Novel features of multiferroic and magnetoelectric ferrites and chromites exhibiting magnetically driven ferroelectricity
Rana
Saha
,
A.
Sundaresan
and
C. N. R.
Rao
*
Chemistry and Physics of Materials Unit and International Centre for Materials Science, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O., Bangalore-560064, India. E-mail: cnrrao@jncasr.ac.in
First published on 17th September 2013
Abstract
A few oxides such as YMnO3, TbMnO3, YMn2O5 and BiFeO3 constituted the small family of well-characterized multiferroics until recently, but this area of research has been enlarged significantly due to the advent of a novel class of oxides exhibiting interesting multiferroic and magnetoelectric properties arising from magnetically induced ferroelectricity. Interestingly, these materials are simple transition metal oxides, most of them possessing the perovskite structure. In this review article, we present the significant features of multiferroic and magnetoelectric ferrites and chromites which owe their ferroelectricity to magnetic interactions. Some of the important systems discussed are BiFeO3 whose properties are affected by magnetic and electric fields, rare-earth orthoferrites LnFeO3 (Ln = Dy, Gd and Sm) and rare-earth orthochromites LnCrO3, where exchange-striction plays a significant role. Perovskite oxides of the type Y(A1−xBx)O3 (A, B = Fe, Cr, Mn) exhibit multiferroic properties, although the existence of these properties in YFeO3 and YCrO3 is in doubt. Such oxides with a non-magnetic rare-earth cation at the A site and two transition metal ions in the B-site permit tuning the transition temperatures by varying the B site ions and their relative proportions or the Ln ion. Multiferroic properties of simple ferrites such as Al(Ga)FeO3 where cation disorder appears to play a role are also discussed. Problems and challenges in this area of research are indicated.
 Rana Saha | Rana Saha obtained his B.Sc. degree in chemistry from the University of Calcutta and M.S. degree in materials science from JNCASR. Currently he is carrying out his Ph.D. studies on multiferroics at JNCASR. |
 A. Sundaresan | A. Sundaresan obtained his Ph.D. degree from Indian Institute of Technology Bombay, and did post-doctoral work in Tsukuba, Caen and Grenoble before joining JNCASR. He is a professor at JNCASR where he works on oxide materials and magnetism. He is the recipient of the bronze medals of the Materials and Chemical Research Societies of India. |
 C. N. R. Rao | C. N. R. Rao is National Research Professor and Linus Pauling Research Professor at Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR) and Honorary Professor at the Indian Institute of Science. His main interests are in the chemistry of materials. He is an honorary fellow of RSC and a member of many of the major academies including the Royal Society and the U. S. National Academy of Sciences. He is the recipient of the Royal Medal of the Royal Society of London, Dan David Prize and Erneslo Illy Trieste Science Prize for materials research and the August Wilhelm von Hoffmann medal of the German Chemical Society. |
Introduction
Although ferroelectricity and magnetism require contradicting characteristics in a material, several multiferroics have indeed been reported in the last few years.1–4 Multiferroic properties can arise from the operation of different mechanisms. In YMnO3, electric polarization arises due to polyhedral tilting, in LnMn2O5, ferroelectricity arises from the magnetostriction induced by collinear spins in the frustrated magnetic structure.2,3 In BiFeO3, the role of the 6s2 lone pair of Bi3+ ion has been invoked.5 Ferroelectricity induced by oxygen octahedral rotation has been predicted in some perovskite oxides.6–11 Occurrence of ferroelectric polarization has been demonstrated experimentally and theoretically in metal organic frameworks (MOF) and organic crystals as well.12–15 Based on first-principles calculations, a new proper ferroelectric state has been predicted in PbNiO3.16 The most intriguing aspect of multiferroics that has emerged in the last few years relates to electric polarization that results from magnetism which is traditionally considered to be incompatible with ferroelectricity.3,17 In this review article, we will examine magnetism-driven ferroelectricity in ferrites and chromites, some of which are magnetoelectric. Magnetoelectric materials are those where there is coupling between magnetic and electric order parameters, wherein electric polarization can be controlled by magnetic fields and magnetization by electric fields.18 All multiferroics are not necessarily magnetoelectric.We shall concentrate our attention on selected oxide materials which exhibit magnetic ordering and ferroelectricity, important examples of such materials being rare-earth orthoferrites, LnFeO3 (Ln = rare earth). It may be recalled that multiferroic properties of TbMnO3, due to the cycloidal magnetic structure which breaks the inversion centre, triggered much interest in such spin-driven polarization phenomena.19 Besides the orthoferrites, we shall examine the evolution of ferroelectric polarization in rare-earth orthochromites, LnCrO3. We then present the case of two simple oxides, AlFeO3 and GaFeO3, where there is cationic disorder. Another aspect of interest is the possibility of tuning ferroelectric and magnetic ordering temperatures in perovskite oxides containing two transition metal ions in the B-site. We shall briefly touch on the challenges in this area.
BiFeO3
We shall first examine the well-studied multiferroic and magnetoelectric properties of BiFeO3 (BFO). It crystallizes in the rhombohedrally distorted perovskite structure (R3c), with relatively high ferroelectric (TC ∼ 1103 K) and antiferromagnetic (TN ∼ 643 K) transition temperatures.20,21 The magnetic structure of BFO is such that the Fe moments couple antiferromagnetically (G-type) in the pseudocubic [111]c direction which is equivalent to the hexagonal [001]h direction, leading to an incommensurately modulated cycloidal spiral spin structure with a long wavelength of 620 ± 20 Å along the hexagonal [110]h direction.22,23 This cycloidal spin structure is destroyed by magnetic fields resulting in a homogenous spin order which allows linear magnetoelectric coupling.24 Along the [111]c direction, the Bi3+ and Fe3+ cations in BFO are displaced from their centrosymmetric positions resulting in spontaneous polarization in this direction.25 The value of spontaneous polarization in the bulk sample is, however, small (∼6 μC cm−2). In epitaxial thin films and single crystals, the polarization is higher (∼100 μC cm−2),26,27 with the films showing a considerable magnetoelectric effect (Fig. 1).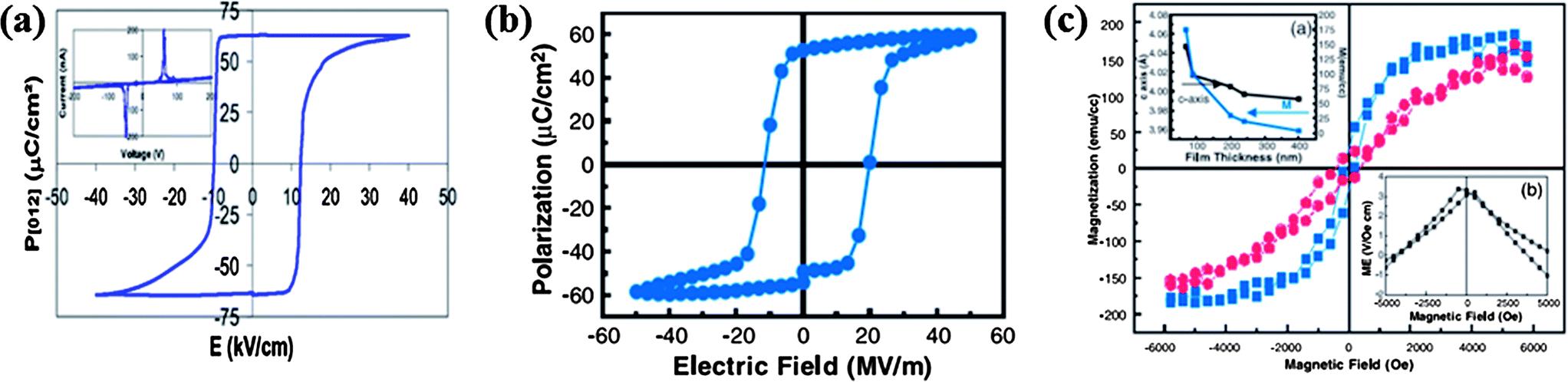 | ||
| Fig. 1 Ferroelectric hysteresis loops of BiFeO3 at room temperature for (a) the single crystal, (b) epitaxial thin film and (c) magnetic hysteresis loop of epitaxial BiFeO3 at room temperature (in-plane loop is in blue and out-of-plane loop is in red) along with the thickness dependence of saturation magnetization (top inset) and magnetoelectric measurements (bottom inset). (reproduced with permission from ref. 26, copyright 2003, by the AAAS; from ref. 27, copyright 2007, by the American Institute of Physics). | ||
Quite recently exchange bias in BFO has been manipulated by electric fields, suggesting possible applications in spintronics, memory devices and sensors.28 With this purpose, epitaxial heterostructures of antiferromagnetic BFO and ferromagnetic La0.7Sr0.3MnO3 (LSMO) have been constructed, which exhibit induced ferromagnetism in the Fe sublattice at the interface due to the interplay between the electronic orbital reconstruction and its coupling to the spin degrees of freedom.29 The moment induced by interfacial coupling together with the antiferromagnetically coupled interfacial and the second Mn layers give rise to the exchange bias (EB)29 on cooling in a magnetic field through a blocking temperature (TB; ∼100–120 K). The effect of the ferroelectric polarization on the exchange bias has been examined below TB to maximize the exchange bias by constructing a multiferroic field effect device employing BFO (200–600 nm)/LSMO (3–10 nm)/STO heterostructures.30,31 Reversible switching of exchange bias between two states with the same (unipolar modulation)30 and opposite polarity (bipolar modulation)31 at 5.5 K without the presence of any magnetic field or field cooling has been observed. The mechanism of bipolar modulation of exchange bias is explained on the basis of a model which considers coupled antiferromagnetic–ferroelectric order in BFO along with the modulation of interfacial exchange interactions due to the displacement of Fe3+ ions in BFO relative to the Mn3+/4+ ions in LSMO.31
Apart from the electrical control of magnetization in BFO thin films, modification of the cycloidal magnetic structure, exchange bias and GMR by epitaxial strain has been investigated.32 Thus, Sando et al.32 have shown that both compressive and tensile strains induced by the substrate can destroy the cycloidal modulation of Fe spins along the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] direction, thereby stabilizing the pseudo-collinear antiferromagnetism. On stretching the film, a net in-plane magnetization develops along the [10] direction, while a progressive reorientation of the spins from in-plane to out-of-plane occurs as the film is subjected to a compressive strain as shown in Fig. 2.33 Sando et al. have also demonstrated that epitaxial strain-induced spin reorientation from in-plane to out-of-plane can be used to tune the exchange bias. Reversible switching between two exchange bias states in the LSMO–BFO heterostructure with opposite exchange bias polarities upon ferroelectric poling of BFO indicates strong coupling of polarization with the magnetization at the interface. Furthermore, strain engineering can play an important role in controlling the magnetization and exchange bias of the BFO film and heterostructures thereby improving the magnetoelectric coupling.
0] direction, thereby stabilizing the pseudo-collinear antiferromagnetism. On stretching the film, a net in-plane magnetization develops along the [10] direction, while a progressive reorientation of the spins from in-plane to out-of-plane occurs as the film is subjected to a compressive strain as shown in Fig. 2.33 Sando et al. have also demonstrated that epitaxial strain-induced spin reorientation from in-plane to out-of-plane can be used to tune the exchange bias. Reversible switching between two exchange bias states in the LSMO–BFO heterostructure with opposite exchange bias polarities upon ferroelectric poling of BFO indicates strong coupling of polarization with the magnetization at the interface. Furthermore, strain engineering can play an important role in controlling the magnetization and exchange bias of the BFO film and heterostructures thereby improving the magnetoelectric coupling.
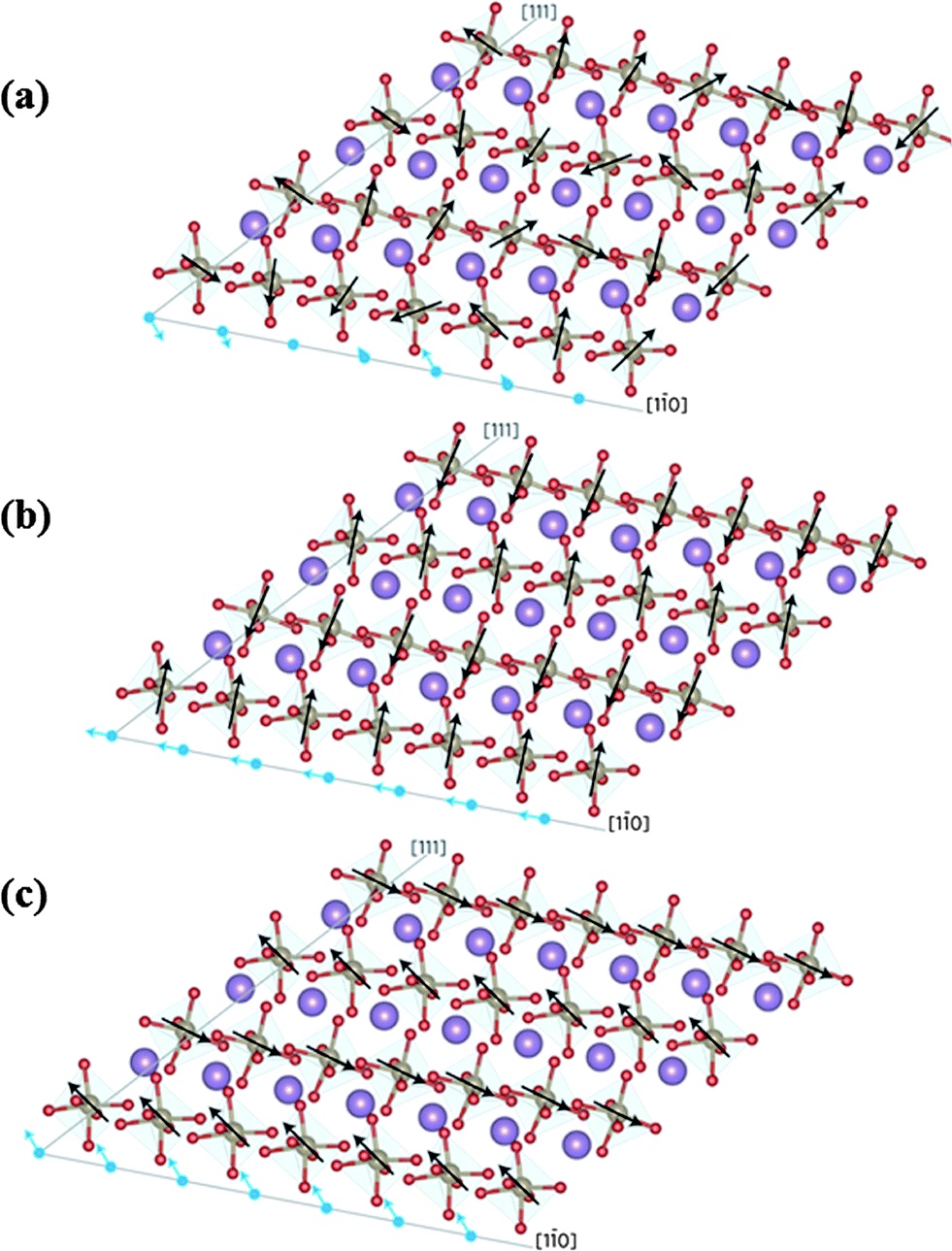 | ||
| Fig. 2 Schematic representation of magnetic moments of BiFeO3 along crystallographic [10 ] and [111] directions for (a) zero strain inducing zero net moment, (b) a tensile strain inducing a net in-plane magnetic moment and (c) a compressive strain inducing a net out-of-plane magnetic moment. The purple, brown and red colours indicate Bi, Fe and O atoms respectively. The black arrows indicate the magnetic moments of Fe; blue arrows indicate the net magnetic moments produced by rows of iron atoms along [111] (reproduced with permission from ref. 33, copyright 2013, by the Nature Publishing Group). | ||
Rare-earth orthoferrites
Rare-earth orthoferrites of the type LnFeO3 (Ln = Gd, Dy, Sm) crystallizing in the orthorhombically distorted perovskite structure (Pbnm) exhibit three types of G-type antiferromagnetic spin structures in the Fe sites, Γ4 (GxAyFz), Γ2 (FxCyGz) and Γ1 (AxGyCz),34 where Gx, Ay, Fz correspond to the components of Fe spins along crystallographic a, b and c directions respectively (Fig. 3). Among Fe–Fe, Ln–Fe and Ln–Ln exchange interactions,35 Fe–Fe ordering is the strongest leading to canted antiferromagnetic ordering of the Fe sublattice at 650–700 K.36 The comparatively weak Ln–Fe interactions cause the Ln sublattice to polarize in the exchange field of weakly ferromagnetic Fe moments. The anisotropic nature of the Ln and Fe moments gives rise to two magnetic phase transitions with temperature: (a) spin reorientation where the direction of antiferromagnetic component (G-type) changes its direction from one crystallographic axis to another and (b) temperature induced magnetization reversal below a compensation point where the rare-earth moment and the weak ferromagnetic moment of Fe sublattice are antiferromagnetically coupled. At low temperatures (<10 K), Ln–Ln interactions lead to antiferromagnetic ordering of rare-earth ions. Orthoferrites with a non-magnetic Ln ion shows only the Γ4 spin structure at all temperatures.36 The above spin structures correspond to nonpolar magnetic point symmetries Γ4 (m′m′m), Γ2 (mm′m′) and Γ1 (mmm), which do not support spontaneous electric polarization.37 Based on symmetry analysis of magnetoelectric interactions in the orthoferrites (and orthochromites), spontaneous polarization or magnetic field induced ferroelectricity is predicted to arise at the antiferromagnetic ordering temperature of the rare-earth ion.38 Indeed experimentally the occurrence of spontaneous polarization was realized at the Néel temperature of Gd3+ ions of GdFeO3 while in DyFeO3 ferroelectricity was discovered at the TN of Dy3+ ions in the presence of a magnetic field.39,40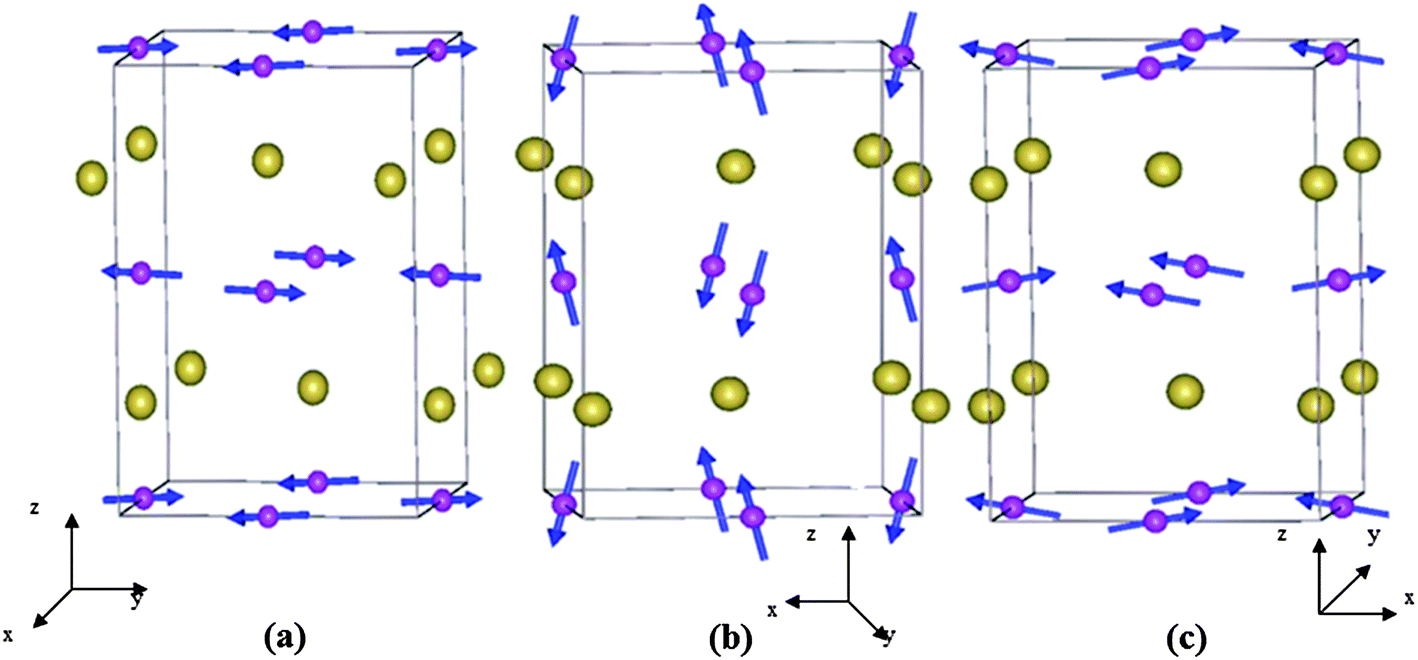 | ||
| Fig. 3 Different types of G-type AFM spins in (a) Γ1 (AxGyCz), (b) Γ2 (FxCyGz) and (c) Γ4 (GxAyFz) configurations for rare-earth orthoferrites and orthochromites with Pbnm symmetry. Fe and Ln are indicated by spheres with and without spins respectively (reproduced with permission from ref. 62, copyright 2012, by the American Physical Society). | ||
GdFeO3 (Pbnm)41 exhibits weak ferromagnetism and spontaneous ferroelectric polarization (0.12 μC cm−2, at 2 K) below the TN of Gd3+ ions in the absence of a magnetic field unlike DyFeO3.39 In contrast to Dy3+, Gd3+ is less anisotropic42 and the ground state magnetic spin configuration is Γ4, with the Fe spins ordering antiferromagnetically at 661 K39 along with weak ferromagnetism due to Dzyaloshinskii–Moriya (DM) interaction.43,44 The Gd moments undergo antiferromagnetic ordering with the spin configuration GxAy (Γ5) below 2.5 K. The magnetic point group of GdFeO3 below the ordering temperature of Gd ion is m′m′2 which allows spontaneous polarization along the crystallographic c axis. The dielectric constant measured at 1 kHz shows a peak at the onset of ferroelectric polarization, coinciding with the magnetic ordering temperature of the Gd ions (Fig. 4). The occurrence of a dielectric anomaly at the Curie temperature (TC = 2.5 K) and the presence of weak ferromagnetism indicate that GdFeO3 is multiferroic. Accordingly, we see from Fig. 4 that with increasing magnetic field along the a axis, concomitantly the dielectric peak shifts towards low temperatures and the ferroelectric polarization is suppressed.
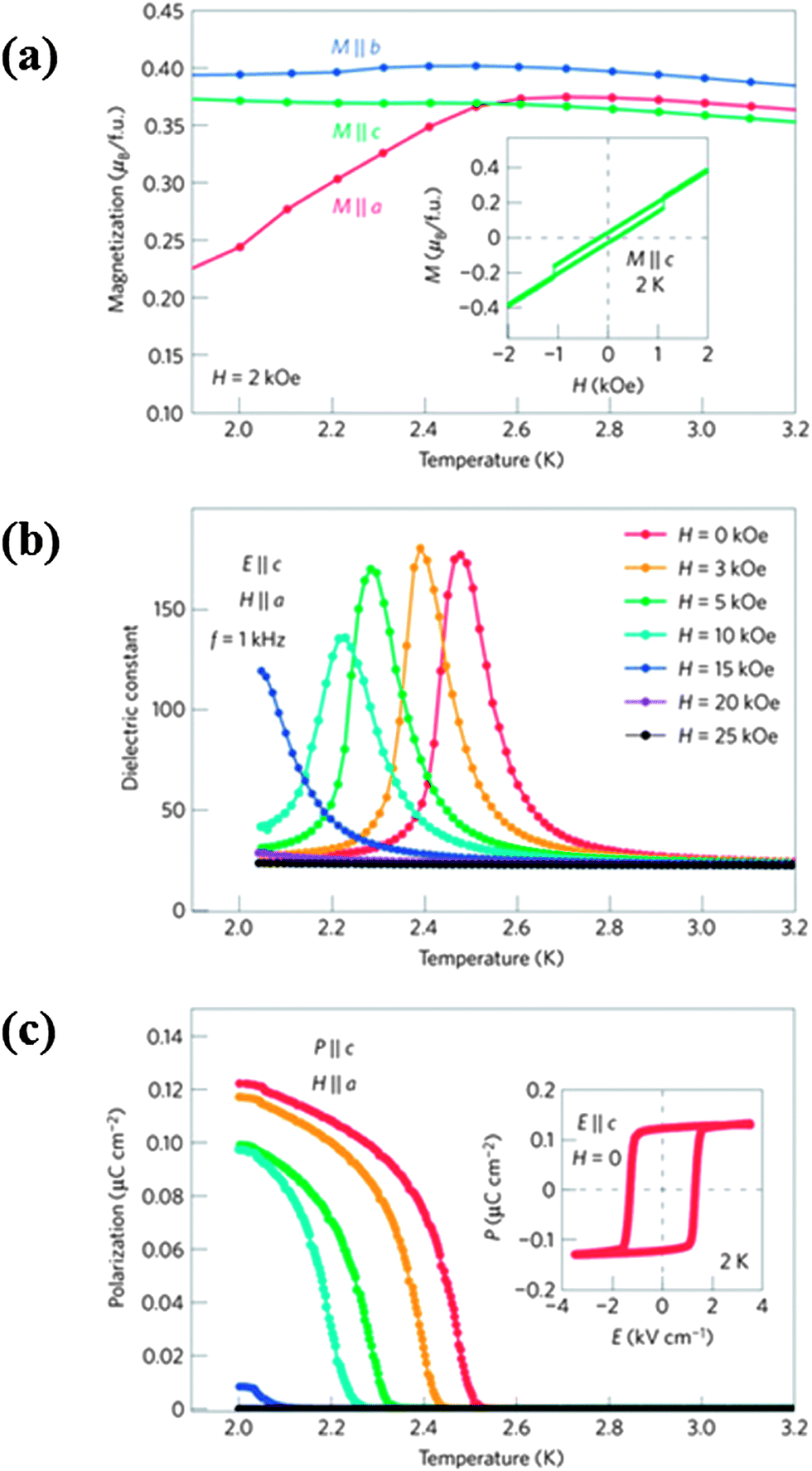 | ||
| Fig. 4 Variation of (a) magnetization at H = 2 kOe and (b) dielectric constant at f = 1 kHz, (c) electric polarization along the c direction for several H ‖ a as a function of temperature in a single crystal of GdFeO3. Insets in (a) and (c) show ferromagnetic and ferroelectric hysteresis loops respectively at 2 K (reproduced with permission from ref. 39, copyright 2009, by the Nature Publishing Group). | ||
The origin of ferroelectricity and the effect of magnetic field on polarization can be understood from the magnetic structure of GdFeO3, where the dominant spin configurations for the Fe and Gd sites are of G type (Fig. 5a). The ground state magnetic configurations of the Fe and Gd sublattice are represented by Γ4 (GxAyFz) and Γ5 (GxAy) respectively as shown in Fig. 5b. In this G type configuration, exchange-striction from the eight nearest neighbour Fe ions on the Gd ions in a cubic cell would be cancelled out (Fig. 5a). On ordering the Gd ions, the total cancellation is lost and therefore exchange-striction force acting on the Gd ions results in displacement along the c direction from the mirror plane giving rise to polarization. On application of a magnetic field (5 < H < 20 kOe) along the a direction the magnetic structure changes to Γ2 (FxCyGz) and Γ7 (Gz) for the Fe and Gd sites respectively (Fig. 5c) resulting in the suppression of polarization. Further application of a magnetic field (H > 20 kOe) along the a direction causes the spin structure of Fe (Γ2) to be retained but the Gd moment goes from Gz to Fx, thereby removing the exchange-striction force from surrounding Fe spins and the disappearance of the polar state as shown in Fig. 5d. However on application of a magnetic field along the c direction, the Fe sublattice retain the Γ4 spin structure but the Gd spins change from GxAy (Γ5) to Fz as shown in Fig. 5e. Exchange-striction force on the Gd ion therefore disappears, resulting in a non-ferroelectric state. The magnetoelectric nature of GdFeO3 can be understood on the basis of composite domain wall clamping of the domain walls for magnetization and polarization.39
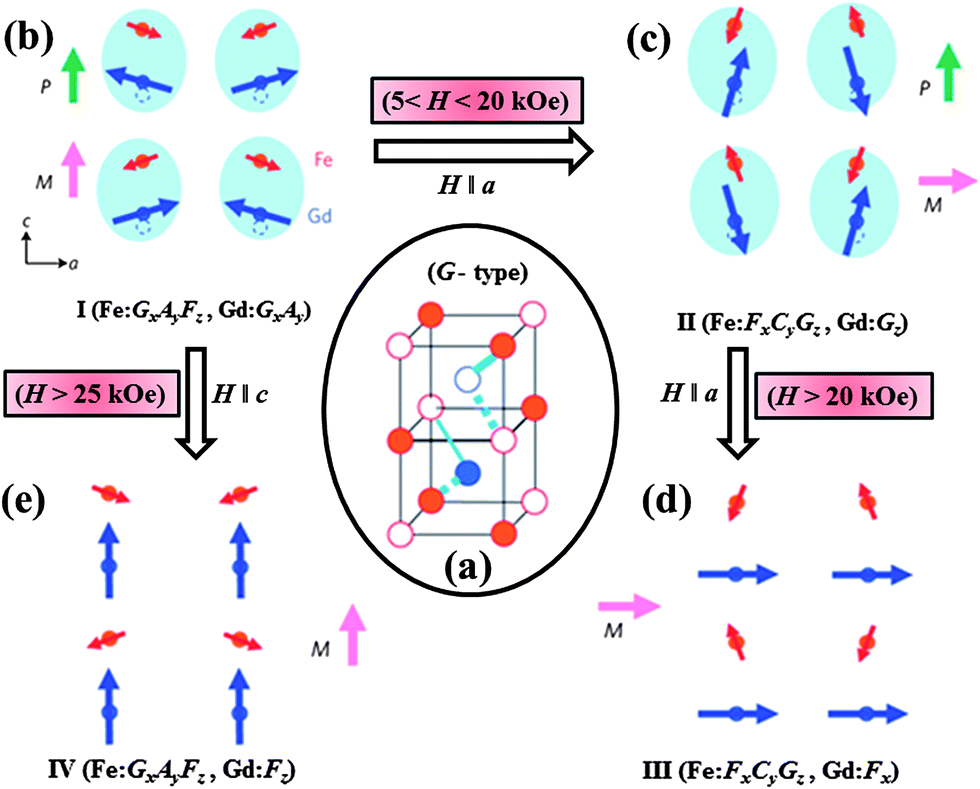 | ||
| Fig. 5 Schematic representation of magnetic structures of GdFeO3 below the magnetic ordering of Gd3+ ions in Pbnm symmetry: (a) exchange striction mechanism with the G-type magnetic structure, (b–e) spin configurations (I–IV) of Fe and Gd ions with varying magnetic field (+ and − symbols indicate the relative spin direction of each ion and blue solid (dotted) lines indicate attractive (repulsive) forces respectively) (modified with permission from ref. 39, copyright 2009, by the Nature Publishing Group). | ||
In the case of DyFeO3, below the Néel temperature (TN = 645 K) of Fe3+ ions, the spin configuration is Γ4 which allows the presence of weak ferromagnetism due to DM interaction along the c direction. On lowering the temperature, the Fe sublattice changes the direction of the antiferromagnetic spin component (G-type) from a to b axis resulting in the Γ1 magnetic structure at 37 K.45 Application of an external magnetic field along the c axis rotates the antiferromagnetic spin component from b to a (Γ1 to Γ4) and a field along the a axis changes the direction from b to c (Γ1 to Γ2).46 Further cooling leads to antiferromagnetic ordering of the Dy moments in the Γ5 (GxAy) configuration with the nonpolar magnetic point symmetry (m′m′m′) at 4 K. The point symmetries of Γ1 and Γ5 correspond to mx, my and mz symmetry, giving rise to nonpolar magnetic point group 222 which does not favour spontaneous ferroelectric polarization at the ground state in the absence of a magnetic field. On application of a magnetic field (30 kOe) along the crystallographic c direction, ferroelectric polarization (≥0.2 μC cm−2) is realized along the c direction at the magnetic ordering temperature (TN = 4 K) of the Dy ions after magnetoelectric poling with E = 2 kV cm−1.40 The occurrence of ferroelectricity at TN of Dy and its appearance on application of a magnetic field indicates the combined role of Dy and Fe spin structures. The magnetic structure of DyFeO3 (Fig. 6) consists of alternate layers of Fe and Dy moments in layered antiferromagnetic fashion.40 On application of a sufficiently high magnetic field, the Γ1 configuration of the Fe sublattice changes to the Γ4 configuration with weak ferromagnetism along the c direction. The Fe and Dy sublattices, now with Γ4 and Γ5 configurations respectively, break the mz symmetry and adopt the polar point group m′m′2 which allows the spontaneous polarization along the c direction. From Fig. 6, we see that the Fe spins remain parallel to one of the nearest Dy spins and antiparallel to other nearest Dy moments, thereby driving the cooperative displacement of the Dy layers to the Fe layers with opposite spins leading to the generation of polarization along the c direction. Density functional theoretical calculations show that coupling between Dy and Fe spins is mediated by Dy-d and O-2p hybridization.47 The existence of a polar state and weak ferromagnetism in the presence of a magnetic field makes DyFeO3 a magnetoelectric multiferroic material at low temperatures (4 K).
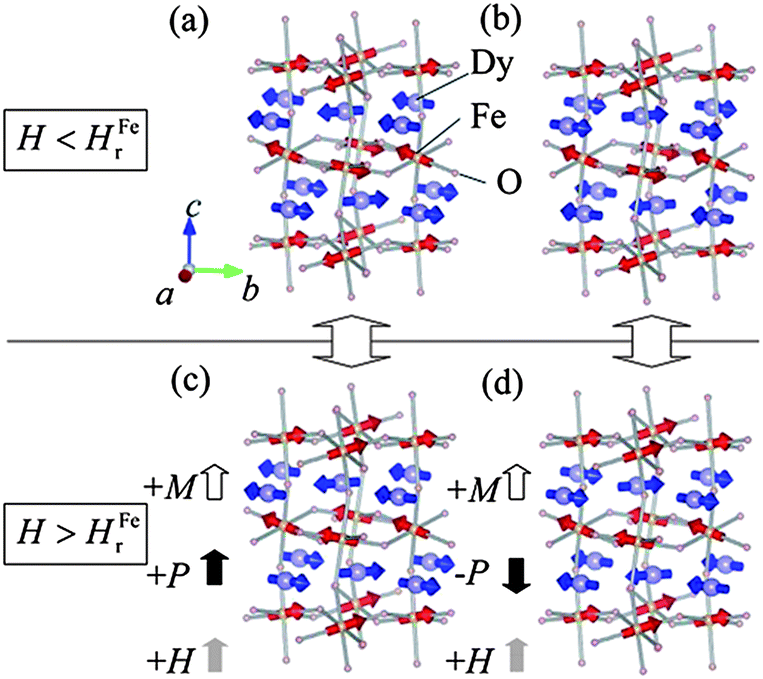 | ||
| Fig. 6 Schematic representation of magnetic structures of DyFeO3 below the magnetic ordering of Dy3+ ion in Pbnm symmetry (a and b) for linear magnetoelectric phase when H < Hr; (c and d) for multiferroic phase when H > Hr (reproduced with permission from ref. 40, copyright 2008, by the American Physical Society). | ||
The presence of electric polarization in the weakly ferromagnetic state of DyFeO3 was reported in a polycrystalline sample,48 wherein ferroelectricity disappears below the spin-reorientation temperature (TSR = 50 K), accompanied by a change in the magnetic structure from Γ4 to Γ1. Temperature dependent magnetization data are shown in Fig. 7a. Results of recent investigations on single crystals by Somnath Ghara are, however, at variance with the results on the polycrystalline material.48 Switchable electric polarization has been observed along the [100] direction (Fig. 7b) when poling was carried out across the spin-reorientation transition from 70 to 10 K, and measured while warming. Further investigations on single crystalline DyFeO3 are clearly necessary to fully understand this ferrite.
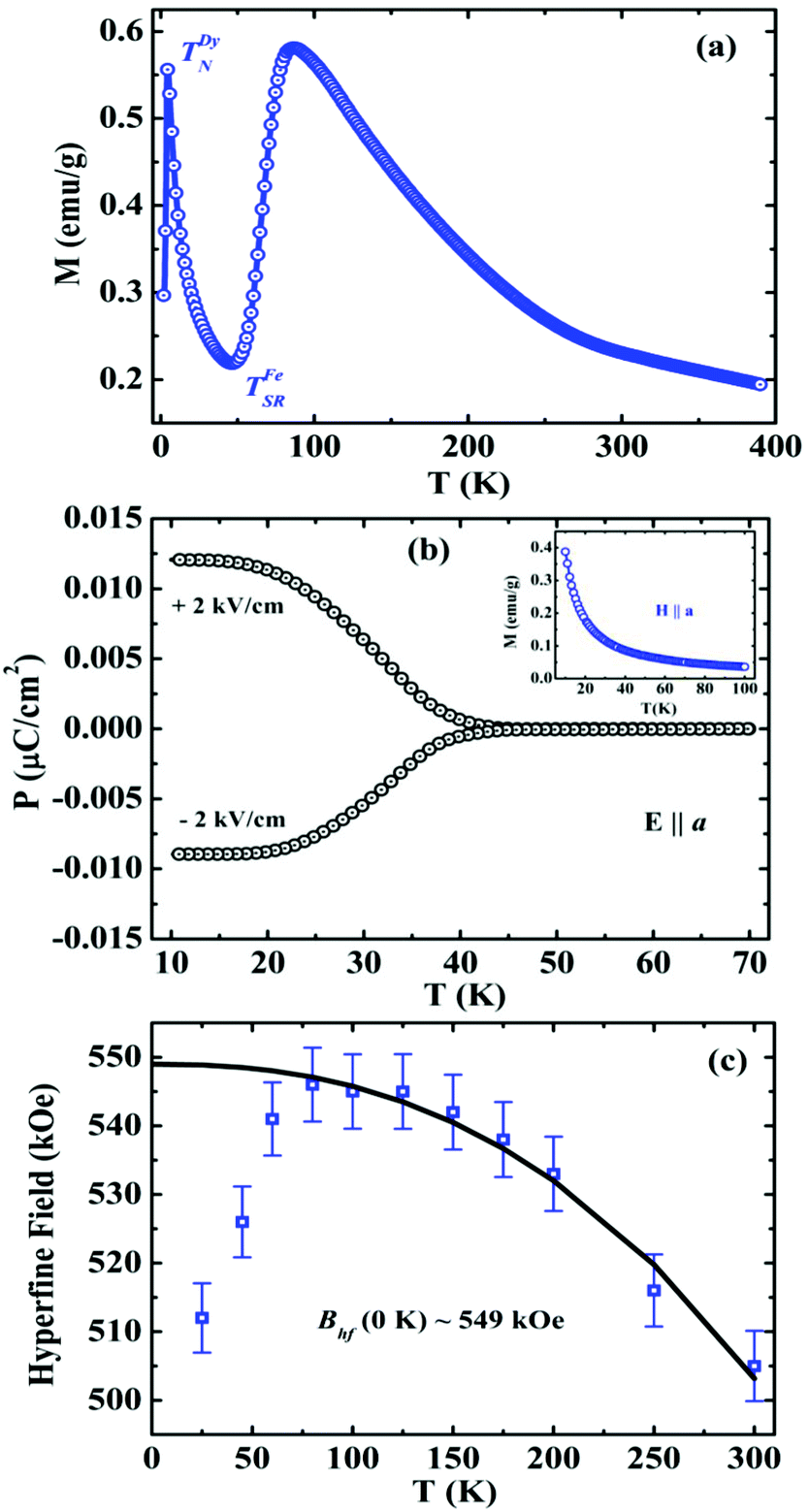 | ||
| Fig. 7 Temperature variation of (a) field-cooled (FC) magnetization in the presence of H = 100 Oe for polycrystalline DyFeO3, (b) electric polarization (E ‖ a) along with field-cooled (FC) magnetization at 100 Oe (inset) for single crystalline DyFeO3 (obtained from L. Herve, Vincent Caignaert and Antoine Maignan) and (c) hyperfine field (Bhf) in the range 25–300 K for polycrystalline DyFeO3 (modified with permission from ref. 48, copyright 2013, by the Institute of Physics). | ||
The importance of the local field induced on Dy ions by the weak ferromagnetic moment of the Fe sublattice in the Γ4 structure is revealed by the zero-field 57Fe Mössbauer spectra of DyFeO3.48 The temperature dependent hyperfine field (Bhf) deviates from the mean field like behaviour as shown in Fig. 7c. The reduction in Bhf at 80 K cannot be accounted as due to a change in the magnetic structure from Γ4 to Γ1, thereby indicating the decoupling of the Dy and Fe moments. A similar change in magnetic structure occurs in YFe0.85Mn0.15O3, but without a change in Bhf below the spin-reorientation temperature.49 The relatively high value of Bhf in the weak ferromagnetic state (Γ4) of DyFeO3 suggests that the iron nucleus experiences a local field that results from the combined effect of Fe and Dy neighbours. When the weak ferromagnetism of the Fe sublattice disappears, the local field at the Dy ions also goes to zero. This study suggests the importance of weak ferromagnetism in the evolution of the polar state in centrosymmetric DyFeO3. In the light of the recent results obtained from single crystals of DyFeO3 it is important to reinvestigate whether the magnetic Ln ion and/or weak ferromagnetism play any decisive role in the ferroelectricity of rare-earth orthoferrites.
While the exchange-striction mechanism explains the appearance of ferroelectricity below the Gd(Dy) ordering temperature, it was surprising to note that single crystalline SmFeO3 (Pbnm) exhibits ferroelectric polarization at the magnetic ordering temperature (670 K) of the Fe spins.50 Similar to LnFeO3 (Ln = Gd and Dy), below the Néel temperatures of Fe3+ ions (670 K) the magnetic structure of Fe sublattices of SmFeO3 is represented by the Γ4 configuration which shows weak ferromagnetic moment along the c axis. On lowering the temperature, the G-type component shows spin-reorientation from a to c at 480 K (TSR), with a small canting along the a direction as represented by Γ2.51 Pyroelectric current measurements show the evolution of spontaneous polarization only along the b direction at 670 K indicating the origin to be spin-driven. The origin of ferroelectricity in this material has been attributed to inverse DM interaction52 involving two non-collinear Fe spin pairs. DFT calculations reveal that spin–orbit coupling is essential to develop this finite polarization and that exchange-striction has no role in the polarization. It has since been pointed out that the neither the Γ4 nor the Γ2 magnetic structure is compatible with the observed ferroelectric polarization.37 From group theoretical analysis, it is shown that both the magnetic structures actually correspond to a nonpolar magnetic point symmetry of the Fe sites (m′m′m for Γ4; mm′m′ for Γ2).42 The magnetic ions (Fe3+) located at the inversion centre cannot therefore break the inversion symmetry to generate macroscopic polarization. The origin of the polar state in SmFeO3 is also explained on the basis of the exchange-striction mechanism between neighbouring Fe spins53 which can lower the symmetry of the material.54 In spite of these developments, the role of the rare-earth element in rare-earth orthoferrites with a magnetic Ln ion is not entirely clear.
AlFeO3 and GaFeO3
It has been shown recently that AlFeO3 and GaFeO3,55 crystallizing in the non-centrosymmetric space group Pna21, exhibit interesting magnetoelectric properties56 due to cationic disorder. Al1−xGaxFeO3 contains four different cation sites namely Fe1, Fe2, Al1 (Ga1) and Al2 (Ga2) where Al1 (Ga1) is present in the tetrahedral environment of oxygen and the other three are present in the octahedral environment of oxygen.57 Neutron diffraction data reveal the presence of considerable disorder among the cations.58 The tetrahedral site is regular in geometry but the octahedral sites are highly distorted due to the mixed occupancy of the cations having different ionic radii. Because of the unequal distribution of Fe3+ ions at the four different sites, it shows ferrimagnetism with a collinear arrangement of spins and a TN in the 200–250 K range.59 These ferrites exhibit spontaneous electric polarization in the vicinity of around 100 K as obtained from pyroelectric current measurements (Fig. 8). The polarization value (±0.3 μC cm−2, at T = 10 K for E = ±2 kV cm−1) from pyroelectric measurements in the case of GaFeO3 is comparable to that in other magnetically induced ferroelectric oxides. The temperature variation of lattice parameters shows the occurrence of broad minima in the vicinity of the pyroelectric peak at 100 K.58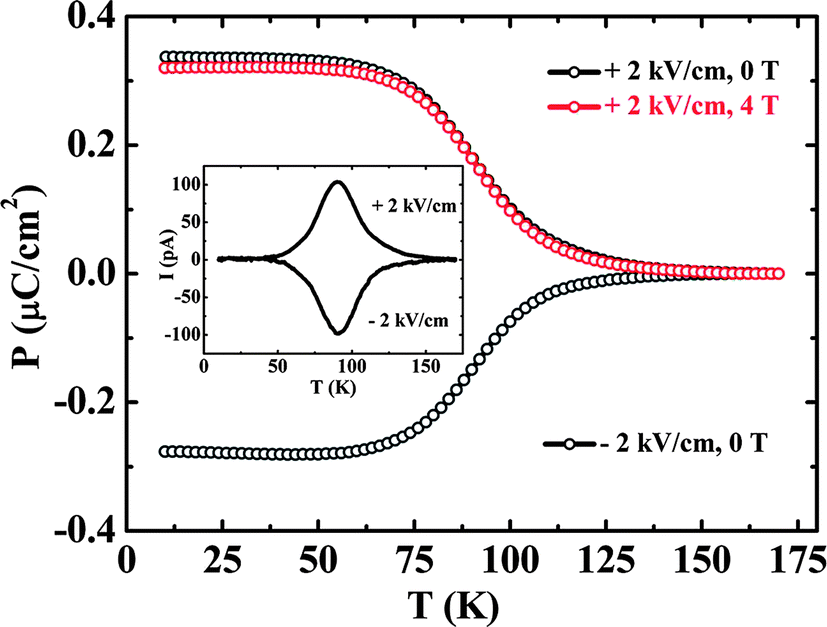 | ||
| Fig. 8 Temperature variation of ferroelectric polarization for polycrystalline GaFeO3. Inset shows the variation of pyroelectric current as a function of temperature with a peak maximum (TP) around 100 K (modified with permission from ref. 60, copyright 2012, by Elsevier). | ||
The origin of the ferroelectric polar state in Al(Ga)FeO3 is considered to arise from the intrinsic anti-site disorder in the crystal structure.60 In the absence of any cationic disorder the ground state magnetic configuration of GaFeO3 is antiferromagnetic because Fe spins at Fe1 and Fe2 sites are opposite in direction but equal in magnitude resulting in cancellation of their magnetic moments as shown in Fig. 9a. Another important point to note is that the interaction between Fe ions at Fe1 sites (or Fe at Fe2 sites) is ferromagnetic in nature but the interaction of Fe ions between neighbouring Fe1 and Fe2 sites is antiferromagnetic in nature resulting in a considerable amount of magnetic frustration. It is known that in TbMnO3, nearest neighbour FM and next nearest neighbour AFM interactions frustrate the magnetic structure, resulting in different magnetic structures with the lowering of magnetic symmetry, thereby favouring spontaneous polarization.2 In GaFeO3 one of the symmetry operations of Pna21, (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) , ȳ, z + 1/2) transforms a pair of Fe1 sites to the other of Fe1 sites. As the magnetic moments of Fe at all the Fe1 sites are equal and opposite to those at Fe2 sites, the magnetic structure preserves C2z rotational symmetry. Thus, it has the planar inversion symmetry in the xy plane and cannot give rise to polarization in the x and y directions. On introduction of anti-site disorder between the Fe and Ga sites, the magnetic moment of the Fe ions at the four Fe1 sites (this holds for Fe2 sites too) no longer remains equal (Fig. 9b) and breaks the spatial inversion symmetry in the ab plane, allowing spontaneous polarization. The switchability of the polarization is explained based on spin–phonon coupling observed in anomalies in their Raman spectra.61 The unequal size of the magnetic moment which develops on ordering of the magnetic ions distorts the crystal structure locally and the resultant non-centrosymmetric structural distortions due to spin–phonon coupling which is switchable with respect to the direction of the electric field generating polarization in the x and y directions. It is noteworthy that unlike LnFeO3 with magnetic Ln ions, GaFeO3 contains only one magnetic ion (Fe3+) but intrinsic disorder makes the two octahedral sites magnetically non-equivalent. The origin of ferroelectricity is therefore associated only with the magnetic Fe3+ ions (non-centrosymmetric structural distortion coupled with spin–phonon coupling at the onset of ferrimagnetic ordering).
, ȳ, z + 1/2) transforms a pair of Fe1 sites to the other of Fe1 sites. As the magnetic moments of Fe at all the Fe1 sites are equal and opposite to those at Fe2 sites, the magnetic structure preserves C2z rotational symmetry. Thus, it has the planar inversion symmetry in the xy plane and cannot give rise to polarization in the x and y directions. On introduction of anti-site disorder between the Fe and Ga sites, the magnetic moment of the Fe ions at the four Fe1 sites (this holds for Fe2 sites too) no longer remains equal (Fig. 9b) and breaks the spatial inversion symmetry in the ab plane, allowing spontaneous polarization. The switchability of the polarization is explained based on spin–phonon coupling observed in anomalies in their Raman spectra.61 The unequal size of the magnetic moment which develops on ordering of the magnetic ions distorts the crystal structure locally and the resultant non-centrosymmetric structural distortions due to spin–phonon coupling which is switchable with respect to the direction of the electric field generating polarization in the x and y directions. It is noteworthy that unlike LnFeO3 with magnetic Ln ions, GaFeO3 contains only one magnetic ion (Fe3+) but intrinsic disorder makes the two octahedral sites magnetically non-equivalent. The origin of ferroelectricity is therefore associated only with the magnetic Fe3+ ions (non-centrosymmetric structural distortion coupled with spin–phonon coupling at the onset of ferrimagnetic ordering).
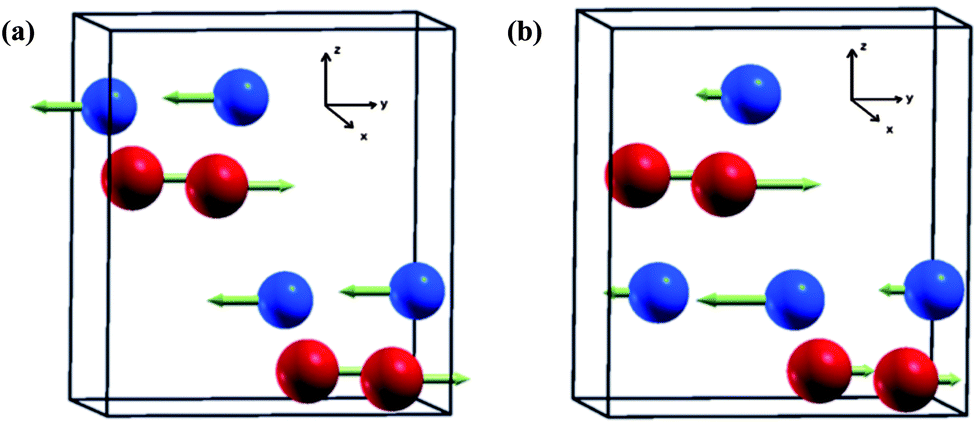 | ||
| Fig. 9 Schematic representation of the magnetic structure of GaFeO3 (a) without anti-site defect (ordered cationic state) which do not support ferroelectric polarization and (b) with anti-site defect (disordered cationic state) which breaks inversion symmetry in the ab plane allowing ferroelectric polarization. (Red and blue colours indicate the Fe1 and Fe2 sites respectively) (reproduced with permission from ref. 60, copyright 2012, by Elsevier). | ||
Rare-earth orthochromites
Evolution of ferroelectricity in rare-earth orthochromites, LnCrO3 which are isostructural with LnFeO3, has been reported. Ferroelectricity appears below the antiferromagnetic ordering temperature of the Cr ions with the sublattice remaining weakly ferromagnetic.62 The magnetic structure of the orthochromites is represented by G-type configurations namely Γ1, Γ2 and Γ4.34 In the case of magnetic rare-earth ions, the high temperature magnetic phase of LnCrO3 is Γ4 or Γ2 depending on the Ln ion, but with lowering temperature it undergoes transformation to another spin configuration namely Γ2 or Γ1 due to anisotropic interaction between magnetic Ln and the canted moment of the Cr ion resulting in spin reorientation.42 LnCrO3 with the non-magnetic Ln ground state remains weakly ferromagnetic (Γ4) and does not show spin-reorientation due to the absence of magnetic interaction between Ln and Cr. On the other hand, the spin-reorientation is induced in YCrO3 and YFeO3 by applying a magnetic field along the a direction.63,64 Polycrystalline SmCrO3 undergoes antiferromagnetic ordering at 197 K with the evolution of weak ferromagnetism (Γ4) followed by a change in the Cr spin configuration from Γ4 to Γ2 below 40 K, while the Gd, Tb and Tm orthochromites show canted antiferromagnetic ordering at 167, 157 and 127 K respectively but no spin-reorientation phenomena down to 15 K.62 Below the magnetic ordering temperature of Gd3+ ion, the Cr3+ and the Gd3+ spins in GdCrO3 show the occurrence of FxCyGz (Γ2) and FxGz configurations respectively.42 ErCrO3 undergoes magnetic ordering at 133 K with the evolution of weak ferromagnetism which disappears below TSR (22 K).65 The orthochromites are found to exhibit ferroelectric polarization which coincides with the magnetic ordering temperature of the Cr sublattice indicating the origin to be magnetism driven. Polycrystalline SmCrO3 shows spontaneous polarization with an anomaly at the onset of canted antiferromagnetic ordering and at the spin-reorientation temperature of Cr spins as shown in Fig. 10.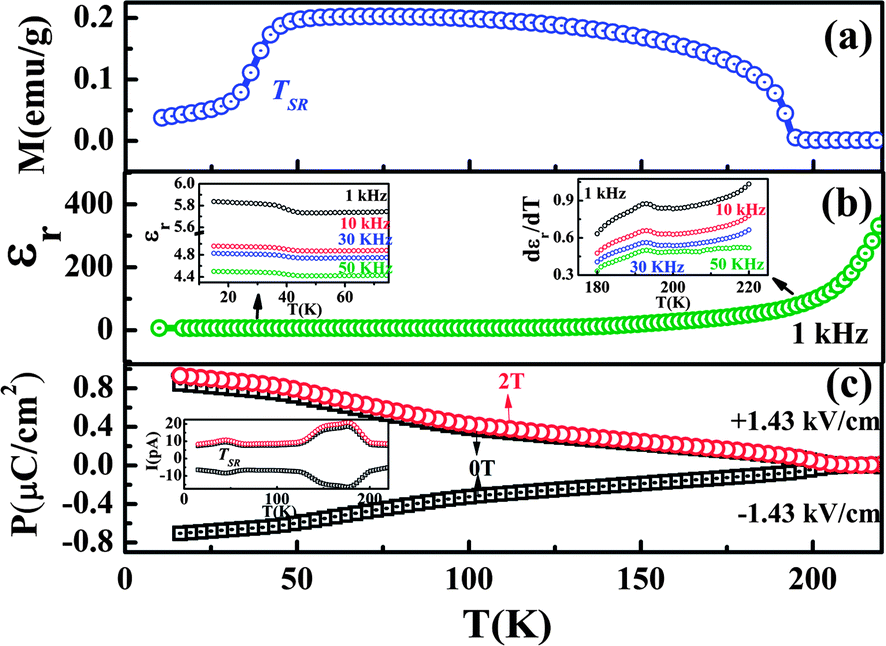 | ||
| Fig. 10 Variation of (a) field-cooled (FC) magnetization at H = 100 Oe, (b) dielectric constant at f = 1 kHz and (c) electric polarization as a function of temperature in polycrystalline SmCrO3. Insets in (b) show the frequency dependence of the dielectric constant and the first derivative of dielectric constant at TSR and TN; inset of (c) shows the temperature dependence of pyroelectric current (modified with permission from ref. 62, copyright 2012, by the American Physical Society). | ||
In polycrystalline ErCrO3, the polar state emerges along with the canted antiferromagnetic structure (Fig. 11). Polycrystalline ErCrO3 has been reported to show the presence of dielectric anomalies at both TN and TSR, indicating its magnetoelectric nature. Recent investigations, however, reveal the appearance of electric polarization at TN which persists down to low temperatures, even below TSR.66 Ferroelectric polarization in GdCrO3 evolves with the onset of canted antiferromagnetic ordering. Polarization in this chromite is shown to be tuneable by an applied magnetic field in the case of GdCrO3.
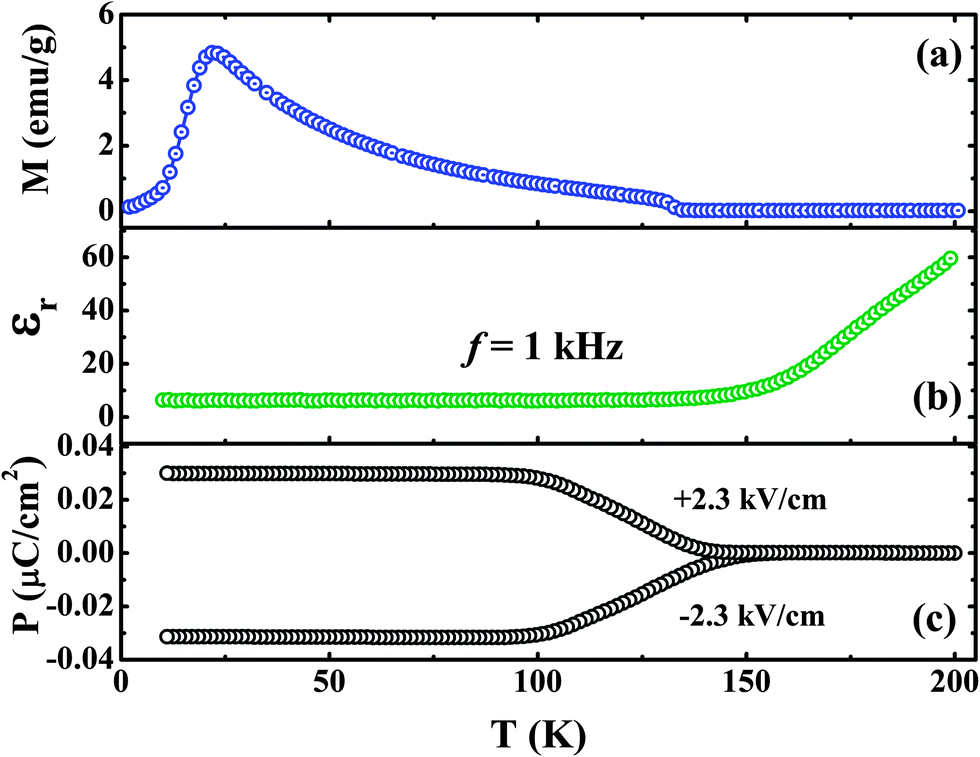 | ||
| Fig. 11 Variation of (a) field-cooled (FC) magnetization at H = 100 Oe showing a spin reorientation from Γ4 to Γ1 at 22 K, (b) dielectric constant at f = 1 kHz and (c) electric polarization as a function of temperature in polycrystalline ErCrO3 (modified with permission from ref. 62, copyright 2012, by the American Physical Society). | ||
Curious cases of YCrO3 and YFeO3
YCrO3 and YFeO3 with a non-magnetic rare-earth ion at the A site crystallize in the orthorhombic perovskite structure with the Pbnm space group.67 YCrO3 is a canted antiferromagnet (G-type) with the magnetic ordering temperature of 140 K.68 At all temperatures, YCrO3 possesses the Γ4 magnetic structure which is not compatible with ferroelectric polarization because of the non-polar magnetic point symmetry. The presence of diamagnetic Y3+ ion at the A site of the perovskite structure rules out the magnetic exchange striction between Cr3+ and Y3+ indicating the absence of a polar state at the magnetic ordering temperature of Cr3+ ion even though weak ferromagnetism exists below the Néel temperature. Surprisingly, YCrO3 is reported to exhibit a ferroelectric state at high temperatures (473 K), accompanied by a leaky hysteresis loop at 300 K.68 It also shows a dielectric anomaly at 473 K which is higher than the magnetic ordering temperature of YCrO3. The frequency-dependent peak maximum of the dielectric phase transition indicates the relaxor nature of the dielectric relaxation. In a centrosymmetric material such as TbMnO3, only a special kind of magnetic structure (spiral or cycloidal) breaks the inversion symmetry leading to ferroelectric polarization.19,69 The polar state in centrosymmetric YCrO3 at a temperature higher than the magnetic ordering temperature can not be associated with magnetic interactions. First principles calculations favour a non-centrosymmetric monoclinic P21 space group over the centrosymmetric Pbnm structure.68 Analysis of the pair distribution functions (PDF) based on neutron diffraction data confirms the presence of local non-centrosymmetry characterized with a Cr off-centring displacement of the order of 0.01 Å along the z direction which is temperature independent.70 Short-range (1–6 Å) neutron diffraction data can be fitted with a non-centrosymmetric P21 space group in the low-temperature ferroelectric phase, while in the high-temperature paraelectric region the data are best fitted with a centrosymmetric structure. The PDF data over the range of 1–22 Å can also be fitted with the centrosymmetric structure. Therefore the origin of the polar state has been considered to be local non-centrosymmetry though globally the structure is centrosymmetric. A recent study of polycrystalline films of YCrO3 grown on single crystalline Rh substrates shows the presence of ferroelectricity with a maximum polarization of 9 μC cm−2 at 300 K and weak ferromagnetism below 150 K.71 The nature and origin of ferroelectricity in YCrO3 are far from being clear. It is noteworthy that LuCrO3 is also suggested to show ferroelectric polarization at TN (Fig. 12) similar to ErCrO3.66 Further study of LuCrO3, however, seems necessary.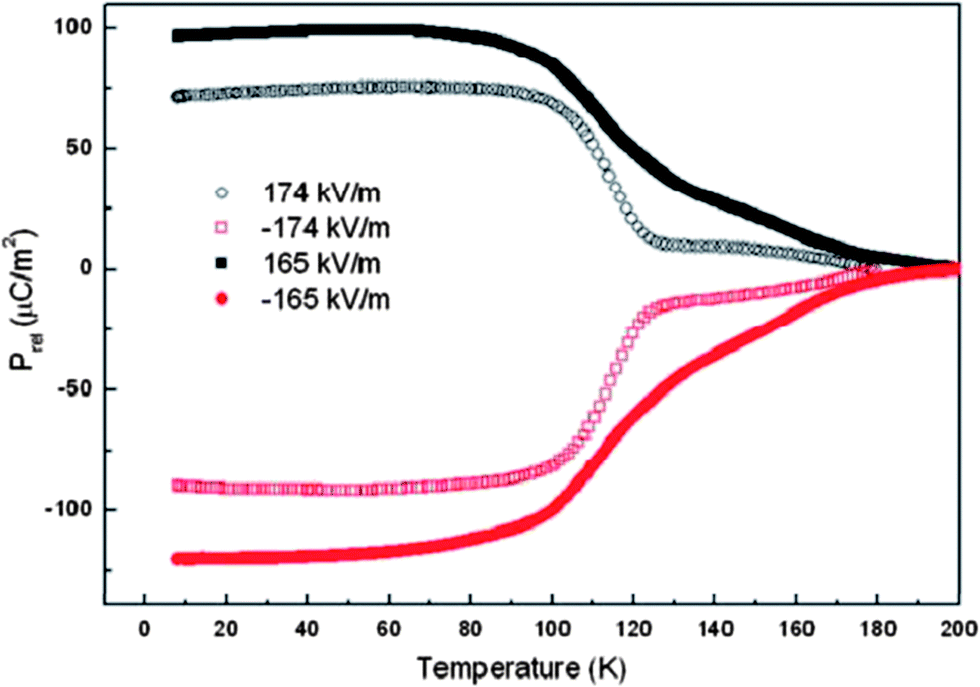 | ||
| Fig. 12 Temperature variation of the ferroelectric polarization for polycrystalline LuCrO3 (solid symbols) and ErCrO3 (open symbols) (reproduced with permission from ref. 66, copyright 2013, by the American Chemical Society). | ||
YFeO3 with a high magnetic ordering temperature (655 K) appears to exhibit ferroelectricity at 420 K.72 The spin configuration of the Fe sublattice of YFeO3 is Γ4 which supports the presence of weak ferromagnetism along the crystallographic c direction. Temperature dependent permittivity data as well as dielectric loss data show a peak feature at 420 K at all frequencies suggesting the occurrence of a ferroelectric to paraelectric phase transition. Although the ferroelectric ordering temperature is lower (or it does not coincide with magnetic ordering of the Fe sublattice) than the magnetic ordering temperature, the polar state is suggested to be magnetic in origin following Lee et al.50 Cheng et al.73 have shown that a single crystal of YFeO3 shows magnetocapacitance and relaxor like dielectric behaviour which is attributed to the combined action of Maxwell–Wagner space charge effect and magnetoresistance, indicating the origin to be extrinsic.73 The occurrence and origin of ferroelectricity in YFeO3 is debatable and needs further investigation.
Solid solutions of YCrO3 and YFeO3, YCr1−xFexO3 with the space group (Pbnm), however, show occurrence of ferroelectricity and magnetoelectric effect at the magnetic ordering temperature of the transition metal ions.74 In YCr1−xFexO3, the Fe3+ and Cr3+ ions are randomly distributed at the 4b crystallographic site leading to Fe–Fe, Cr–Cr and Cr–Fe superexchange interactions. YCr0.5Fe0.5O3 undergoes antiferromagnetic ordering at 260 K where Fe and Cr moments are coupled antiferromagnetically followed by temperature induced magnetization reversal with a compensation temperature of 248 K.75 Unlike YFeO3 and YCrO3, the spin configuration of YCr0.5Fe0.5O3 is represented by a magnetic structure which is in between Γ2 and Γ4 where spin reorientation is incomplete, magnetic moments are pointing in between a and c axes below the Néel temperature. With lowering temperature, magnetic moments start moving from the a axis, but do not reach the c axis down to 2 K resulting in an incomplete spin-reorientation phenomenon. A complete spin reorientation to the c axis would give rise to a Γ2 spin configuration. Pyroelectric current measurements show the occurrence of spontaneous polarization at the magnetic ordering temperature (TN = 260 K) as shown in Fig. 13. YCr0.5Fe0.5O3 shows the presence of a dielectric anomaly at TN indicating the magnetodielectric nature of this material. The magnetoelectric effect of this material was confirmed by studying the effect of magnetic fields on the pyroelectric current. Tunable ferroelectric transition temperatures in YCr1−xFexO3 with varying proportions of Fe3+ and Cr3+ ions at the B site of the perovskite are demonstrated in Fig. 13. The effect of disordered cations of non-equivalent spins in inducing ferroelectricity is understood based on a spin-current model found in canted antiferromagnetic oxide.52,69 According to this model, when there is only one transition metal ion at the 4b site of the Pbnm structure, the local polarization induced by inverse DM interaction would be cancelled out due to the alternating nature of the spin pairs (Fig. 14a). Upon introducing another transition metal ion of non-equivalent spin at the 4b site, two plausible situations can arise: (a) an ordered distribution of cations of non-equivalent spins giving rise to local polarization following inverse DM interaction which gets cancelled due to a change in the direction of the induced local polarization (Fig. 14b);69 (b) A disordered arrangement of cations of non-equivalent spins generating a net polarization because the oxygen displacement need not be equivalent (Fig. 14c). Significant polarization is observed for x ∼ 0.5, because it leads to less cluster formation of Fe–Fe and Cr–Cr resulting in cancellation of the ferroelectric polarization due to alternate spin pairs. Though this model explains the origin of ferroelectric polarization, microscopic insights would be helpful to understand the phenomena properly. Replacing Y3+ with another non-magnetic rare earth also generates ferroelectric polarization at the magnetic ordering temperature of Cr–Fe interactions, indicating that the origin of polarization is associated only with the disordered transition metal ions of non-equivalent spins at the 4b sites of the Pbnm structure. Replacing the transition metal ions at the B site (e.g. YCr1−xMnxO3) retains the weak ferromagnetism and ferroelectricity at the magnetic ordering temperature. This behaviour enables tuning of the ferroelectric transition temperature by varying the A and B site cations. It is, however, curious that YFe1−xMnxO3 exhibits magnetodielectric effects at both spin-reorientation and Néel temperatures (TSR < TN) and ferroelectricity at even lower temperatures.76
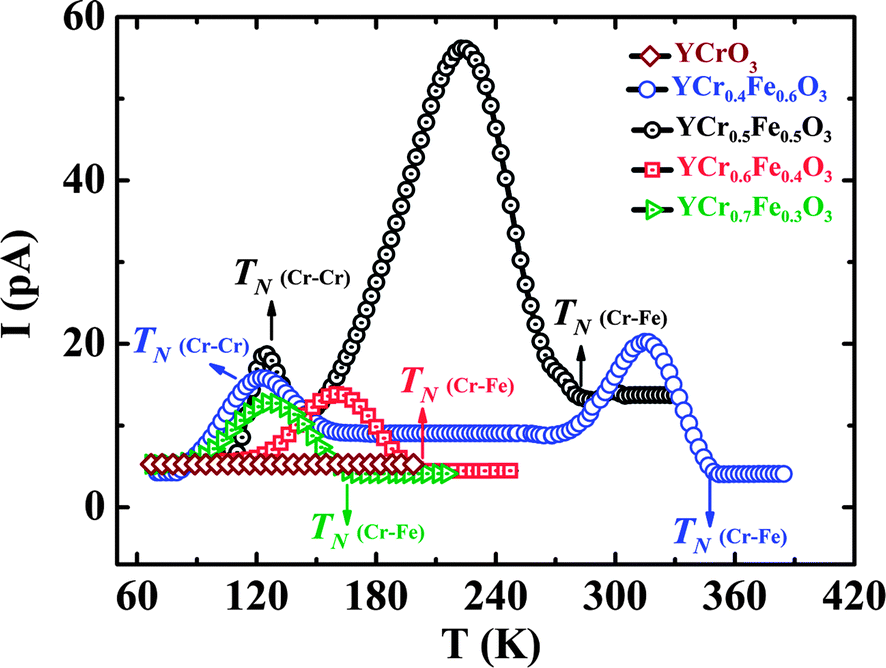 | ||
| Fig. 13 Variation of onset of pyroelectric current with composition of YCr1−xFexO3 (x = 0.3, 0.4, 0.5, 0.6) demonstrating the occurrence of polarization at TN (modified with permission from ref. 74, copyright 2012, by the American Chemical Society). | ||
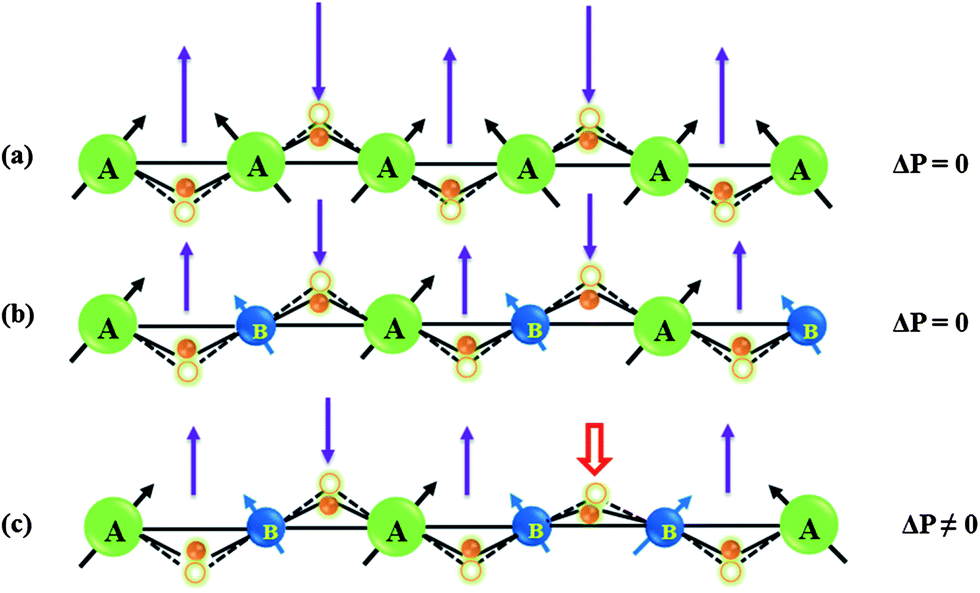 | ||
| Fig. 14 Schematic representation of the origin of ferroelectric polarization in a canted AFM system for (a) an invariant B-site cation, (b) an ordered and (c) a disordered arrangement of two cations of non-equivalent spins at the B-site (modified with permission from ref. 74, copyright 2012, by the American Chemical Society). | ||
Problems and challenges
Traditionally ferroelectricity in a material is established by recording the P–E hysteresis loop using the Sawyer–Tower circuit. This method has been seriously questioned since most materials seem to show hysteresis loops even if they are leaky.77 Clearly, one should employ other methods to establish intrinsic ferroelectricity. Most of the transition metal oxides, for example ferrites, show leaky hysteresis loops. Pyroelectric current measurements after electric poling78 have been employed to characterize the polar state, but this method is also not free of artifacts due to space-charge effects induced by poling bias, which can be one of the causes of reversal behaviour of the depolarization current in non-ferroelectric materials.79 Thus measurement of ferroelectric polarization in a material having residual conductivity remains problematic. The PUND (Positive-Up-Negative-Down) or DWM (Double-Wave Method)80 method has been used by several workers recently to extract information of the intrinsic ferroelectric polarization from the non-hysteretic and leakage part.50,81,82 Practitioners continue to face serious difficulty in establishing the intrinsic ferroelectricity in a material.Research on multiferroics is often carried out on polycrystalline materials where leakage problems are severe. It would be most useful to carry out measurements on single crystals and good epitaxial thin films. In terms of materials, it will be of great value if one can discover a material which is ferromagnetic and magnetoelectric at room temperature. Tuning the spin and charge ordering as well as orbital degrees of freedom at the interface of thin film heterostructures would be of great interest.29,30 Controlling interface magnetism using electric fields would be interesting as shown by a recent work on thin film heterostructure of BiFeO3–LSMO.31 It seems that electric field control of magnetism can provide better insight on the magnetoelectric coupling. Fully understanding the role of interfaces in films deposited on substrates requires further investigations.
Concluding remarks
In the preceding sections, we have shown how ferrites and chromites exhibit interesting multiferroic and magnetoelectric properties arising from magnetically induced ferroelectricity. Thus, in thin films of BiFeO3 magnetoelectric coupling and the exchange bias can be tuned by epitaxial strain.32 It must be noted that in TbMnO3 ferroelectricity arises from the non-collinear cycloidal spin structure which breaks the spatial inversion symmetry to induce polarization.19 In rare-earth orthoferrites and orthochromites with a magnetic rare-earth ion, exchange striction between the magnetic rare-earth and Fe3+ (Cr3+) ions is suggested to cause electric polarization.40,48 Interestingly, in the simple ferrimagnetic ferrites, Al(Ga)FeO3, cation disorder driven non-equivalence of the octahedral Fe3+ ions alone appears to be sufficient to give rise to ferroelectricity.60 The multiferroic nature of ferrites such as LuFe2O4 has been reported83 but some doubt has been expressed about this observation.84 There is some interest in examining properties of Fe3O4 in view of the recent findings related to its structure.85 Ferroelectric features reported in orthorhombic YCrO3 and YFeO3 are not fully understood. In this context, ferroelectricity in epitaxially constrained hexagonal LuFeO3 becomes noteworthy.86 Another noteworthy observation which needs proper understanding is that of ferroelectricity at the magnetic ordering temperature of Ln(Fe,Cr)O3 containing two magnetic transition metal ions at the B-site, allowing the tuning of the ordering temperature by varying the Ln ion at the A-site or the composition at the B-site.74 Clearly there is a need for further investigations to fully understand the multiferroic and magnetoelectric properties of many of the ferrites and chromites. Problems and challenges in the area of multiferroic and magnetoelectric oxides were mentioned in the previous section of this article but what is worthy of note is that the subject itself is of great interest.Acknowledgements
We would like to thank L. Herve, Vincent Caignaert and Antoine Maignan for helping and providing the facilities to grow the single crystals of DyFeO3.References
- N. A. Hill, J. Phys. Chem. B, 2000, 104, 6694 CrossRef CAS.
- S.-W. Cheong and M. Mostovoy, Nat. Mater., 2007, 6, 13 CrossRef CAS PubMed.
- C. N. R. Rao and C. R. Serrao, J. Mater. Chem., 2007, 17, 4931 RSC.
- N. A. Spaldin, S.-W. Cheong and R. Ramesh, Phys. Today, 2010, 63, 38 CrossRef.
- R. Seshadri and N. A. Hill, Chem. Mater., 2001, 13, 2892 CrossRef CAS.
- J. M. Rondinelli and C. J. Fennie, Adv. Mater., 2012, 24, 1961 CrossRef CAS PubMed.
- A. T. Mulder, N. A. Benedek, J. M. Rondinelli and C. J. Fennie, Adv. Funct. Mater., 2013 DOI:10.1002/adfm.201300210.
- T. Fukushima, A. Stroppa, S. Picozzi and J. M. Perez-Mato, Phys. Chem. Chem. Phys., 2011, 13, 12186 RSC.
- N. A. Benedek and C. J. Fennie, Phys. Rev. Lett., 2011, 106, 107204 CrossRef PubMed.
- P. Ghosez and J.-M. Triscone, Nat. Mater., 2011, 10, 269 CrossRef CAS PubMed.
- G. Gou and J. M. Rondinelli, 2013, arXiv:1304.4911.
- A. Stroppa, P. Jain, P. Barone, M. Marsman, J. M. Perez-Mato, A. K. Cheetham, H. W. Kroto and S. Picozzi, Angew. Chem., Int. Ed., 2011, 50, 5847 CrossRef CAS PubMed.
- A. Stroppa, D. Di Sante, S. Horiuchi, Y. Tokura, D. Vanderbilt and S. Picozzi, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 014101 CrossRef.
- G. Giovannetti, S. Kumar, J.-P. Pouget and M. Capone, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 205146 CrossRef.
- D.-W. Fu, H.-L. Cai, Y. Liu, Q. Ye, W. Zhang, Y. Zhang, X.-Y. Chen, G. Giovannetti, M. Capone, J. Li and R.-G. Xiong, Science, 2013, 339, 425 CrossRef CAS PubMed.
- X. F. Hao, A. Stroppa, S. Picozzi, A. Filippetti and C. Franchini, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 014116 CrossRef.
- C. N. R. Rao, A. Sundaresan and R. Saha, J. Phys. Chem. Lett., 2012, 3, 2237 CrossRef CAS PubMed.
- W. Eerenstein, N. D. Mathur and J. F. Scott, Nature, 2006, 442, 759 CrossRef CAS PubMed.
- T. Kimura, T. Goto, H. Shintani, K. Ishizaka, T. Arima and Y. Tokura, Nature, 2003, 426, 55 CrossRef CAS PubMed.
- Y. N. Venevtsev, G. Zhdanov and S. Solov'ev, Sov. Phys. Crystallogr., 1960, 4, 538 Search PubMed.
- G. Smolenskii, V. Isupov, A. Agranovskaya and N. Krainik, Sov. Phys. Solid State, 1961, 2, 2651 Search PubMed.
- I. Sosnowskat, T. Peterlin-Neumaier and E. Steichele, J. Phys. C: Solid State Phys., 1982, 15, 4835 CrossRef.
- I. Sosnowska, M. Loewenhaupt, W. I. F. David and R. M. Ibberson, Phys. B, 1992, 180–181, 117 CrossRef CAS.
- B. Ruette, S. Zvyagin, A. P. Pyatakov, A. Bush, J. F. Li, V. I. Belotelov, A. K. Zvezdin and D. Viehland, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 064114 CrossRef.
- C. Ederer and N. A. Spaldin, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 060401 CrossRef.
- J. Wang, J. B. Neaton, H. Zheng, V. Nagarajan, S. B. Ogale, B. Liu, D. Viehland, V. Vaithyanathan, D. G. Schlom, U. V. Waghmare, N. A. Spaldin, K. M. Rabe, M. Wuttig and R. Ramesh, Science, 2003, 299, 1719 CrossRef CAS PubMed.
- D. Lebeugle, D. Colson, A. Forget and M. Viret, Appl. Phys. Lett., 2007, 91, 022907 CrossRef.
- J. F. Scott, J. Mater. Chem., 2012, 22, 4567 RSC.
- P. Yu, J. S. Lee, S. Okamoto, M. D. Rossell, M. Huijben, C. H. Yang, Q. He, J. X. Zhang, S. Y. Yang, M. J. Lee, Q. M. Ramasse, R. Erni, Y. H. Chu, D. A. Arena, C. C. Kao, L. W. Martin and R. Ramesh, Phys. Rev. Lett., 2010, 105, 027201 CrossRef CAS PubMed.
- S. M. Wu, S. A. Cybart, P. Yu, M. D. Rossell, J. X. Zhang, R. Ramesh and R. C. Dynes, Nat. Mater., 2010, 9, 756 CrossRef CAS PubMed.
- S. M. Wu, S. A. Cybart, D. Yi, J. M. Parker, R. Ramesh and R. C. Dynes, Phys. Rev. Lett., 2013, 110, 067202 CrossRef CAS PubMed.
- D. Sando, A. Agbelele, D. Rahmedov, J. Liu, P. Rovillain, C. Toulouse, I. C. Infante, A. P. Pyatakov, S. Fusil, E. Jacquet, C. Carrétéro, C. Deranlot, S. Lisenkov, D. Wang, J. M. Le Breton, M. Cazayous, A. Sacuto, J. Juraszek, A. K. Zvezdin, L. Bellaiche, B. Dkhil, A. Barthélémy and M. Bibes, Nat. Mater., 2013, 12, 641 CrossRef CAS PubMed.
- A. M. Mulders, Nat. Phys., 2013, 9, 398 CrossRef CAS.
- E. F. Bertaut, in Magnetism III, ed. G. T. Rado and H. Suhl, Academic, New York, 1968, p. 149 Search PubMed.
- T. Yamaguchi, J. Phys. Chem. Solids, 1974, 35, 479 CrossRef CAS.
- R. L. White, J. Appl. Phys., 1969, 40, 1061 CrossRef CAS.
- R. D. Johnson, N. Terada and P. G. Radaelli, Phys. Rev. Lett., 2012, 108, 219701 CrossRef CAS PubMed.
- A. K. Zvezdin and A. A. Mukhin, JETP Lett., 2009, 88, 505 CrossRef.
- Y. Tokunaga, N. Furukawa, H. Sakai, Y. Taguchi, T.-h. Arima and Y. Tokura, Nat. Mater., 2009, 8, 558 CrossRef CAS PubMed.
- Y. Tokunaga, S. Iguchi, T. Arima and Y. Tokura, Phys. Rev. Lett., 2008, 101, 097205 CrossRef CAS PubMed.
- S. Geller, J. Chem. Phys., 1956, 24, 1236 CrossRef CAS.
- T. Yamaguchi and K. Tsushima, Phys. Rev. B: Solid State, 1973, 8, 5187 CrossRef CAS.
- I. Dzyaloshinsky, J. Phys. Chem. Solids, 1958, 4, 241 CrossRef CAS.
- T. Moriya, Phys. Rev., 1960, 120, 91 CrossRef CAS.
- G. Gorodetsky, B. Sharon and S. Shtrikman, J. Appl. Phys., 1968, 39, 1371 CrossRef CAS.
- L. A. Prelorendjo, C. E. Johnson, M. F. Thomas and B. M. Wanklyn, J. Phys. C: Solid State Phys., 1980, 13, 2567 CrossRef.
- A. Stroppa, M. Marsman, G. Kresse and S. Picozzi, New J. Phys., 2010, 12, 093026 CrossRef.
- B. Rajeswaran, D. Sanyal, M. Chakrabarti, Y. Sundarayya, A. Sundaresan and C. N. R. Rao, Europhys. Lett., 2013, 101, 17001 CrossRef.
- Y. Sundarayya, P. Mandal, A. Sundaresan and C. N. R. Rao, J. Phys.: Condens. Matter, 2011, 23, 436001 CrossRef CAS PubMed.
- J.-H. Lee, Y. K. Jeong, J. H. Park, M.-A. Oak, H. M. Jang, J. Y. Son and J. F. Scott, Phys. Rev. Lett., 2011, 107, 117201 CrossRef PubMed.
- G. Gorodetsky and L. M. Levinson, Solid State Commun., 1969, 7, 67 CrossRef CAS.
- H. Katsura, N. Nagaosa and A. V. Balatsky, Phys. Rev. Lett., 2005, 95, 057205 CrossRef PubMed.
- J.-H. Lee, Y. K. Jeong, J. H. Park, M.-A. Oak, H. M. Jang, J. Y. Son and J. F. Scott, Phys. Rev. Lett., 2012, 108, 219702 CrossRef.
- S. Goshen, D. Mukamel, H. Shaked and S. Shtrikman, Phys. Rev. B: Solid State, 1970, 2, 4679 CrossRef.
- J. P. Remeika, J. Appl. Phys., 1960, 31, S263 CrossRef.
- T. Arima, D. Higashiyama, Y. Kaneko, J. P. He, T. Goto, S. Miyasaka, T. Kimura, K. Oikawa, T. Kamiyama, R. Kumai and Y. Tokura, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 064426 CrossRef.
- A. Shireen, R. Saha, P. Mandal, A. Sundaresan and C. N. R. Rao, J. Mater. Chem., 2011, 21, 57 RSC.
- R. Saha, A. Shireen, A. K. Bera, S. N. Shirodkar, Y. Sundarayya, N. Kalarikkal, S. M. Yusuf, U. V. Waghmare, A. Sundaresan and C. N. R. Rao, J. Solid State Chem., 2011, 184, 494 CrossRef CAS.
- R. Saha, A. Shireen, S. N. Shirodkar, U. V. Waghmare, A. Sundaresan and C. N. R. Rao, J. Solid State Chem., 2011, 184, 2353 CrossRef CAS.
- R. Saha, A. Shireen, S. N. Shirodkar, U. V. Waghmare, A. Sundaresan and C. N. R. Rao, Solid State Commun., 2012, 152, 1964 CrossRef CAS.
- P. Kumar, A. Bera, D. V. S. Muthu, S. N. Shirodkar, R. Saha, A. Shireen, A. Sundaresan, U. V. Waghmare, A. K. Sood and C. N. R. Rao, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 134449 CrossRef.
- B. Rajeswaran, D. I. Khomskii, A. K. Zvezdin, C. N. R. Rao and A. Sundaresan, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 214409 CrossRef.
- I. S. Jacobs, H. F. Burne and L. M. Levinson, J. Appl. Phys., 1971, 42, 1631 CrossRef CAS.
- G. W. Durbin, C. E. Johnson and M. F. Thomas, J. Phys. C: Solid State Phys., 1975, 8, 3051 CrossRef CAS.
- Y. Su, J. Zhang, B. Li, B. Kang, Q. Yu, C. Jing and S. Cao, Ceram. Int., 2012, 38, S421 CrossRef CAS.
- K. R. S. P. Meher, C. Martin, V. Caignaert, F. Damay and A. Maignan, Chem. Mater., 2013 DOI:10.1021/cm4020546.
- S. Geller and E. A. Wood, Acta Crystallogr., 1956, 9, 563 CrossRef CAS.
- C. R. Serrao, A. K. Kundu, S. B. Krupanidhi, U. V. Waghmare and C. N. R. Rao, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 220101 CrossRef.
- T. Kimura, Annu. Rev. Mater. Res., 2007, 37, 387 CrossRef CAS.
- K. Ramesha, A. Llobet, Th. Proffen, C. R. Serrao and C. N. R. Rao, J. Phys.: Condens. Matter, 2007, 19, 102202 CrossRef.
- J. D. Seo and J. Y. Son, J. Cryst. Growth, 2013, 375, 53 CrossRef CAS.
- M. Shang, C. Zhang, T. Zhang, L. Yuan, L. Ge, H. Yuan and S. Feng, Appl. Phys. Lett., 2013, 102, 062903 CrossRef.
- Z. X. Cheng, H. Shen, J. Y. Xu, P. Liu, S. J. Zhang, J. L. Wang, X. L. Wang and S. X. Dou, J. Appl. Phys., 2012, 111, 034103 CrossRef.
- B. Rajeswaran, P. Mandal, R. Saha, E. Suard, A. Sundaresan and C. N. R. Rao, Chem. Mater., 2012, 24, 3591 CrossRef CAS.
- N. Dasari, P. Mandal, A. Sundaresan and N. S. Vidhyadhiraja, Europhys. Lett., 2012, 99, 17008 CrossRef.
- P. Mandal, V. S. Bhadram, Y. Sundarayya, C. Narayana, A. Sundaresan and C. N. R. Rao, Phys. Rev. Lett., 2011, 107, 137202 CrossRef CAS PubMed.
- J. F. Scott, J. Phys.: Condens. Matter, 2008, 20, 021001 CrossRef.
- I. Lubomirsky and O. Stafsudd, Rev. Sci. Instrum., 2012, 83, 051101 CrossRef PubMed.
- M. Maglione and M. A. Subramanian, Appl. Phys. Lett., 2008, 93, 032902 CrossRef.
- M. Fukunaga and Y. Noda, Jpn. J. Appl. Phys., 2008, 77, 064706 CrossRef.
- S. Horiuchi, Y. Tokunaga, G. Giovannetti, S. Picozzi, H. Itoh, R. Shimano, R. Kumai and Y. Tokura, Nature, 2010, 463, 789 CrossRef CAS PubMed.
- S. M. Feng, Y. S. Chai, J. L. Zhu, N. Manivannan, Y. S. Oh, L. J. Wang, Y. S. Yang, C. Q. Jin and K. H. Kim, New J. Phys., 2010, 12, 073006 CrossRef.
- N. Ikeda, H. Ohsumi, K. Ohwada, K. Ishii, T. Inami, K. Kakurai, Y. Murakami, K. Yoshii, S. Mori, Y. Horibe and H. Kito, Nature, 2005, 436, 1136 CrossRef CAS PubMed.
- M. Angst, Phys. Status Solidi RRL, 2013, 7, 383 CrossRef CAS.
- M. S. Senn, J. P. Wright and J. P. Attfield, Nature, 2011, 481, 173 CrossRef PubMed.
- Y. K. Jeong, J.-H. Lee, S.-J. Ahn and H. M. Jang, Chem. Mater., 2012, 24, 2426 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |