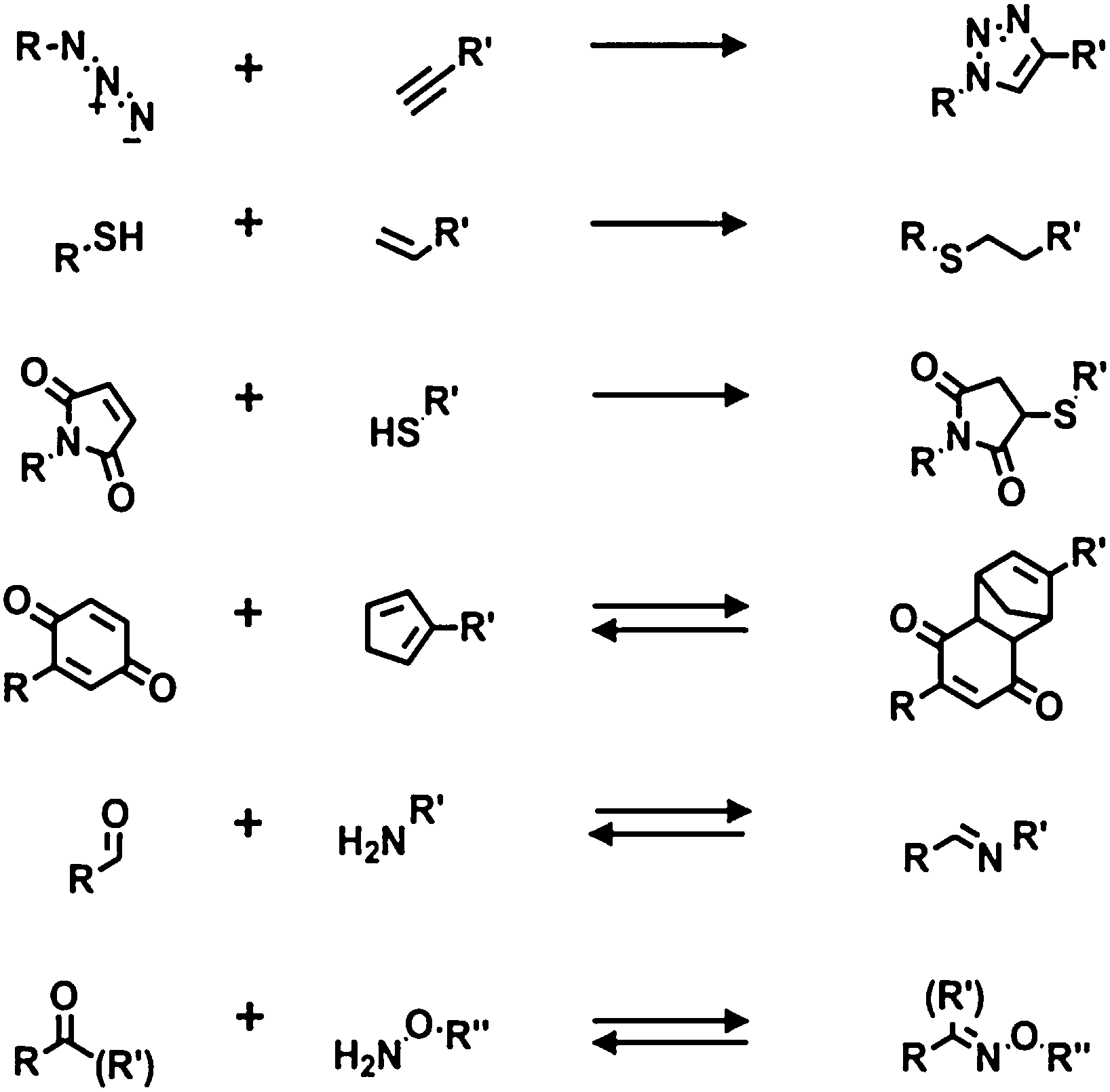 | ||
| Scheme 4 Modification of terminal groups of monolayers by means of surface-confined reactions. Examples of “click” reactions employed for monolayer modification, from top to bottom: Huisgen 1,3-cycloaddition, thiol-ene reaction, Michael addition, Diels–Alder cycloaddition, and imine and oxime formation. | ||
2.1 Azide–alkyne cycloaddition
The Huisgen reaction is a [3 + 2] cycloaddition that occurs between an organic azide (1,3-dipolar molecule) and an alkyne (dipolarophile). Owing to the kinetic stability of alkynes and azides, the reaction usually requires elevated temperatures and long reaction times with the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1,4- and 1,5-regioisomers. This reaction has obtained substantial attention only after the introduction of the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC or “click” chemistry),24,43,44 providing regioselectivity towards the 1,4-regioisomer and an extraordinary enhancement of reactivity (increase in reaction rate up to 107 times) under mild reaction conditions.45,46 Cu(I) can be provided to the reaction mixture by means of different methods: (i) the most common approach is by chemical reduction of Cu(II) salts (usually Cu(II) sulfate pentahydrate in the presence of an excess of sodium ascorbate);24 (ii) by direct addition of a Cu(I) salt;47 (iii) by comproportionation of Cu(II) salts with copper metal;43 or (iv) by electrochemical reduction of a Cu(II) salt.48 Furthermore, although CuAAC works effectively under “ligand-free” conditions, a further acceleration of the reaction has been observed upon addition of certain chelates (e.g. amine triazoles), allowing a drastic reduction of the amount of the loaded catalyst.49 The numerous examples in the literature confirm that a wide variety of reaction conditions can be successfully employed in the CuAAC.
:1 mixture of 1,4- and 1,5-regioisomers. This reaction has obtained substantial attention only after the introduction of the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC or “click” chemistry),24,43,44 providing regioselectivity towards the 1,4-regioisomer and an extraordinary enhancement of reactivity (increase in reaction rate up to 107 times) under mild reaction conditions.45,46 Cu(I) can be provided to the reaction mixture by means of different methods: (i) the most common approach is by chemical reduction of Cu(II) salts (usually Cu(II) sulfate pentahydrate in the presence of an excess of sodium ascorbate);24 (ii) by direct addition of a Cu(I) salt;47 (iii) by comproportionation of Cu(II) salts with copper metal;43 or (iv) by electrochemical reduction of a Cu(II) salt.48 Furthermore, although CuAAC works effectively under “ligand-free” conditions, a further acceleration of the reaction has been observed upon addition of certain chelates (e.g. amine triazoles), allowing a drastic reduction of the amount of the loaded catalyst.49 The numerous examples in the literature confirm that a wide variety of reaction conditions can be successfully employed in the CuAAC.
The use of the CuAAC reaction has found particular value in the selective and efficient modification of SAMs. In two early reports, Collman and coworkers employed CuAAC to functionalize azide monolayers on gold electrodes (prepared mixing azidoundecanethiol with decanethiol as diluent) with alkyne-modified ferrocene in solution.50,51 The reaction, in terms of azide consumption, was monitored via infrared (IR) and X-ray photoelectron spectroscopies (XPS) while the extent of formation of triazole was assessed via electrochemistry, exploiting the redox-active ferrocene substituents. Furthermore, using the same building blocks, they demonstrated the selective functionalization of independently addressed microelectrodes by means of the control of the CuAAC via electrochemical activation/deactivation of a copper(II) complex (Scheme 5).50 These experiments demonstrated the potential of CuAAC for the electrochemically driven local functionalization of monolayers on metals.
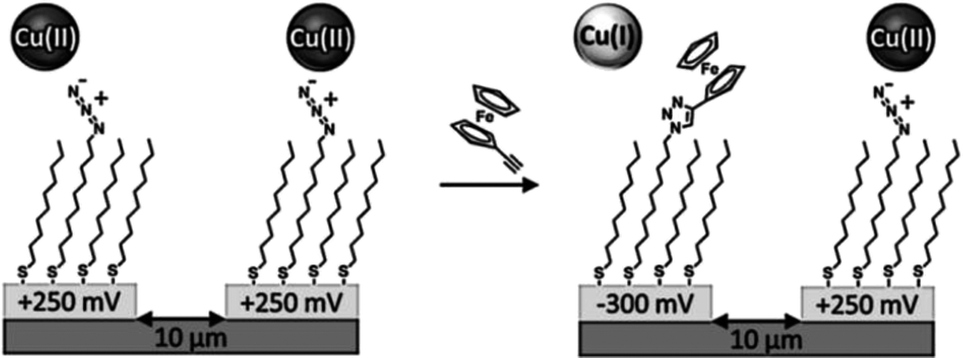 | ||
| Scheme 5 Selective functionalization of independently addressed microelectrodes by electrochemical activation and deactivation of a copper catalyst for the CuAAC reaction. Adapted from ref. 50. | ||
Electrochemically driven CuAAC via scanning electrochemical microscopy (SECM) was presented by Ku et al. as a method to anchor small molecules to an insulating substrate.52 A gold ultra microelectrode (UME) was brought close to an azido-terminated monolayer on glass and the Cu(I) active catalyst was locally generated via electrochemical reduction of a Cu(II) salt and employed for the patterning of an alkyne-functionalized fluorescent molecule via click reaction. This study thus presents a valuable method to extend the surface patterning properties of SECM.
Also alkyne-functionalized SAMs have been used as a platform for click modification. Lee et al. explored the reactivity of ethynyl-terminated SAMs on gold towards “click” chemistry using an extensive surface characterization: IR and XP spectroscopies, ellipsometry, and contact angle goniometry were employed to demonstrate that also ethynyl-terminated SAMs are useful for the introduction of functional groups on surfaces via CuAAC.53 Chaikof and coworkers prepared alkyne-terminated monolayers by the Diels–Alder reaction of an α,ω-poly(ethylene glycol) (PEG) linker with alkyne and cyclopentadiene terminal groups on a N-(ε-maleimidocaproyl)-functionalized glass slide.54 This platform was employed to immobilize, by CuAAC, a wide range of azide-containing biomolecules (biotin, carbohydrates and proteins).
Soft lithographic techniques were employed in combination with “click” chemistry for spatially resolved functionalization of monolayers. Ravoo and coworkers demonstrated fast triazole formation induced via microcontact printing of an alkyne-inked PDMS stamp onto an azide-functionalized monolayer on glass.55 Surprisingly the reaction proceeded without Cu(I) catalysis presumably owing to the high local alkyne concentration. Further studies established that addition of Cu(I) or the use of Cu(0)-coated PDMS stamps improve the efficiency and surface density (Scheme 6).56,57 “Click” chemistry by μCP was conveniently employed by Bertozzi and co-workers57 and Ravoo et al.58 to pattern microarrays of carbohydrates on azide monolayers as a probe of glycan-binding receptors, antibodies, and enzymes.
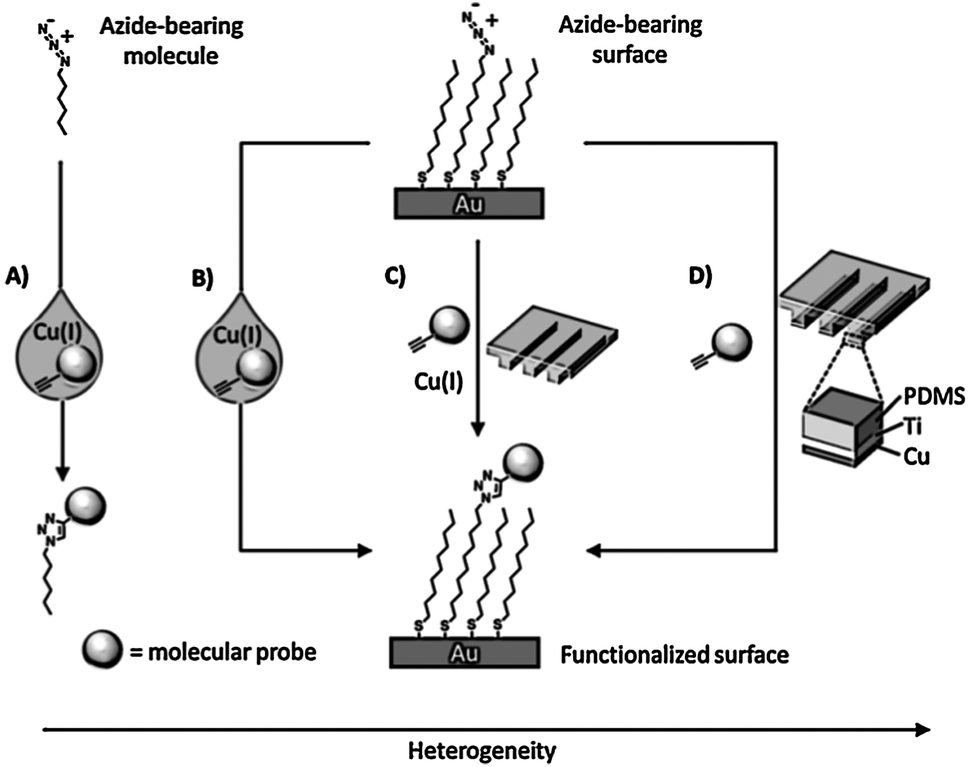 | ||
| Scheme 6 Various routes to achieve the Cu(I)-catalyzed azide–alkyne coupling (CuAAC) in different environments: (A) solution: a solution reaction where the azide, alkyne, and catalyst participate in a homogeneous reaction; (B) solution–surface: a heterogeneous reaction where the dissolved alkyne and catalyst react with a surface bound azide; (C) reagent-stamping: a heterogeneous reaction where the alkyne and catalyst are brought into contact with a surface-bound azide in the condensed phase; (D) StampCat: a heterogeneous reaction where immobilized copper catalyzes the reaction of an alkyne with a surface bound azide in the condensed phase. Adapted from ref. 56. | ||
Nanometer-scale patterning was obtained by Paxton et al. by a constructive scanning probe lithography method using copper-coated atomic force microscopy tips to catalyze the CuAAC between small alkyne molecules in solution and azide-terminated monolayers on silicon surfaces.59 The click reaction occurred only in the areas where the Cu-coated tip was brought into contact with the monolayer.
A rapid reaction under mild conditions has also been obtained in a Cu-free system by means of strained dipolarophiles such as cyclo-octynes60 and dibenzocyclo-octynes.61 Recently a Cu-free click chemistry62,63 (strain-promoted azide–alkyne cycloaddition, SPAAC) was introduced as a surface immobilization strategy ideal for biological applications due to the elimination of copper salts that are potentially cytotoxic, in combination with the high retention of activity in comparison with CuAAC.64 Orski and coworkers employed the Cu-free click chemistry using a photoactive protected strained cyclo-octyne to achieve the selective spatial immobilization of azide-functionalized fluorescent dyes.65 A silicon wafer was functionalized with poly(N-hydroxysuccidimide 4-vinyl benzoate) brushes for the coupling of a cyclopropenone-masked dibenzocyclooctynes (Scheme 7). UV irradiation promoted the fast decarbonylation of the cyclopropenone to the alkyne that became available for the Cu-free click chemistry with azide-modified fluorescent molecules. By means of UV irradiation in the presence of a shadow mask they demonstrated the fabrication of multicomponent surfaces with spatially resolved chemical functionalities.
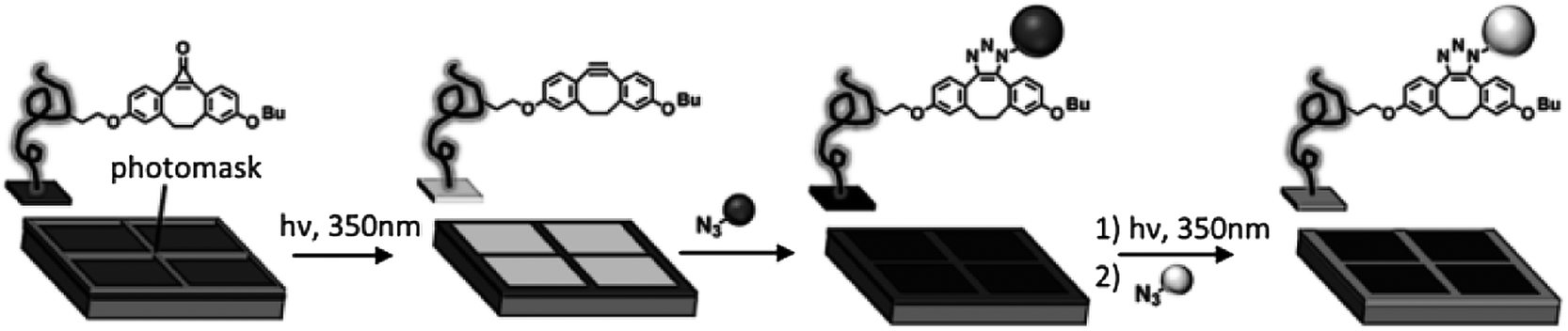 | ||
| Scheme 7 Stepwise photodecarbonylation of cyclopropenone-masked dibenzocyclooctynes for the local immobilization of azide-functionalized dyes via Cu-free click chemistry. Adapted from ref. 65. | ||
Furthermore, μCP and SPAAC were employed by Ravoo and coworkers for a fast and efficient modification of azide monolayers on glass for the immobilization of multiple biomolecules on the same substrate.66 In this work they demonstrated the orthogonality of SPAAC with other interfacial click reactions (e.g. nitrile oxide–alkene/alkyne cycloaddition) for the fabrication of protein microarrays.
2.2 Michael addition and thiol-ene reactions
The two most common thiol click reactions are the base-catalyzed Michael addition reaction and the radical-mediated thiol-ene reaction. The thiolate anion and the thiyl radical are highly reactive species leading to extremely rapid conjugation reactions with maleimides and alkenes (or alkynes), respectively.Using thiols as reactive building blocks for the functionalization and/or patterning of surfaces has a unique biological benefit. Cysteine (Cys) is the only naturally occurring amino acid containing a thiol group in its side chain, and its relative abundance in proteins is small (less than 1%). Cys residues can be introduced in a protein through site-specific mutation of, for example, Ser or Ala residues, preferably in a remote solvent-accessible part of the protein. Gaub and coworkers genetically modified an enzyme to carry an accessible C-terminal cysteine residue, which was then shown to selectively bind to a maleimide-functionalized surface.67
Among other advantages, complex and diverse biologically active arrays can be prepared using large numbers of cysteine-functionalized peptides generated in solid-phase schemes. Owing to the high yield and excellent selectivity for immobilization of the maleimide–thiol reaction, Mrksich et al. demonstrated that SAMs presenting a maleimide functional group can be conveniently used for the preparation of biochips upon reaction with thiol-modified biologically active ligands (e.g. peptides and carbohydrates, Scheme 8).68 An interesting application was developed by Magnusson and coworkers for the fabrication of surfaces with specific effects on cell behavior.69 In particular they used a maleimide-functionalized SAM to immobilize a Cys-modified peptide that triggers cellular chemotaxis and a calcium-dependent oxidative metabolism.
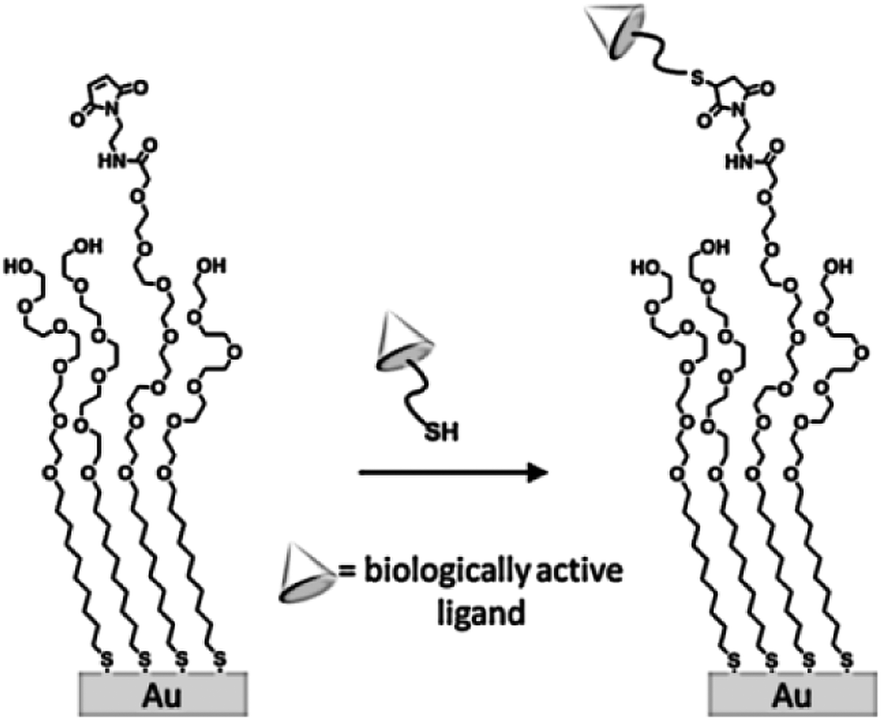 | ||
| Scheme 8 Structure of a self-assembled monolayer used to immobilize thiol-terminated ligands. Adapted from ref. 68. | ||
Jonkheijm et al. employed the thiol-ene reaction to pattern proteins onto a surface using the biotin/streptavidin (SAv) approach.70 An alkene-modified biotin was patterned on a thiol-modified silicon surface upon exposure to UV light at 365 nm through a micro-featured photomask or at 411 nm by means of laser-assisted nanopatterning. The biotin pattern was incubated with Cy5-labeled SAv yielding fluorescently visible protein patterns employed in a SAv sandwich approach, to immobilize bioactive enzymes. In a similar approach, Escorihuela and coworkers used UV-promoted thiol-ene coupling for the fabrication of DNA microarrays and the implementation of hybridization assays on silicon.71 The selective attachment of DNA occurred through a multistep process including the preparation of a thiol-functionalized silicon slide, the UV-promoted thiol-ene coupling of an alkene-modified biotin and the subsequent immobilization of SAv and biotinylated DNA. A photochemical μCP method was employed by Ravoo and coworkers to pattern bioactive thiols on alkene- or alkyne-terminated SAMs on silicon oxide (Scheme 9).72 An oxidized PDMS stamp was incubated in a diluted solution of thiol and a radical initiator (α,α-dimethoxy-α-phenylacetophenone, Irgacure 651), dried and brought into conformal contact with the functionalized substrate. Successful immobilization was achieved upon short time (5–600 s) UV irradiation at 365 nm. This technique, in combination with orthogonal contact chemistry for amide and triazole formation, was employed by the same group for the fabrication of multifunctional platforms for the immobilization of biomolecules in microarrays.73
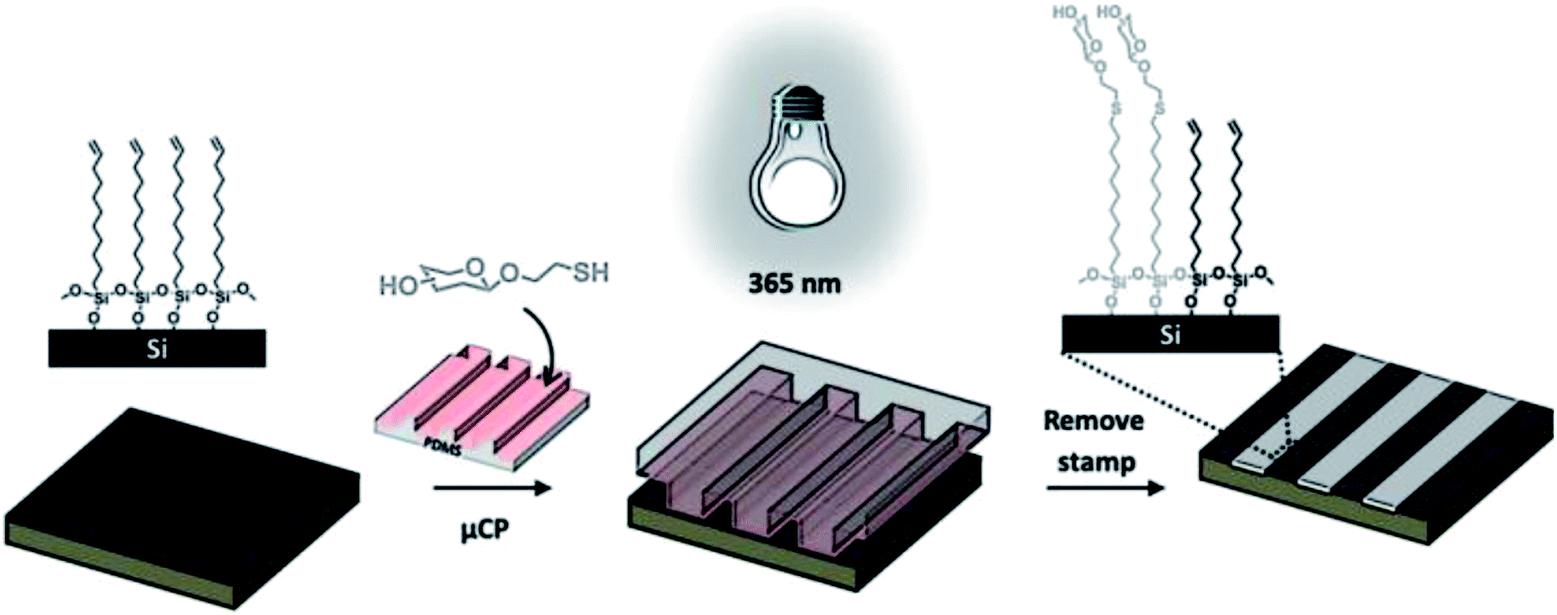 | ||
| Scheme 9 Schematic illustration of photochemical μCP by thiol-ene chemistry: an oxidized PDMS stamp inked with a thiol and a radical initiator is placed on an alkene-terminated SAM and irradiated with UV light (365 nm). Immobilization of the thiol occurs exclusively in the area of contact. Adapted from ref. 72. | ||
2.3 Diels–Alder reaction
The Diels–Alder (D–A) reaction is a reversible [4 + 2] cycloaddition occurring between a conjugated diene (in the cis configuration) and an electron-deficient dienophile. The reaction is orthogonal, efficient, atom conservative, does not require a catalyst and is insensitive to the reaction conditions (e.g. solvent, air).The pioneers to investigate the D–A reaction for the immobilization of biologically active molecules on SAMs were Yousaf and Mrksich.74 Hydroxyl and hydroquinone mixed SAMs on gold were prepared and the D–A reaction with cyclopentadiene-modified ligands was electrochemically modulated via oxidation of the hydroquinone to the active quinone. The D–A reaction between the quinone monolayer and a cyclopentadiene-modified biotin in solution was monitored via cyclic voltammetry, resulting in a loss in current due to the cycloaddition (the quinone reagent is electrochemically active while the product is not). Furthermore they used the association of biotin/streptavidin as a model system for the D–A-mediated immobilization of proteins. In this work and in follow-up studies they demonstrated that the interfacial reaction occurs following a pseudo-first-order rate law (with excess of cyclopentadiene in solution) that strongly depends on the nature of the microenvironment surrounding the quinone moieties in the monolayer.75–77 Moreover, exploiting the local electrochemical activation of the hydroquinone groups, this reactive monolayer was conveniently and elegantly employed to direct the selective stepwise attachment and microscale patterning of two different cell types,78 to switch on cell migration79 (Fig. 1) and to fabricate peptide chips to quantify the enzymatic activity of protein kinase.80
 | ||
| Fig. 1 Strategy for the design of a substrate that can be electrically switched to turn on cell adhesion. Adapted with permission from ref. 79. © 2001 Wiley-VCH Verlag GmbH, Weinheim, Germany. | ||
Mrksich and coworkers introduced a variation of the system for the photopatterned immobilization of ligands.81 The hydroquinone unit was equipped with a nitroveratryloxycarbonyl (NVOC) group resulting in a photoactive monolayer. Upon UV irradiation through a microfiche mask or using the light through an optical microscope, the hydroquinone was locally deprotected and extended for the subsequent electrochemical oxidation to quinone and the D–A-mediated immobilization of cyclopentadiene-modified ligands.
More recently, Ravoo et al. performed the D–A reaction via reactive μCP.82 Cyclopentadiene- or furan-modified carbohydrates were immobilized on maleimide-functionalized glass and silicon substrates by means of fast cycloaddition locally induced via μCP. Microarrays containing up to three different carbohydrates were prepared using this method and the binding of lectins was assessed.
Photochemically activated D–A reactions were recently developed for the spatially controlled cycloaddition on SAMs.83,84 Barner-Kowollik et al. achieved spatial control by immobilization of the photoactive component and subsequent direct UV activation (Fig. 2).83 The strategy is based on the immobilization of a triethoxysilane-functionalized o-methylphenyl aldehyde on a silicon substrate followed by the photoisomerization to photoenol that undergoes a fast D–A reaction in the presence of a dienophile (e.g. maleimide) (Fig. 2). A selective local surface-confined reaction was confirmed by the photopatterning of a small-molecule ATRP initiator (Fig. 2B), a polymer, and a peptide.
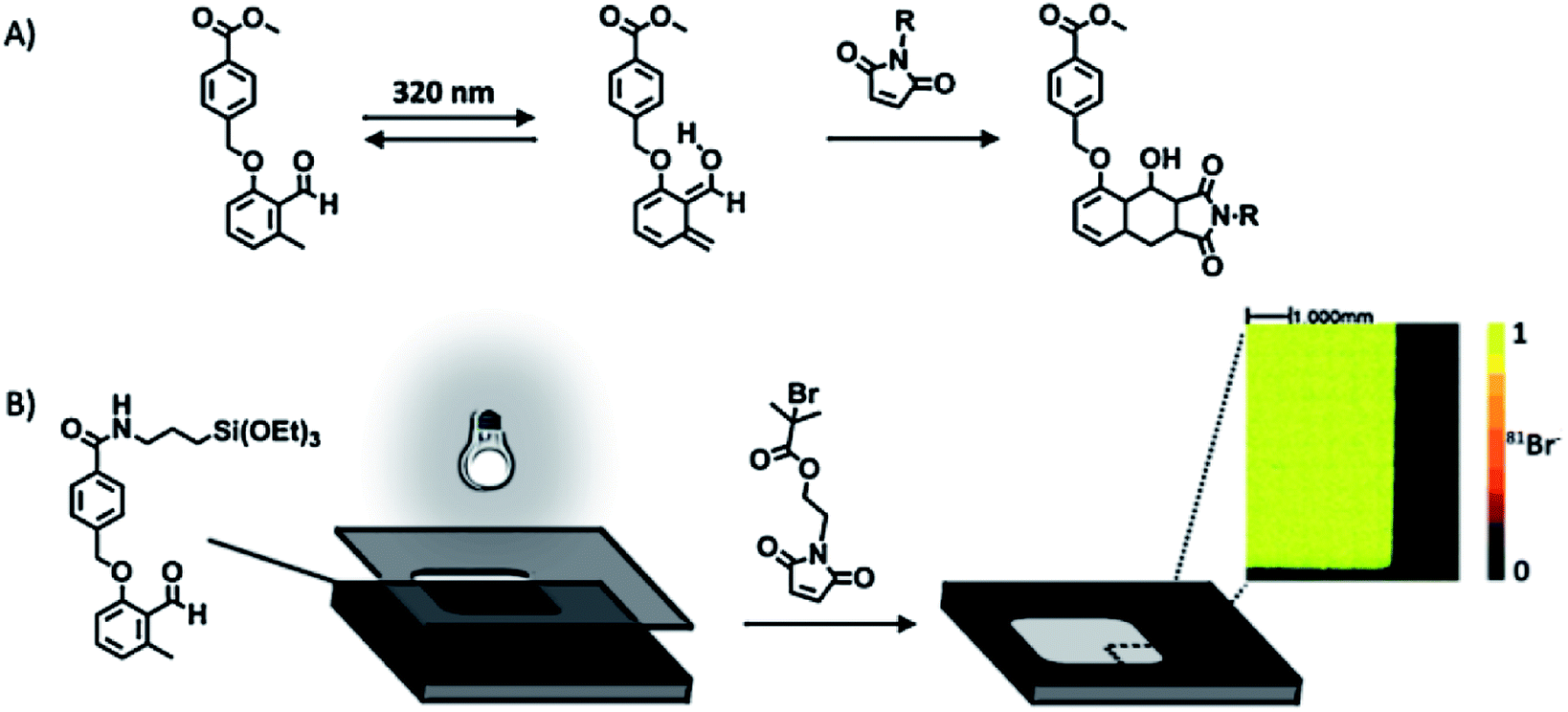 | ||
| Fig. 2 (A) Photoinduced isomerization of a 2-formyl-3-methylphenoxy (FMP) derivative and subsequent Diels–Alder [4 + 2] cycloaddition with a dienophile. (B) Structure of the FMP-functionalized monolayer and representation of the phototriggered Diels–Alder surface grafting of a bromine-containing maleimide derivative through a shadow mask. The inset shows a ToF-SIMS image of the patterned silicon wafers. Adapted with permission from ref. 83. © 2012 Wiley-VCH Verlag GmbH, Weinheim, Germany. | ||
Arumugam and Popik employed a photochemically inert surface and a light-sensitive compound in solution to perform a hetero-D–A addition of 2-napthoquinone-3-methides (oNQMs) to a vinyl ether-functionalized substrate (Fig. 3).84 Irradiation of a 3-(hydroxymethyl)-2-naphthol (oNQM precursor) resulted in efficient and fast dehydration to oNQM that underwent a fast and quantitative D–A cycloaddition with vinyl groups on the surface. The facile D–A reaction combined with the short lifetime of the photoactivated species allowed the spatial control of surface derivatization. The interface reaction was visualized by the immobilization of a biotin-modified oNQM and subsequent co-localization of avidin-FITC (Fig. 3B).
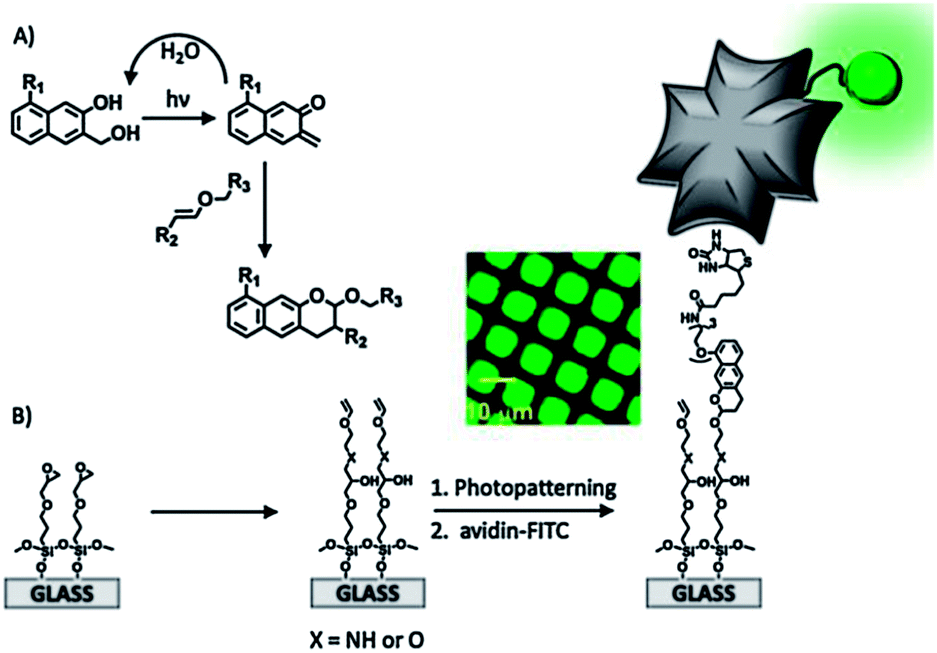 | ||
| Fig. 3 (A) Mechanism of the dehydration of the substrate and the formation of oNQM and quantitative Diels–Alder cycloaddition to yield photostable benzo[g]chromans. (B) Schematic representation of the preparation and light-directed biotinylation of vinyl ether-coated slides followed by immobilization of FITC-avidin. The inset shows a fluorescence microscopy image of a vinyl ether-coated surface irradiated through a 12.5 μm pitch copper grid. Adapted with permission from ref. 84. © 2011, American Chemical Society. | ||
2.4 Imine and oxime formation
The reaction between an amine or an aminooxy moiety and an aldehyde (or a ketone) for the preparation of an imine or oxime, respectively, is widely employed in the fabrication of monolayers in which the interactions between the molecules in solution and the counterpart immobilized on the surface occur through the formation of reversible molecular bonds. Myles and coworkers were pioneers of the immobilization of amines on aldehyde-terminated monolayers on gold substrates, providing a comprehensive characterization of the imine formation by means of FTIR, XPS and contact angle measurements.85Ravoo et al. have described a method for the reversible formation of imines from the reaction of amines/aldehydes with aldehyde/amine monolayers on gold and silicon oxide.86 An amino-terminated monolayer on gold or silicon was directly reacted with aldehydes in solution or via μCP to form full or patterned imine monolayers. Alternatively, the amine-reactive functionality was switched to aldehyde via reaction with terephthaldehyde to allow the reaction with aliphatic amines or the fluorescent Lucifer Yellow for the optical readout of the imine formation. Contact angle goniometry, FT-IRRAS, AFM and fluorescence microscopy attested the reversibility of the obtained imine monolayers under acid-catalyzed hydrolysis. In a following study, aldehyde-terminated monolayers were employed for the microcontact printing-mediated covalent immobilization of collagen-type protein col3a1 for studies on adhesion, proliferation and migration of HeLa cells.87
Barner-Kowollik and coworkers combined the phototriggered deprotection of an o-nitrobenzyl derivative to obtain spatial and temporal control of the oxime reaction.88 Silicon wafers were coated with a 2-[(4,5-dimethoxy-2-nitrobenzyl)oxy]tetrahydro-2H-pyranyl (NOTP) silane derivative that experienced fast photocleavage upon irradiation at 370 nm, yielding a nitrosobenzaldehyde-terminated monolayer. When the silicon wafer was covered with a photomask, the photo-deprotection led to the formation of a pattern of aldehyde groups. Next, the oxime formation was demonstrated by means of the reaction with O-[(perfluorophenyl)methyl] hydroxylamine hydrochloride (fluoro marker) and GRGSGR peptide, and subsequent surface imaging via time-of-flight secondary ion mass spectrometry (ToF-SIMS).
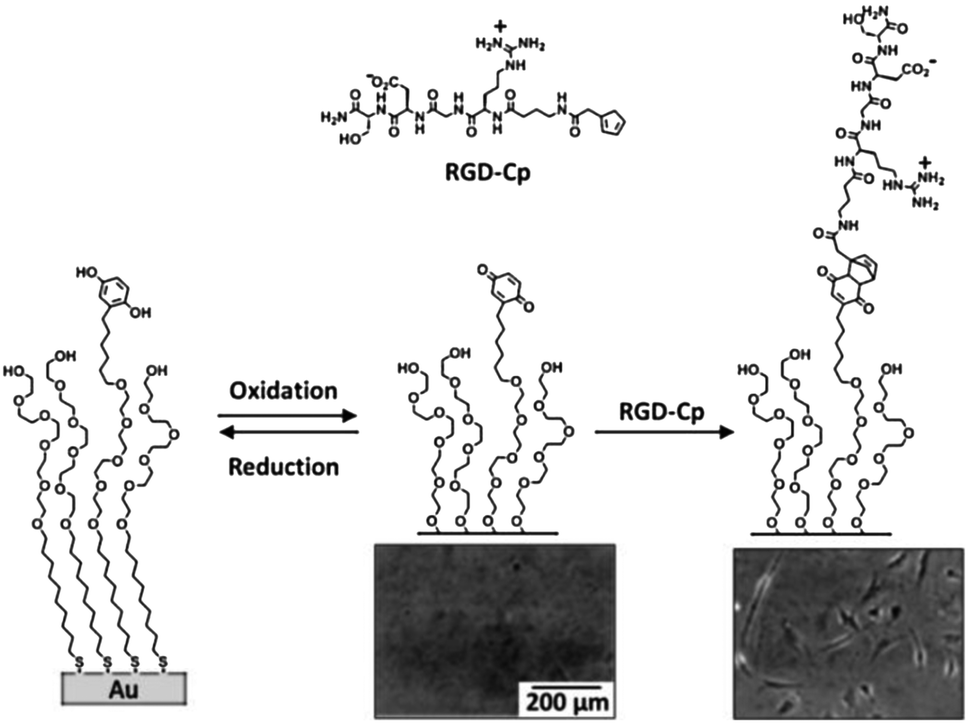
Yousaf et al. proposed, in a set of studies, a system for the reversible and tunable attachment of aminooxy-functionalized ligands: a redox-active hydroquinone monolayer on gold was electrochemically oxidized to benzoquinone, which was subsequently reacted with aminooxy-containing molecules to form the corresponding oxime (Fig. 4A).89 Since both quinone and oxime are electrochemically active (characterized by different redox potentials) the yield of the reaction and the density of the immobilized ligand were determined and modulated (Fig. 4B). The versatility of this method was demonstrated by the immobilization of peptides for protein binding89 and for cell adhesion89 (Fig. 4C) and differentiation90 studies, and by the fabrication of renewable microarrays.91
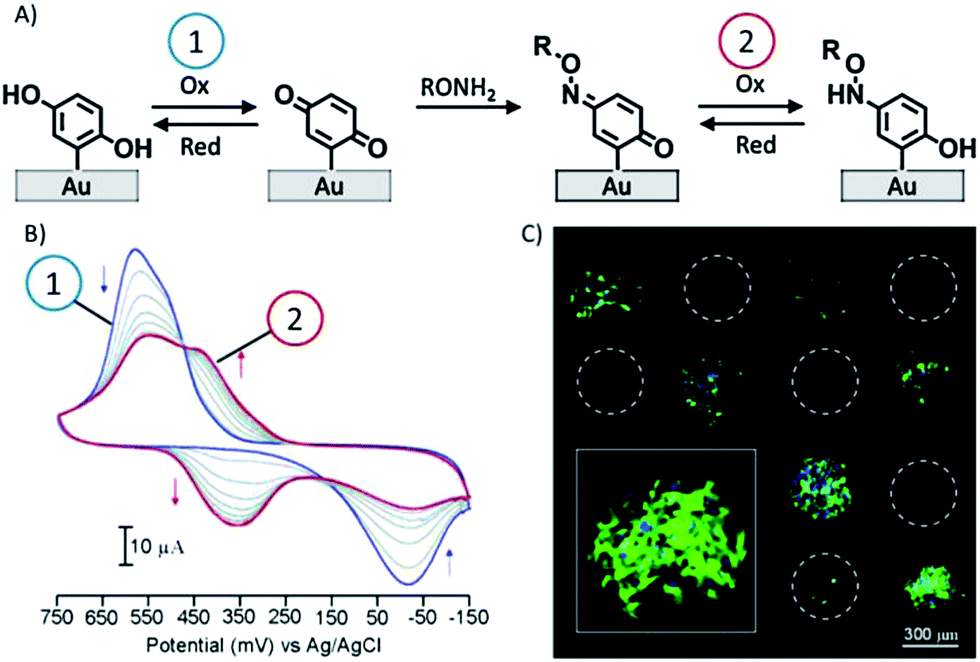 | ||
| Fig. 4 (A) Redox-active hydroquinone monolayer undergoes electrochemical oxidation to benzoquinone. The resulting quinone then reacts chemoselectively with aminooxy acetic acid to give the corresponding oxime. (B) Cyclic voltammograms showing the extent of the interfacial reaction between soluble aminooxy acetic acid and a quinone monolayer. (C) A fluorescence microscopy image of attached cells on a surface arrayed with immobilized aminooxy-modified RGD and GRD peptides. Cells attached only to the spots presenting the RGD peptide. Adapted with permission from ref. 89. © 2006, American Chemical Society. | ||
2.5 Fluorogenic monolayers
Fluorescence-based technologies are employed in materials science and (bio)sensing since they allow the fast, simple, sensitive and non-destructive detection, diagnosis and investigation of (bio)chemical processes.92–95 Many fluorescent probes have been designed and employed to be selective and sensitive towards various analytes operating through specific chemical reactions.Fluorogenic molecules have been employed as reactive monolayers for the fabrication of microarrays96–98 and for the simultaneous immobilization and detection of bio- and macro-molecules.99,100 To this end, Salisbury et al. synthesized a wide range of fluorogenic peptidyl coumarin substrates, 7-amino-4-carbamoylmethyl coumarin peptides, to study protease activity.97 A set of peptide-modified fluorogenic coumarins were spotted and immobilized via oxime ligation onto an aldehyde-terminated monolayer. The microarrays were incubated with a variety of serine proteases and the fluorescence intensity recorded after proteolysis was used to quantify the extent of the cleavage, giving direct information on the enzyme/peptide specificity. In a similar approach, Zhu et al. described the synthesis of different coumarin-based fluorogenic molecules and their use in microarrays to quantitatively and specifically detect the activity of four classes of enzyme hydrolases.98
Also in our group technologies based on the construction of fluorogenic platforms were recently developed. Huskens et al. described the immobilization of a pyrylium derivative on a glass substrate for the anchoring of amines (e.g. aliphatic amines, a fluorescent protein and a lissamine rhodamine B ethylenediamine) through μCP and dip-pen nanolithography (Scheme 10).99 Upon reaction with a primary amine the initially intense fluorescence of the pyrylium monolayer faded out proving the actual covalent immobilization.
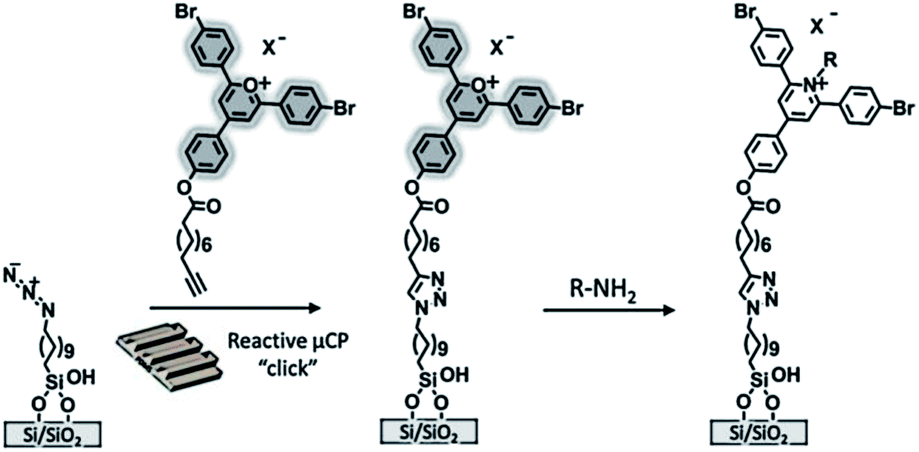 | ||
| Scheme 10 Preparation of a pyrylium-terminated monolayer. The printing of the alkyne-functionalized pyrylium is followed by the covalent immobilization and detection of amines. Adapted from ref. 99. | ||
Velders and coworkers demonstrated the selectivity and specificity of orthogonal covalent and noncovalent functionalization for small molecules.100 In their work bifunctional alkyne-cyclodextrin patterned surfaces were prepared via reactive μCP of an azido-modified β-cyclodextrin on a fluorogenic alkyne-modified coumarin monolayer. The fluorescence enhancement upon alkyne–azide cycloaddition was used to monitor the effective bond formation and to localize the β-cyclodextrin monolayer.
Recently Huskens and Jonkheijm described the fabrication of a thiol-sensitive fluorogenic reactive platform that allowed reporting of the immobilization of thiols by fluorescent signaling using an orthogonally modified coumarin (Fig. 5).101–103 The fluorogenic coumarin was equipped with an alkyne moiety for the immobilization on azide monolayers on glass via CuAAC and a methyl-4-oxo-2-butenoate group for the fluorogenic Michael addition of thiols. The fluorogenic platform allowed for the spatial identification and coverage determination of the thiol immobilization. A powerful aspect of the platform is the visualization of binding events onto the bound thiol ligand by colocalized fluorescence imaging, witnessing local binding events onto immobilized ligands which are signalled by the underlying coumarin layer. This system was employed for biological applications, such as the anchoring of cell adhesion101 and cell differentiation102 promoting peptides, and the orthogonal immobilization of fluorescent proteins103 (Fig. 5). In the latter case, a patterned fluorescent protein array was fabricated through the combination of covalent and non-covalent chemistry. Oriented protein immobilization was achieved using a cysteine-engineered fluorescent protein (Clavularia cyan fluorescent protein, TFP) for the direct formation of a covalent bond with the surface-confined coumarin and a thiol-modified nitrilotriacetate ligand for the supramolecular binding of hexahistidine-tagged red-fluorescing protein (Entacmaea quadricolor, TagRFP) in the presence of NiCl2.
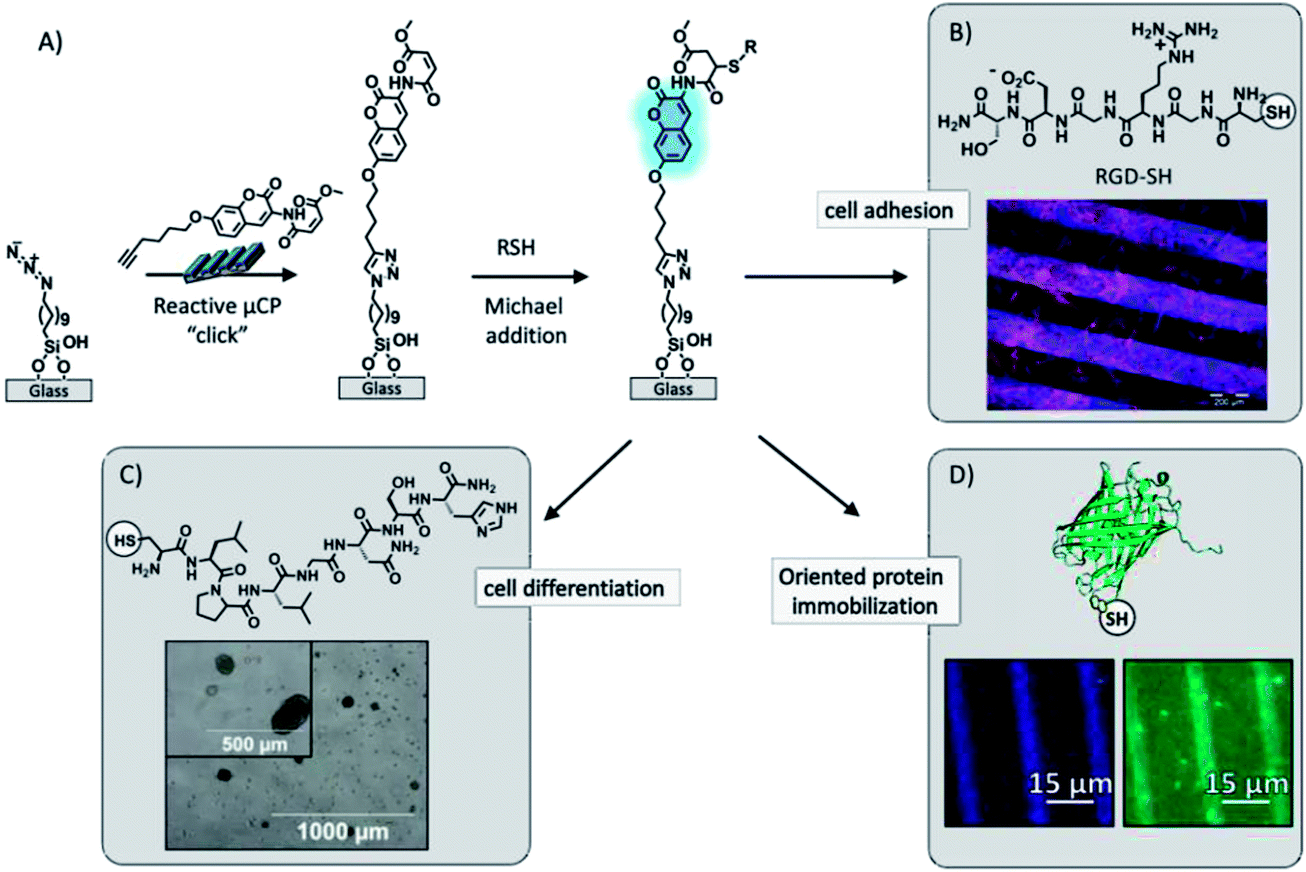 | ||
| Fig. 5 (A) Schematic procedure of the surface functionalization by printing the bi-functionalized coumarin via “click” chemistry onto an azide monolayer and followed by simultaneous covalent immobilization and detection of thiols by means of the fluorogenic Michael addition to the methyl-4-oxo-2-butenoate moiety. (B) Fluorescence microscopy image after incubation of the fluorogenic platform in a RGD-SH solution and subsequent C2C12 (mouse myoblast) cell culture. Adapted with permission from ref. 101. © 2012, Wiley-VCH Verlag GmbH, Weinheim, Germany. (C) Bright field microscopy image after reported immobilization of a cysteine-modified peptide for binding and delivery of growth factors (TGF-β1) for chondrogenic differentiation in bone marrow mesenchymal progenitor cells. Adapted with permission from ref. 102. © 2013, The Royal Society of Chemistry. (D) Fluorescence microscopy images after the detection of immobilization of a cysteine-engineered green fluorescent protein. Adapted with permission from ref. 103. © 2013, American Chemical Society. | ||
3 Surface chemical gradients of self-assembled monolayers
Surface chemical gradients are surfaces with gradual, continuous or discrete, variation in space and/or time of physicochemical properties. Surface gradients have been successfully employed for the study of interfacial phenomena in the areas of, among others, biology to investigate cell migration (haptotaxis) and polarization, materials science, e.g. for the study of the driven motion of liquid droplets,104,105 and combinatorial/analytical chemistry.106–108The first study describing the fabrication of surface chemical gradients was illustrated by Elwing et al. in 1987.109 The gradient was the result of controlled silane diffusion in liquids. A hydrophilic silicon plate was placed in a cuvette filled with a biphasic solution of dimethyldichlorosilane in trichloroethylene covered with xylene. In this system, organosilane molecules diffuse to the xylene phase and deposit on the silicon substrate yielding surface gradients. The resulting gradient was employed to study the wettability-driven adsorption and interaction of proteins and polymers at the liquid–solid interface. From that very first study, a wide variety of methods and techniques were developed for the generation of surface chemical gradients mainly based on the controlled adsorption/desorption of monolayers on substrates.
In 1992 Chaudhury and Whitesides prepared wettability surface gradients by vapor diffusion of decyltrichlorosilane along a silicon substrate, one of the most commonly used techniques to prepare silane-based gradients in the millimeter–centimeter scale.38 These gradients were employed to study the motion of water droplets based on the surface tension acting on the liquid–solid interface on the two opposite sides of the drop.
An exhaustive description of gradient fabrication methods has been reported in recent comprehensive reviews.39–42 Therefore we focus here on the preparation of surface chemical gradients by means of local chemical modification of reactive terminal functional groups of SAMs. Below are described flexible and dynamic methods where the formation of surface gradients is driven by interfacial chemical reactions and interactions with the possibility of tailoring and controlling the functionalization of arbitrary surfaces in space and time.
3.1 Photochemically controlled surface reactions
A common strategy to develop surface patterns is based on the photochemical deprotection of terminal photosensitive groups of SAMs. Yousaf and coworkers have employed this methodology to pattern ligands and cells in gradients on inert surfaces.110,111 A nitroveratryloxycarbonyl (NVOC)-protected hydroquinone ethylene glycol-terminated alkanethiol monolayer on gold underwent photochemical deprotection upon UV illumination to reveal the electrochemically active hydroquinone unit. When the irradiation was performed in the presence of a grayscale photomask, a surface gradient of hydroquinone moieties was obtained. An aminooxy reactive quinone was obtained upon electrochemical oxidation of the hydroquinone while the NVOC-protected units remained completely redox inactive. The quinone gradient was reacted with soluble aminooxy-tagged ligands to form a stable oxime conjugate via chemoselective ligation. A rhodamine–oxyamine was used to visualize the surface gradient while an RGD–oxyamine peptide was immobilized to study cell migration and proliferation along the gradient. Interestingly the dynamic character of the monolayers allowed the electrochemical release of the ligands by means of the reduction of the oxyamine bond and the restoration of the surface for a further ligand immobilization step.Ito and coworkers prepared a surface chemical gradient via photodegradation of an octadecylsilane (ODS) monolayer on silicon.112 The photodegradation was performed using a vacuum UV light (VUV) setup with an excitation wavelength of 172 nm. The VUV light was absorbed by the ODS layer with formation of radicals due to the dissociation of C–C, C–H and C–Si bonds that can react with oxygen and water to give surface oxidized species. A millimeter-scale surface gradient of oxidized groups (e.g. carboxy, aldehyde and hydroxy) was obtained by moving the substrate, positioned on a sample holder, at a controlled velocity of 50–100 μm s−1. The formation of the gradient was confirmed by water contact angle goniometry and fluorescence microscopy after labelling the carboxylate groups with fluoresceinamine. Furthermore, the obtained surface gradient was employed to investigate the motion of water micro-droplets from the hydrophobic to the hydrophilic side. A similar approach was described by Gallant and coworkers.113 An ODS monolayer on silicon was gradually oxidized by placing the substrate on a motorized stage next to the slit aperture of a UV lamp. The exposure time-dependent ozone-derived oxidation of the monolayer resulted in the formation of surface gradients of oxidized species (alcohols, aldehydes, and carboxylic acids). A bifunctional propargyl-derivatized amino linker was attached to the acid gradient by using standard amidation methods to yield a surface possessing varying coverages of alkyne groups: a useful platform for the subsequent “click” modification. In this way an RGD peptide surface gradient was fabricated to investigate cell adhesion and spreading behavior.
3.2 Electrochemically driven surface chemical reactions
Control over the length-scale, shape and functionality of surface chemical gradients was recently achieved by means of electrochemically mediated reactions, in particular the electrochemically activated copper(I) azide–alkyne cycloaddition (“e-click”) and atom transfer radical polymerization (“e-ATRP”).By means of stenciled114 or bipolar115 “e-click”, surface gradients of covalently bound alkyne-bearing molecules were created on azide-functionalized conductive polymers. Hansen et al. fabricated surface gradients of fluorine-rich and bioactive alkyne-modified molecules using a stenciled electro-click process, by tuning the amount of electrochemically generated Cu(I) (by reduction of CuSO4) by spatial confinement of the active electrodes.114 The shape of the gradient obtained on the conductive polymer (poly-3,4-(1-azidomethylethylene)-dioxythiophene (PEDOT-N3)) was defined by the geometry of the insulating layer positioned on the copper counter electrode. The distance between the counter electrode and the reactive surface dictates the speed of generation of the catalyst while the reaction conditions (e.g. concentration of reagents and catalyst, applied potential and reaction time) control the steepness and density of the gradient. The parameters affecting the formation of the surface gradient were first investigated by immobilization of a fluorine-rich alkyne-bearing molecule and characterization by XPS. Thereafter, biological applications were demonstrated by fabrication of cell adhesion peptide and protein gradients.
Huskens and coworkers described a method for the investigation of the reactivity of interfacial reactions in space and time.116 Electrochemically derived solution gradients of a reaction parameter (pH) and of a catalyst (Cu(I)) were employed to fabricate micron-scale surface chemical gradients and to study the kinetics of the surface-confined imine hydrolysis and the CuAAC (Fig. 6). This method suggested the possibility of investigating the effect of the reaction parameters of a wide range of reactions on the reaction kinetics in space and time.
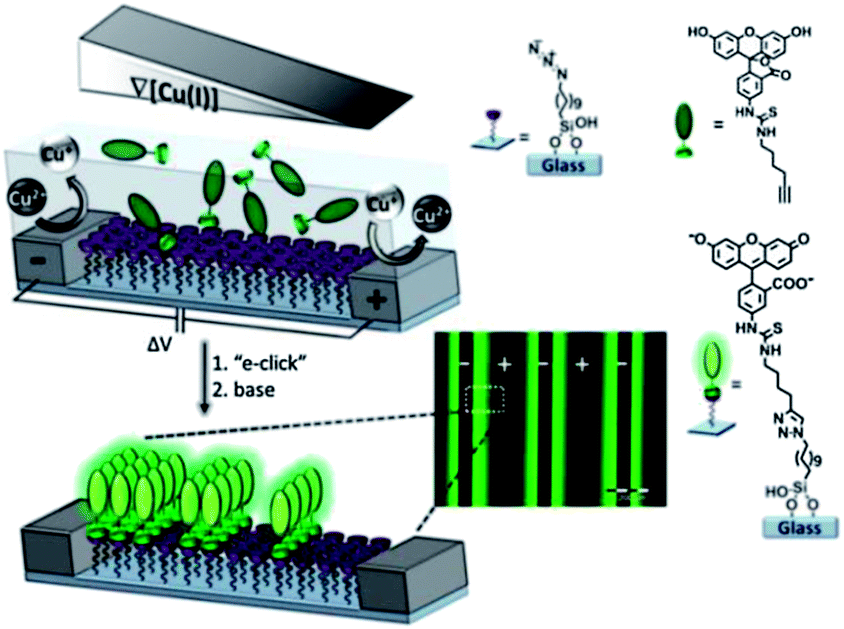 | ||
| Fig. 6 Illustration of the fabrication of surface chemical gradients via electrochemically promoted CuAAC of an alkyne-modified fluorescein on an azide monolayer on glass between platinum microelectrode arrays. Inset: a fluorescence microscopy image of the surface chemical gradients. Adapted with permission from ref. 116. © 2013, Nature Publishing Group. | ||
Li and coworkers demonstrated that a solution concentration gradient of Cu(I) can be exploited to initiate ATRP on non-conducting substrates (silicon) for the preparation of grafted gradient polymer brushes.117 A stable diffusion gradient of activator Cu(I) and deactivator Cu(II) was formed at the gap between the working electrode and the initiator-terminated substrate. The ratio of Cu(I)/Cu(II) was tuned on different locations of the surface by placing the substrate at a tilted angle along the Cu(II)/Cu(I) gradient, thereby creating different polymerization rates at different areas, leading to a gradient in the polymer brush length (Scheme 11).
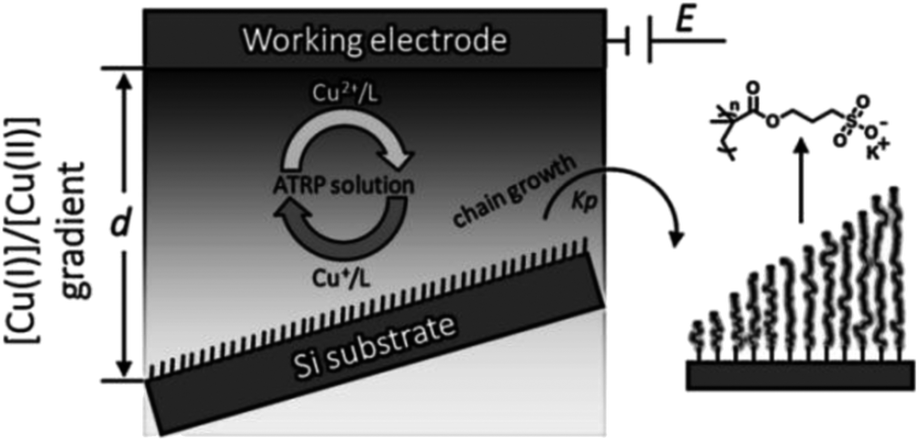 | ||
| Scheme 11 Illustration of the diffusion-controlled eATRP for the fabrication of a surface gradient of polymer brushes. Adapted from ref. 117. | ||
3.3 Non-covalent interactions and dynamic chemical reactions
Huskens and coworkers analyzed the directional spreading of multivalent ligands along self-developing gradients on a receptor platform.118 To this end, fluorescent ligands bearing one, two or three legs functionalized with adamantyl units were microcontact printed onto a cyclodextrin monolayer on glass. The surface diffusion of the ligands was driven by the developing concentration gradient of free surface receptors. The multivalent surface diffusion was monitored by fluorescence microscopy and the mechanisms involved were investigated using different concentrations of a soluble receptor (cyclodextrin) as a competitor: in pure water the divalent ligand was strongly engaged with the surface receptor monolayer and thus it walked slowly down the receptor concentration gradient; at moderate concentrations of soluble cyclodextrin, the system experienced weakening of the multivalent interaction and one of the ligand's feet was capable of making a 'solution' complex, allowing the ligand to hop along the surface using the second foot; when the cyclodextrin concentration was increased, the probability of both feet binding to soluble cyclodextrin increased, and the ligand became free to fly further across the surface leading to more rapid spreading of the fluorescent ligands (Fig. 7).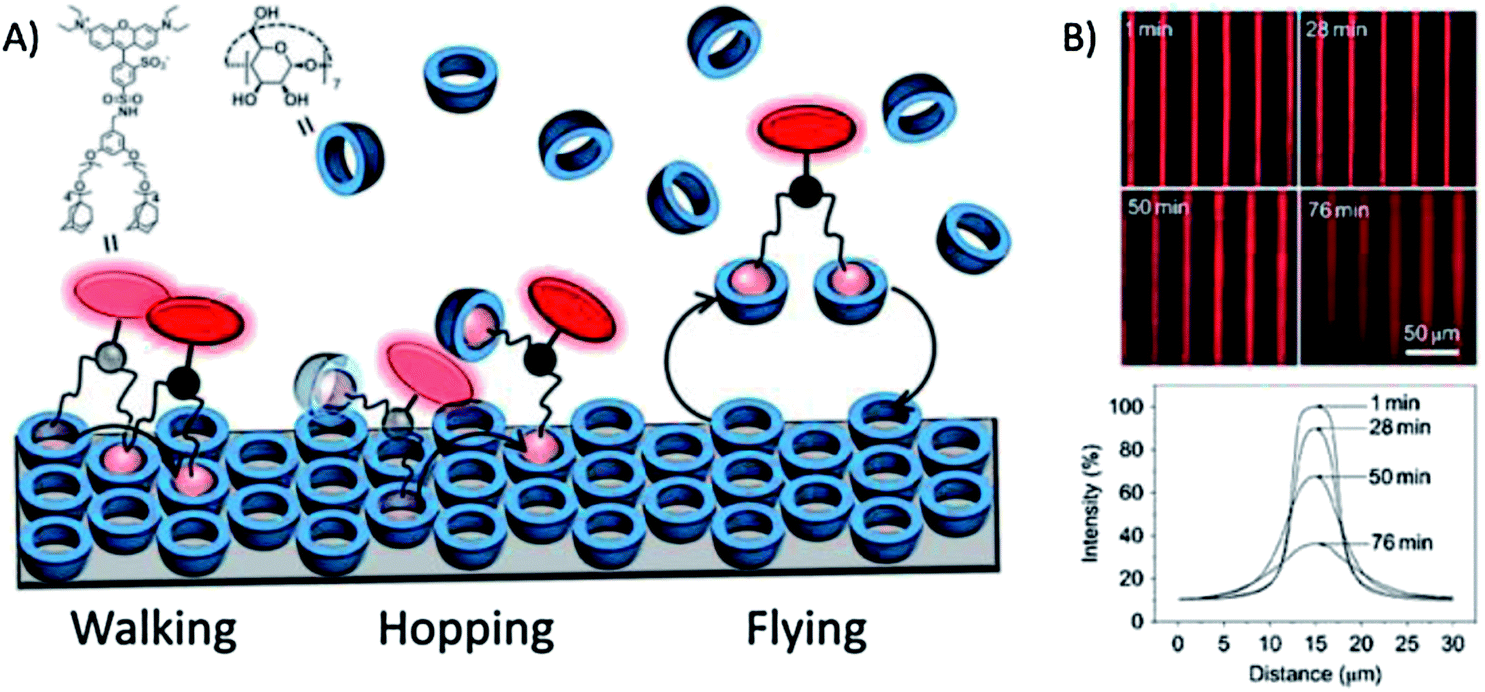 | ||
| Fig. 7 (A) Schematic representation of the basic mechanisms involved in multivalent surface diffusion. (B) Fluorescence microscopy images (top) and integrated line profiles (bottom) of printed lines of a bis-adamantyl fluorescent ligand on a β-cyclodextrin-terminated surface after incubation for given amounts of time in a solution with 2 mM β-cyclodextrin. Adapted with permission from ref. 118. © 2011, Nature Publishing Group. | ||
Giuseppone and coworkers developed dynamic self-assembled monolayers for the fabrication of mixed surface gradients of small molecules and proteins.119 In particular they used dynamic covalent chemistry as a tool to control the selective functionalization of surfaces in space and time: an aldehyde-terminated monolayer on silicon was incubated in a solution of various amine-functionalized fluorophores of different pKa values (e.g. benzylamine (9.5) and alkylamine (10.5)) with simultaneous modulation of the pH (time-dependent parameter) and withdrawal of the sample at constant speed (space-dependent parameter). As a result, highly modular (bio)functional surface imine gradients were obtained with applications for the fabrication of protein and wettability gradients. This example represents a method to design new responsive interfacial systems that can adapt their constituents to external parameters.
4 Conclusions
Reactive monolayers constitute a powerful tool for the modification of surfaces with the introduction of new functionalities for the preparation of novel materials with crucial relevance in the fields of biological microarrays, surface science and molecular discovery. A vast number of reactions can be successfully applied on monolayers to tailor the nature of terminal functional groups. The yield or rate of reactions at the surface can be limited by steric hindrance and diffusion at the solid–liquid interface. Moreover, since purification of the monolayer is impossible, high-yielding, efficient, selective and clean reactions are required. To this end, the introduction of the click chemistry paradigm has given beneficial propulsion over the last decade to materials science with the implementation of simple, orthogonal and highly efficient reactions. The development of strategies to control and switch the surface chemistry and properties by means of the integration of dynamic monolayers (e.g. electro- and photo-active SAMs) has allowed numerous applications for the investigation of the behavior of biological systems at interfaces, in particular for cell adhesion and migration studies.Control of the local surface composition of monolayers appeared remarkably important for the systematic investigation of physicochemical phenomena at the interface in space and time, laying the foundation for gradient surfaces. In the last two decades this field exhibited an incredible growth. More importantly, surface chemical gradients recently evolved in combination with reactive monolayer technologies with the development of novel flexible and powerful strategies allowing the exploration of new surface properties. Existing space and time-dependent surface reactions are good candidates for the development of powerful dynamic surface chemical gradients. We expect that these highly efficient and flexible reactions will be used for the development and implementation of new functional surfaces with tailored surface properties and performances for important applications, among others, in the fields of biology and surface science.
Acknowledgements
The work was supported by the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (NWO-CW, Vici grant 700.58.443).Notes and references
- R. D. Astumian, Phys. Chem. Chem. Phys., 2007, 9, 5067 RSC.
- G. M. Whitesides, Small, 2005, 1, 172 CrossRef CAS PubMed.
- R. D. Astumian, Science, 1997, 276, 917 CrossRef CAS.
- A. Ulman, Chem. Rev., 1996, 96, 1533 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103 CrossRef CAS PubMed.
- S. Onclin, B. J. Ravoo and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2005, 44, 6282 CrossRef CAS PubMed.
- J. Sagiv, J. Am. Chem. Soc., 1980, 102, 92 CrossRef CAS.
- L. Netzer and J. Sagiv, J. Am. Chem. Soc., 1983, 105, 674 CrossRef CAS.
- C. Haensch, S. Hoeppener and U. S. Schubert, Chem. Soc. Rev., 2010, 39, 2323 RSC.
- R. G. Nuzzo and D. L. Allara, J. Am. Chem. Soc., 1983, 105, 4481 CrossRef CAS.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805 RSC.
- H. Hakkinen, Nat. Chem., 2012, 4, 443 CrossRef PubMed.
- C. D. Bain, J. Evall and G. M. Whitesides, J. Am. Chem. Soc., 1989, 111, 7155 CrossRef CAS.
- T. P. Sullivan and W. T. S. Huck, Eur. J. Org. Chem., 2003, 17 CrossRef CAS.
- P. Jonkheijm, D. Weinrich, H. Schroder, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 9618 CrossRef CAS PubMed.
- V. Chechik, R. M. Crooks and C. J. M. Stirling, Adv. Mater., 2000, 12, 1161 CrossRef CAS.
- N. Balachander and C. N. Sukenik, Langmuir, 1990, 6, 1621 CrossRef CAS.
- N. Balachander and C. N. Sukenik, Tetrahedron Lett., 1988, 29, 5593 CrossRef CAS.
- G. E. Fryxell, P. C. Rieke, L. L. Wood, M. H. Engelhard, R. E. Williford, G. L. Graff, A. A. Campbell, R. J. Wiacek, L. Lee and A. Halverson, Langmuir, 1996, 12, 5064 CrossRef CAS.
- D. A. Hutt and G. J. Leggett, Langmuir, 1997, 13, 2740 CrossRef CAS.
- J. M. Brockman, A. G. Frutos and R. M. Corn, J. Am. Chem. Soc., 1999, 121, 8044 CrossRef CAS.
- L. Yan, C. Marzolin, A. Terfort and G. M. Whitesides, Langmuir, 1997, 13, 6704 CrossRef CAS.
- S. H. Hsu, D. N. Reinhoudt, J. Huskens and A. H. Velders, J. Mater. Chem., 2008, 18, 4959 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- H. Nandivada, X. Jiang and J. Lahann, Adv. Mater., 2007, 19, 2197 CrossRef CAS.
- B. J. Adzima and C. N. Bowman, AIChE J., 2012, 58, 2952 CrossRef CAS.
- L. Nebhani and C. Barner-Kowollik, Adv. Mater., 2009, 21, 3442 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620 CrossRef CAS PubMed.
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550 CrossRef CAS.
- A. Kumar, H. A. Biebuyck and G. M. Whitesides, Langmuir, 1994, 10, 1498 CrossRef CAS.
- A. Perl, D. N. Reinhoudt and J. Huskens, Adv. Mater., 2009, 21, 2257 CrossRef CAS.
- L. Yan, W. T. S. Huck, X.-M. Zhao and G. M. Whitesides, Langmuir, 1999, 15, 1208 CrossRef CAS.
- B. J. Ravoo, J. Mater. Chem., 2009, 19, 8902 RSC.
- C. Wendeln and B. J. Ravoo, Langmuir, 2012, 28, 5527 CrossRef CAS PubMed.
- M. K. Chaudhury and G. M. Whitesides, Science, 1992, 256, 1539 CAS.
- A. Pulsipher and M. N. Yousaf, ChemBioChem, 2010, 11, 745 CrossRef CAS PubMed.
- J. Genzer and R. R. Bhat, Langmuir, 2008, 24, 2294 CrossRef CAS PubMed.
- S. Morgenthaler, C. Zink and N. D. Spencer, Soft Matter, 2008, 4, 419 RSC.
- C. G. Simon and S. Lin-Gibson, Adv. Mater., 2011, 23, 369 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed.
- V. D. Bock, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2006, 2006, 51 CrossRef.
- F. Fazio, M. C. Bryan, O. Blixt, J. C. Paulson and C.-H. Wong, J. Am. Chem. Soc., 2002, 124, 14397 CrossRef CAS PubMed.
- V. Hong, A. K. Udit, R. A. Evans and M. G. Finn, ChemBioChem, 2008, 9, 1481 CrossRef CAS PubMed.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853 CrossRef CAS PubMed.
- J. P. Collman, N. K. Devaraj, T. P. A. Eberspacher and C. E. D. Chidsey, Langmuir, 2006, 22, 2457 CrossRef CAS PubMed.
- J. P. Collman, N. K. Devaraj and C. E. D. Chidsey, Langmuir, 2004, 20, 1051 CrossRef CAS.
- S. Y. Ku, K. T. Wong and A. J. Bard, J. Am. Chem. Soc., 2008, 130, 2392 CrossRef CAS PubMed.
- J. K. Lee, Y. S. Chi and I. S. Choi, Langmuir, 2004, 20, 3844 CrossRef CAS.
- X.-L. Sun, C. L. Stabler, C. S. Cazalis and E. L. Chaikof, Bioconjugate Chem., 2005, 17, 52 CrossRef PubMed.
- D. I. Rozkiewicz, D. Janczewski, W. Verboom, B. J. Ravoo and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2006, 45, 5292 CrossRef CAS PubMed.
- J. M. Spruell, B. A. Sheriff, D. I. Rozkiewicz, W. R. Dichtel, R. D. Rohde, D. N. Reinhoudt, J. F. Stoddart and J. R. Heath, Angew. Chem., Int. Ed., 2008, 47, 9927 CrossRef CAS PubMed.
- K. Godula, D. Rabuka, K. T. Nam and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 4973 CrossRef CAS PubMed.
- O. Michel and B. J. Ravoo, Langmuir, 2008, 24, 12116 CrossRef CAS PubMed.
- W. F. Paxton, J. M. Spruell and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 6692 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS PubMed.
- X. H. Ning, J. Guo, M. A. Wolfert and G. J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793 CrossRef CAS PubMed.
- A. Kuzmin, A. Poloukhtine, M. A. Wolfert and V. V. Popik, Bioconjugate Chem., 2010, 21, 2076 CrossRef CAS PubMed.
- S. V. Orski, A. A. Poloukhtine, S. Arumugam, L. Mao, V. V. Popik and J. Locklin, J. Am. Chem. Soc., 2010, 132, 11024 CrossRef CAS PubMed.
- C. Wendeln, I. Singh, S. Rinnen, C. Schulz, H. F. Arlinghaus, G. A. Burley and B. J. Ravoo, Chem. Sci., 2012, 3, 2479 RSC.
- K. Blank, J. Morfill and H. E. Gaub, ChemBioChem, 2006, 7, 1349 CrossRef CAS PubMed.
- B. T. Houseman, E. S. Gawalt and M. Mrksich, Langmuir, 2002, 19, 1522 CrossRef.
- J. Wetterö, T. Hellerstedt, P. Nygren, K. Broo, D. Aili, B. Liedberg and K.-E. Magnusson, Langmuir, 2008, 24, 6803 CrossRef PubMed.
- P. Jonkheijm, D. Weinrich, M. Koehn, H. Engelkamp, P. C. M. Christianen, J. Kuhlmann, J. C. Maan, D. Nuesse, H. Schroeder, R. Wacker, R. Breinbauer, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 4421 CrossRef CAS PubMed.
- J. Escorihuela, M. Jose Banuls, R. Puchades and A. Maquieira, Chem. Commun., 2012, 48, 2116 RSC.
- C. Wendeln, S. Rinnen, C. Schulz, H. F. Arlinghaus and B. J. Ravoo, Langmuir, 2010, 26, 15966 CrossRef CAS PubMed.
- C. Wendeln, S. Rinnen, C. Schulz, T. Kaufmann, H. F. Arlinghaus and B. J. Ravoo, Chem.–Eur. J., 2012, 18, 5880 CrossRef CAS PubMed.
- M. N. Yousaf and M. Mrksich, J. Am. Chem. Soc., 1999, 121, 4286 CrossRef CAS.
- M. N. Yousaf, E. W. L. Chan and M. Mrksich, Angew. Chem., Int. Ed., 2000, 39, 1943 CrossRef CAS.
- E. W. L. Chan, M. N. Yousaf and M. Mrksich, J. Phys. Chem. A, 2000, 104, 9315 CrossRef CAS.
- Y. Kwon and M. Mrksich, J. Am. Chem. Soc., 2002, 124, 806–812 CrossRef CAS PubMed.
- M. N. Yousaf, B. T. Houseman and M. Mrksich, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5992 CrossRef CAS PubMed.
- M. N. Yousaf, B. T. Houseman and M. Mrksich, Angew. Chem., Int. Ed., 2001, 40, 1093 CrossRef CAS.
- B. T. Houseman, J. H. Huh, S. J. Kron and M. Mrksich, Nat. Biotechnol., 2002, 20, 270 CrossRef CAS PubMed.
- W. S. Dillmore, M. N. Yousaf and M. Mrksich, Langmuir, 2004, 20, 7223 CrossRef CAS PubMed.
- C. Wendeln, A. Heile, H. F. Arlinghaus and B. J. Ravoo, Langmuir, 2010, 26, 4933 CrossRef CAS PubMed.
- T. Pauloehrl, G. Delaittre, V. Winkler, A. Welle, M. Bruns, H. G. Boerner, A. M. Greiner, M. Bastmeyer and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2012, 51, 1071 CrossRef CAS PubMed.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2011, 133, 15730 CrossRef CAS PubMed.
- R. C. Horton Jr, T. M. Herne and D. C. Myles, J. Am. Chem. Soc., 1997, 119, 12980 CrossRef.
- D. I. Rozkiewicz, B. J. Ravoo and D. N. Reinhoudt, Langmuir, 2005, 21, 6337 CrossRef CAS PubMed.
- D. I. Rozkiewicz, Y. Kraan, M. W. T. Werten, F. A. de Wolf, V. Subramaniam, B. J. Ravoo and D. N. Reinhoudt, Chem.–Eur. J., 2006, 12, 629 CrossRef PubMed.
- T. Pauloehrl, G. Delaittre, M. Bruns, M. Meissler, H. G. Boerner, M. Bastmeyer and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2012, 51, 9181 CrossRef CAS PubMed.
- E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2006, 128, 15542 CrossRef CAS PubMed.
- W. Luo, E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2010, 132, 2614 CrossRef CAS PubMed.
- A. Pulsipher and M. N. Yousaf, Chem. Commun., 2011, 47, 523 RSC.
- M. Eun Jun, B. Roy and K. Han Ahn, Chem. Commun., 2011, 47, 7583 RSC.
- O. S. Wolfbeis, J. Mater. Chem., 2005, 15, 2657 RSC.
- H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2009, 110, 2620 CrossRef PubMed.
- X. Qian, Y. Xiao, Y. Xu, X. Guo, J. Qian and W. Zhu, Chem. Commun., 2010, 46, 6418 RSC.
- S. Y. Lim, W.-Y. Chung, H. K. Lee, M. S. Park and H. G. Park, Biochem. Biophys. Res. Commun., 2008, 376, 633 CrossRef CAS PubMed.
- C. M. Salisbury, D. J. Maly and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 14868–14870 CrossRef CAS PubMed.
- Q. Zhu, M. Uttamchandani, D. Li, M. L. Lesaicherre and S. Q. Yao, Org. Lett., 2003, 5, 1257 CrossRef CAS PubMed.
- F. A. Scaramuzzo, A. Gonzalez-Campo, C.-C. Wu, A. H. Velders, V. Subramaniam, G. Doddi, P. Mencarelli, M. Barteri, P. Jonkheijm and J. Huskens, Chem. Commun., 2010, 46, 4193 RSC.
- A. Gonzalez-Campo, S. H. Hsu, L. Puig, J. Huskens, D. N. Reinhoudt and A. H. Velders, J. Am. Chem. Soc., 2010, 132, 11434 CrossRef CAS PubMed.
- C. Nicosia, J. Cabanas-Danes, P. Jonkheijm and J. Huskens, ChemBioChem, 2012, 13, 778 CrossRef CAS PubMed.
- J. Cabanas-Danes, C. Nicosia, E. Landman, M. Karperien, J. Huskens and P. Jonkheijm, J. Mater. Chem. B, 2013, 1, 1903 RSC.
- D. Wasserberg, C. Nicosia, E. E. Tromp, V. Subramaniam, J. Huskens and P. Jonkheijm, J. Am. Chem. Soc., 2013, 135, 3104 CrossRef CAS PubMed.
- R. B. van Dover, L. F. Schneemeyer and R. M. Fleming, Nature, 1998, 392, 162 CrossRef CAS PubMed.
- S. Suresh, Science, 2001, 292, 2447 CrossRef CAS PubMed.
- J. Genzer, D. A. Fischer and K. Efimenko, Appl. Phys. Lett., 2003, 82, 266 CrossRef CAS.
- C. M. Stafford, C. Harrison, K. L. Beers, A. Karim, E. J. Amis, M. R. VanLandingham, H.-C. Kim, W. Volksen, R. D. Miller and E. E. Simonyi, Nat. Mater., 2004, 3, 545 CrossRef CAS PubMed.
- D. Julthongpiput, M. J. Fasolka, W. Zhang, T. Nguyen and E. J. Amis, Nano Lett., 2005, 5, 1535 CrossRef CAS PubMed.
- H. Elwing, S. Welin, A. Askendal, U. Nilsson and I. Lundstrom, J. Colloid Interface Sci., 1987, 119, 203 CrossRef CAS.
- E.-J. Lee, E. W. L. Chan and M. N. Yousaf, ChemBioChem, 2009, 10, 1648 CrossRef CAS PubMed.
- E. W. L. Chan and M. N. Yousaf, Mol. BioSyst., 2008, 4, 746 RSC.
- Y. Ito, M. Heydari, A. Hashimoto, T. Konno, A. Hirasawa, S. Hori, K. Kurita and A. Nakajima, Langmuir, 2007, 23, 1845 CrossRef CAS PubMed.
- N. D. Gallant, K. A. Lavery, E. J. Amis and M. L. Becker, Adv. Mater., 2007, 19, 965 CrossRef CAS.
- T. S. Hansen, J. U. Lind, A. E. Daugaard, S. Hvilsted, T. L. Andresen and N. B. Larsen, Langmuir, 2010, 26, 16171 CrossRef CAS PubMed.
- N. Shida, Y. Ishiguro, M. Atobe, T. Fuchigami and S. Inagi, ACS Macro Lett., 2012, 1, 656 CrossRef CAS.
- S. O. Krabbenborg, C. Nicosia, P. Chen and J. Huskens, Nat. Commun., 2013, 4, 1667 CrossRef PubMed.
- B. Li, B. Yu, W. T. S. Huck, W. Liu and F. Zhou, J. Am. Chem. Soc., 2013, 135, 1708 CrossRef CAS PubMed.
- A. Perl, A. Gomez-Casado, D. Thompson, H. H. Dam, P. Jonkheijm, D. N. Reinhoudt and J. Huskens, Nat. Chem., 2011, 3, 317 CrossRef CAS PubMed.
- L. Tauk, A. P. Schroeder, G. Decher and N. Giuseppone, Nat. Chem., 2009, 1, 649 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |