Impact of collagen confinement vs. ionic substitutions on the local disorder in bone and biomimetic apatites†
Yan
Wang
,
Stanislas
Von Euw
,
Guillaume
Laurent
,
Charlène
Crevant
,
Laure
Bonhomme-Coury
,
Marie-Madeleine
Giraud-Guille
,
Florence
Babonneau
,
Nadine
Nassif
* and
Thierry
Azaïs
*
UPMC Univ Paris 06 & CNRS, UMR 7574, Chimie de la Matière Condensée de Paris, Collège de France, 11 Place Marcelin Berthelot, F-75005 Paris, France. E-mail: nadine.nassif@upmc.fr; thierry.azais@upmc.fr
First published on 10th October 2013
Abstract
The aim of the study is to identify some key physico-chemical parameters that influence the structural characteristics of carbonated hydroxyapatite and, in particular, the so-called “poorly crystallized” nature of bone hydroxyapatite. Here, we investigate this feature by 1D and 2D 1H–31P solid-state NMR experiments through the evaluation of the local order/disorder around the phosphate and the hydroxyl ions in the crystalline apatitic core as well as the quantification of the amorphous layer. The study relies on a large variety of apatite models, i.e. from pure highly crystalline apatites to tissue-like models, as well as on fresh intact bone. We show that both collagen confinement and ionic substitutions have an impact on the structural characteristics of apatite. Nevertheless, while collagen confinement appears predominantly in the initial stages of apatite formation, these characteristics are later dominated by the ionic substitutions, specifically from carbonate ions. We show that an amount of CO32− around 7–8 wt% is necessary to set biomimetic apatite models possessing similar structural characteristics to those found for bone minerals.
Introduction
Skeletal materials found in living organisms offer a variety of complex and subtle architectures with various specific properties. One remarkable example of a natural composite material is bone. The bone mineral platelets are located in an organic matrix where Type I collagen (i.e. its main component) assembles into fibrils that further organize to form a dense tri-dimensional network.1 The c-axis of apatite crystals aligns with the collagen fibril direction. The initial steps of mineralization are first expected to occur within the fibril in confined well-distributed gaps (∼40 nm) of axial periodicity (67 nm), described as nucleation sites (i.e. intra-fibrillar mineralization),2,3 then continue along the collagen fibrils. Nevertheless, crystals may also nucleate from inter-fibrillar collagen sites.4 Inside this organic 3D restricted volume, confinement effects may occur on protein's activities as well as on mineral formation (nucleation and growth). Noticeably, non-collagen proteins (NCPs) can show opposite activities depending on their surrounding state, either in diluted or condensed conditions.5,6Bone mineral consists of an inorganic calcium phosphate phase for which the composition derives from the stoichiometric hydroxyapatite (Hap), Ca10(PO4)6(OH)2. Bone apatite is structurally disordered, and compositionally nonstoichiometric due to the incorporation of a substantial amount of anionic (CO32−) and cationic substitutions (Na+, Mg2+) into the crystalline structure.7 The most noticeable substitution is the replacement of the PO43− (B-type substitution) and/or OH− (A-type substitution) anions by carbonate ions CO32− (up to 5–8 weight%). Recently, it was shown that bone nanosized platelets (∼10 × 25 × 50 nm)1 display a crystalline apatitic core and is characterized by a so-called “hydrated disordered surface layer” that is mainly composed of divalent species such as Ca2+, HPO42−, CO32− and structural H2O.8 Overall, this explains the proposed general formula in the literature: Ca8.3□1,7(PO4)4.3(HPO4 or CO3)1.7(OH or 1/2CO3)0,3□1.7.9 Hence, in contrast to stoichiometric Hap, bone minerals are often described as “poorly crystallized” calcium-deficient apatite. More recently, it has been shown that this hydrated disordered layer identified as an amorphous calcium phosphate (ACP) related phase is involved in the stacking of the apatite platelets along their c-axis in the presence of water.8 The final structural features of the mineral platelets provide specific properties to bone, in particular: (i) the poorly crystallized nature of the bone mineral particles favors the resorption/remodelling process;1 (ii) the hydrated disordered surface layer is proposed to act as an ion reservoir;10 (iii) the intimate 3D association between the mineral, the organic part and water enhances the mechanical properties.11,12
In the literature, various analytical tools are used to characterize the intrinsic structural disorder of carbonated Hap. Among those, X-ray diffraction provides a typical pattern of bone apatite displaying very broad reflections compared to stoichiometric Hap.1 Solid state NMR (ssNMR) is also widely used for the investigation of the bone structure,13,14 its hydration level,15 and its organo-mineral interface.16–19 This spectroscopy is sensitive to the structural disorder since it is also directly related to the line width of the resonances, which are, in that case, dominated by a distribution of chemical shifts. Thus, 1D ssNMR of 1H and 31P nuclei allows the investigation of the local environment around the phosphate20 and the hydroxyl ions21 of bone apatite, respectively. In particular, the 31P spectrum of bone mineral displays a typical broad resonance22 reflecting a highly disordered environment for the phosphate groups compared to stoichiometric Hap.
For a deeper understanding of bone biomineralization mechanisms, in vitro models are still needed since the study of living bone remains difficult without chemical and/or biological treatments. Nevertheless, synthesis of biomimetic Hap particles that exhibit the main structural characteristics of bone minerals (i.e. nanosized platelets incorporating a high amount of CO32− and displaying a hydrated disordered surface layer) is not straightforward. Nanorods or elongated particles vs. Hap platelets are usually precipitated in vitro.23 Moreover, ionic substitutions in hydroxyapatite, in particular by carbonate ions, are difficult to control and hard to obtain in large amounts.23 The presence of a hydrated disordered surface layer is also difficult to establish. Overall, it partly explains why the physico-chemical parameters influencing the structural characteristics of bone apatite are not well-defined.
In previous work, the use of appropriate models,24i.e. a bone tissue-like matrix, studied by ssNMR was useful to show that the collagen matrix not only controls the size and the three-dimensional distribution of apatite at different length scales, but also has a further impact on the local hydrated environment of phosphate ions.22,25 It was shown that the proportion of the hydrated amorphous calcium phosphate layer vs. the crystalline apatitic core depends on the 3D spatial confinement set by the dense fibrillar scaffold.22
Here, we investigate through ssNMR the impact of such collagen confinement vs. the impact of ionic substitutions (in particular, the role of carbonate ions) on the structural characteristics of apatite. We show that both influence the so-called “poorly crystallized” nature of the particles as well as the extent of the hydrated disordered layer. However, the confinement effect is minimized with the increase of ionic substitutions. Hence, the structural characteristics of bone apatite seem to be determined by the incorporation of carbonate ions both in the crystalline core (as substitution ions) and in the amorphous layer while the impact of the 3D confinement in the dense collagen matrix may be of importance in the first stage of particles' nucleation.
Our studies rely on the use of different models including the bone tissue-like matrices (Coll/CHA and Coll/CHA(SBF))22 and pure synthetic apatites from biomimetic (CHA and CHA–SBF)8,26 to more crystalline ones (HA, cHA-3.6, cHA-5.2, cHA-8.1).27 The extended sample array provides a larger variety of particles' morphologies, carbonate content, and crystalline structures (according to 1D and 2D multinuclear (1H, 31P) solid-state NMR, powder XRD and TEM) that are compared to a fresh bone sample to investigate the importance of the related effects (confinement vs. ionic substitutions) in vivo. In particular, we demonstrate that the specific spectral features of bone minerals can be restored as soon as the proportion of CO32− ions is increased up to 7–8 wt%, a value close to the proportion found in bone minerals.1 This is principally established here through 2D 1H–31P ssNMR experiments based on the 1H–31P dipolar coupling that gives access to the spatial proximity between the two nuclei.28 2D {1H}31P HetCor experiment is used to correlate phosphate with its respective proton environment.29–31 This method leads to the spectral differentiation of the 31P and 1H resonances coming from the apatitic core (essentially composed of PO43− and OH− species) from those related to the hydrated disordered layer (essentially composed of HPO42− and H2O species) as previously shown for bone8,22 and synthetic apatite.32
Results and discussion
Confinement effects in collagen on apatite formation
To probe the impact of spatial confinement induced by collagen, two models were first compared: Hap particles were precipitated from the same mother solution in the absence (CHA) or in the presence of collagen (Coll/CHA). The Hap particles nucleate starting from an aqueous solution containing ionic precursors of carbonated Hap (Ca2+, PO42− and CO32−). CHA was identified as biomimetic in a previous study.26 Coll/CHA is characterized by (i) a 3D dense and hierarchically organized collagenous fibrillar network33 mimicking the collagen organization in compact bone (cholesteric, aligned and isotropic domains) and (ii) a low mineralization degree (∼5 wt%).22 TEM observations of unstained samples (Fig. 1a) show that this matrix exhibits both (i) lateral packing of small crystals (white arrow) across the diameter of the collagen fibrils that resemble the early stages of intrafibrillar mineralization, as described in the 2D channel model by Hodge and Petruska;34 and (ii) the continuous apatite crystal deposition into both the intermolecular spaces between collagen molecules and the interfibrillar spaces of collagen fibrils (yellow arrow) that may illustrate the later stages of calcification. The benefit of using such a matrix here is the presence of very small “dot-like” crystals within the collagen fibrils remarkably similar to the very early stages of normal in vivo calcification of embryonic chick bone reported by Fitton-Jackson2 or in vitro mineralization of decalcified bone.1 In contrast, the CHA sample displays a platelet's morphology (Fig. 1b).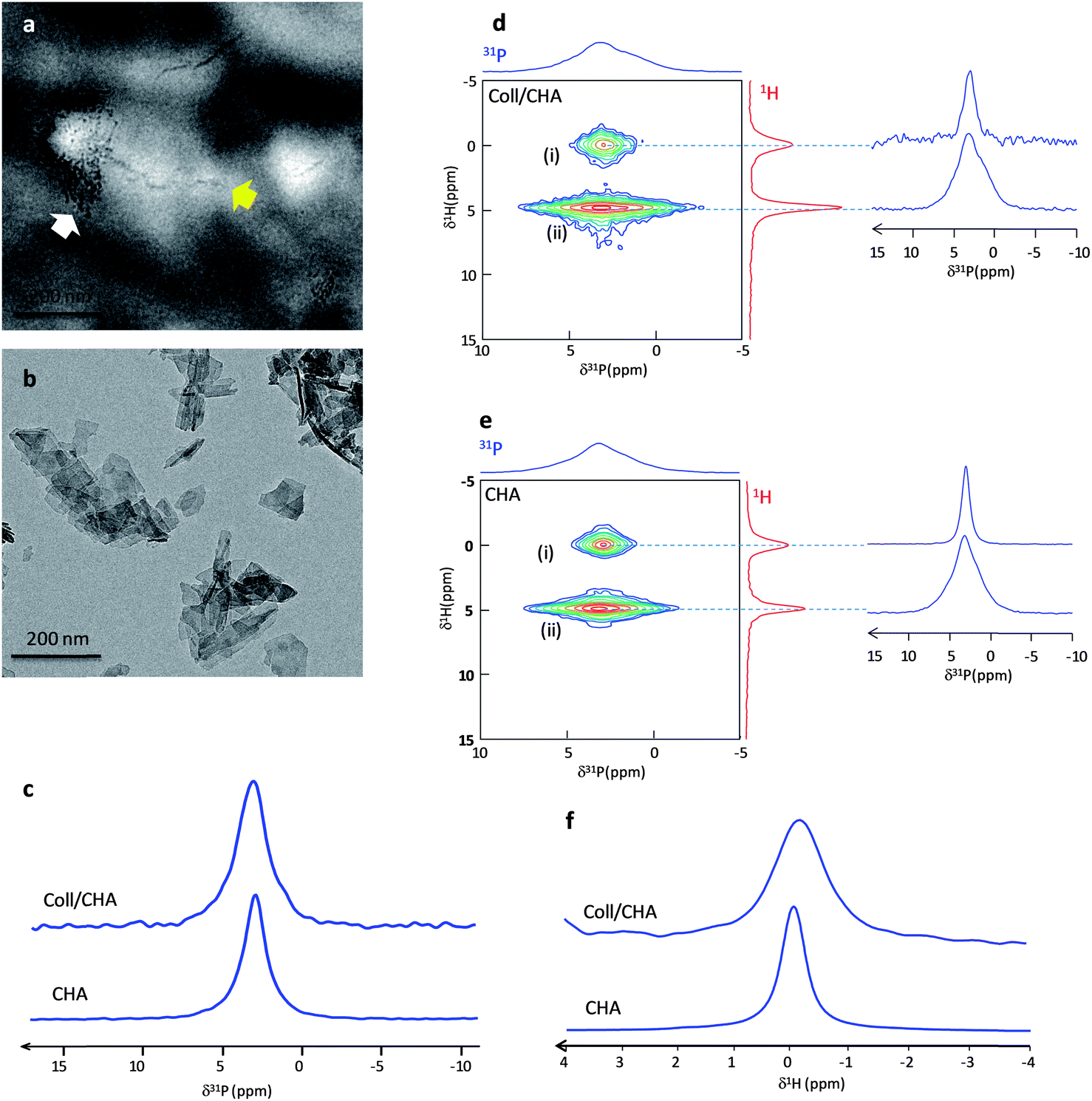 | ||
| Fig. 1 (a and b) TEM micrographs of Coll/CHA and CHA. (c) 31P MAS NMR quantitative spectra of Coll/CHA and CHA. (d and e) 2D {1H}31P HetCor spectra of Coll/CHA and wet CHA displaying two distinct domains corresponding to (i) the apatitic core and (ii) the hydrated disordered layer. (f) {1H–31P}1H double CP spectra of Coll/CHA and CHA. | ||
Previous results emphasized the need to keep bone in its native hydration state to avoid artifacts coming from dehydration and thus, misleading interpretations. Hence, the Coll/CHA matrix was kept in its hydrated state while CHA powder was wetted for safe comparison. The analyses were performed after the same time of maturation (∼6 days). Fig. 1c displays the 1D 31P MAS spectrum of Coll/CHA and CHA that exhibits a resonance at 2.9 ppm characteristic of carbonated apatite.26 We note that the resonance of Coll/CHA is significantly broader than CHA (LW = 260 Hz vs. 180 Hz, respectively). The 2D {1H}31P HetCor spectra of the two samples are also similar (Fig. 1d and e) and exhibit two cross-peaks (δ(31P) ∼ 2.9 ppm) that correspond to (i) apatitic phosphate correlation with OH− ions (δ(1H) = 0 ppm) and (ii) phosphate from the outer layer correlating with water molecules (δ(1H) = 4.85 ppm). This feature demonstrates that the Hap particles in both samples are composed of an apatitic core surrounded by a hydrated disordered layer as found in bone minerals.22 Nevertheless, the 31P projections relative to each P group (apatitic core and outer layer) confirm the tendency observed above since they are globally broader for Coll/CHA (LW 31P apatitic core = 200 Hz; LW 31P disordered layer = 490 Hz) compared to CHA (LW 31P apatitic core = 140 Hz; LW 31P disordered layer = 430 Hz) (Table 1). Concerning the apatitic core, this reflects a higher degree of disorder of the apatitic lattice for Coll/CHA which is somehow intriguing since the Hap particles of both samples are precipitated from the same mother solution (Table S1†).
| Sample | 31P apatitic core | 31P amorphous layer | Ratio apatitic/amorphous 31P (±5%) | 1H apatitic core | |||
|---|---|---|---|---|---|---|---|
| δ(31P) ± 0.1 ppm | LW ± 20 Hz | δ(31P) ± 0.1 ppm | LW ± 20 Hz | δ(1H) ± 0.1 ppm | LW ± 10 Hz | ||
| a Asymmetric line shape. b Global linewidth. | |||||||
| CHA | 3.0 | 140 | 3.1 | 430 | 65/35 | −0.2 | 160 |
| Coll/CHA | 3.1 | 200 | 3.1 | 490 | 40/60 | −0.2 | 330 |
| Bone | 3.1 | 270 | 3.2 | 640 | 55/45 | 0.0a | 400b |
| Coll/CHA(SBF) | 3.1 | 285 | 3.2 | 660 | 55/45 | 0.0a | 430b |
| CHA–SBF | 3.1 | 290 | 3.2 | 660 | 55/45 | 0.0a | 430b |
| HA | 2.8 | 80 | — | — | 100/0 | −0.3 | 140 |
| cHA-3.6 | 2.8 | 110 | 2.9 | 280 | 70/30 | −0.3 | 170 |
| cHA-5.2 | 2.9 | 115 | 3.1 | 330 | 50/50 | −0.3 | 175 |
| cHA-8.1 | 3.0 | 210 | 3.0 | 370 | 35/65 | 0.0a | 342 |
Due to the presence of collagen and water that dominates the 1H spectrum, the direct acquisition of a 1H MAS spectrum of Coll/CHA does not allow the identification of the OH− resonance. Thus, we used 31P-filtered experiments to record direct 1H spectra through a {1H–31P}1H double CP experiment.35 The use of long contact times (10 ms) allows the specific spectral edition of the hydroxyl resonance. Fig. 1f displays the {1H–31P}1H double CP spectra of CHA and Coll/CHA. A similar trend for the 1H apatitic LW is evidenced by the OH− resonance that is twice broader for Coll/CHA compared to CHA (Table 1). This also highlights the consequence of the confinement on the structural characteristics of Hap particles within the 3D dense collagenous matrix that increases the local disorder in the apatitic lattice.
The proportion of surface vs. core species detected by 2D NMR is higher for Coll/CHA compared to CHA as seen on the 1H projection (the HetCor experiments have been recorded with the same contact time tCP = 10 ms). The estimation of the relative amount of surface vs. apatitic species can be achieved by the fitting of the 1D 31P spectra using the 31P line shapes extracted from the 2D HetCor spectra (Fig. S1†). A greater amount of phosphate ions is found in the outer layer vs. in the apatitic core for Coll/CHA compared to CHA (60/40 ± 5 and 35/65 ± 5, respectively; Table 1). This result can be directly related to the size of the Hap particles. CHA platelets (Fig. 1a) appear bigger in size than those located within the dense collagen matrix (Coll/CHA) (Fig. 1b). This size effect enhances the proportion of surface species for the small particles. It is worth mentioning that a kinetic effect on the crystal's growth and maturation may occur in the presence of collagen although Coll/CHA samples appear stable for a few months.
These results emphasize the impact of confinement by the surrounding 3D collagen matrix on the initial steps of Hap formation in Coll/CHA that may occur in intra- (∼40 nm) and/or interfibrillar spaces (∼1.5 nm).
Influence of ionic substitutions on apatite formation
When Coll/CHA is immersed under ‘dynamic’ conditions in a concentrated simulated body fluid (SBF),22i.e. a serum-like solution,36 the resulting matrix Coll/CHA(SBF) possesses a mineral content (∼50 wt%) close to that described for mature bone. The Hap particles in Coll/CHA(SBF) appear with irregular edges and in the same size range as that found in bone.3 They are aligned with a preferred (002) orientation along the main axis of the collagen fibrils (Fig. 2a) similarly to bone minerals (Fig. 2b). Significantly, it was shown that the 1D 31P spectra of Coll/CHA(SBF) and mature (intact) fresh bone are identical (Fig. 2d).22 They exhibit a single slightly asymmetric resonance at 3.2 ppm relatively broad compared to CHA and Coll/CHA (LW = 340 vs. 180 and 260 Hz, respectively). Hence, the Coll/CHA(SBF) matrix is an appropriate model to assess the later stages of bone Hap particle formation. | ||
| Fig. 2 (a, b and c) TEM micrographs of Coll/CHA(SBF), bone and CHA–SBF, respectively. (d) 31P MAS quantitative spectra of CHA–SBF, Coll/CHA(SBF) and fresh bone. The arrows display the characteristic shoulder of the bone mineral 31P spectrum. (e) {1H–31P}1H double CP spectra of CHA–SBF, Coll/CHA(SBF) and intact bone (CT1 = CT2 = 10 ms for CHA–SBF, CT1 = CT2 = 15 ms for bone and Coll/CHA(SBF)). | ||
To distinguish a probable effect of ionic substitutions vs. the effect of collagen confinement on apatite formation, Coll/CHA(SBF) and (intact) fresh bone were compared to Hap precipitated directly from SBF (CHA–SBF). Under such conditions, the Ca2+, PO42− and CO32− precursors are at a lower concentration than previously used for Coll/CHA and CHA. The advantage of using CHA–SBF (Fig. 2c) is twofold: (i) the size of the particles appears closer to bone apatite compared to CHA and (ii) the in vitro simulations appear closer to the in vivo conditions thanks to the ionic composition of the mother solution. Here, the effect of collagen confinement is excluded since CHA–SBF is precipitated without collagen.
Interestingly, the 1D 31P NMR spectra of fresh bone and Coll/CHA(SBF) are identical to that of CHA–SBF (Fig. 2d). This result hints that the spatial confinement which affects the Hap platelets during their formation inside the 3D dense collagen matrix does not influence the 31P line shape in contrast to Coll/CHA and CHA. Thus, it seems that the line shape is strictly determined by the ionic composition of the mother solution i.e. the biological serum for the bone and the SBF (for which the ionic composition is close to the biological serum) for Coll/CHA(SBF) and CHA–SBF. The three spectra display a characteristic shoulder (arrows) on the left side of the resonance whereas it is not observed for Coll/CHA and CHA. It is worth mentioning that such a line shape is also observed for different mammalian bone samples (including bovine, rat and human bone).29,37 Hence, this may be the hallmark of in vivo-like bone minerals for future studies.
Fig. 2e displays the {1H–31P}1H double CP spectra of bone, Coll/CHA(SBF) and CHA–SBF. The signal of hydroxyl ions is broad in accordance with 31P results. However, interestingly, the samples display a complex line shape composed of at least 3 components with variable relative intensities. At this stage of study, it is difficult to assign precisely these signals that correspond to OH− ions in diverse environments. Surprisingly, no difference was observed through 31P NMR, while it is interesting to point out that they exhibit different 1H spectra. This means that 1H NMR is more sensitive than 31P NMR regarding the local disorder induced by the precipitation conditions of Hap.
Fig. 3a displays the 2D {1H}31P HetCor spectrum of wetted CHA–SBF. The two phosphate resonances are detected: (i) apatitic PO43− ions (δ(31P) = 3.0 ppm) correlating with OH− ions (δ(1H) = 0 ppm) and (ii) phosphates from the outer disordered layer (δ(31P) = 3.2 ppm) that correlate with water molecules (δ(1H) = 4.85 ppm). The phosphate signal from the outer disordered layer is slightly shifted and associated with a line width significantly broader than the apatitic one (LW 31P apatitic core = 290 Hz; LW 31P disordered layer = 660 Hz). The close chemical shifts observed between these different species may be determined by the establishment of some H-bonds as proposed for octacalcium phosphate.38 Thus, the shoulder on the left side of the 31P MAS spectrum is arising from the resonance of the surface layer phosphates ions (arrow on the 31P HetCor projections). The 31P line shapes extracted from the 31P projections at δ(1H) = 0 and 4.85 ppm allow the fitting of the 1D 31P spectrum. The relative amount of phosphate ions in both regions is (31P apatitic/31P surface) ∼55/45 (Table 1). The 2D {1H}31P HetCor spectra of Coll/CHA(SBF) (Fig. 3b) and intact fresh bone (Fig. 3c) are similar.
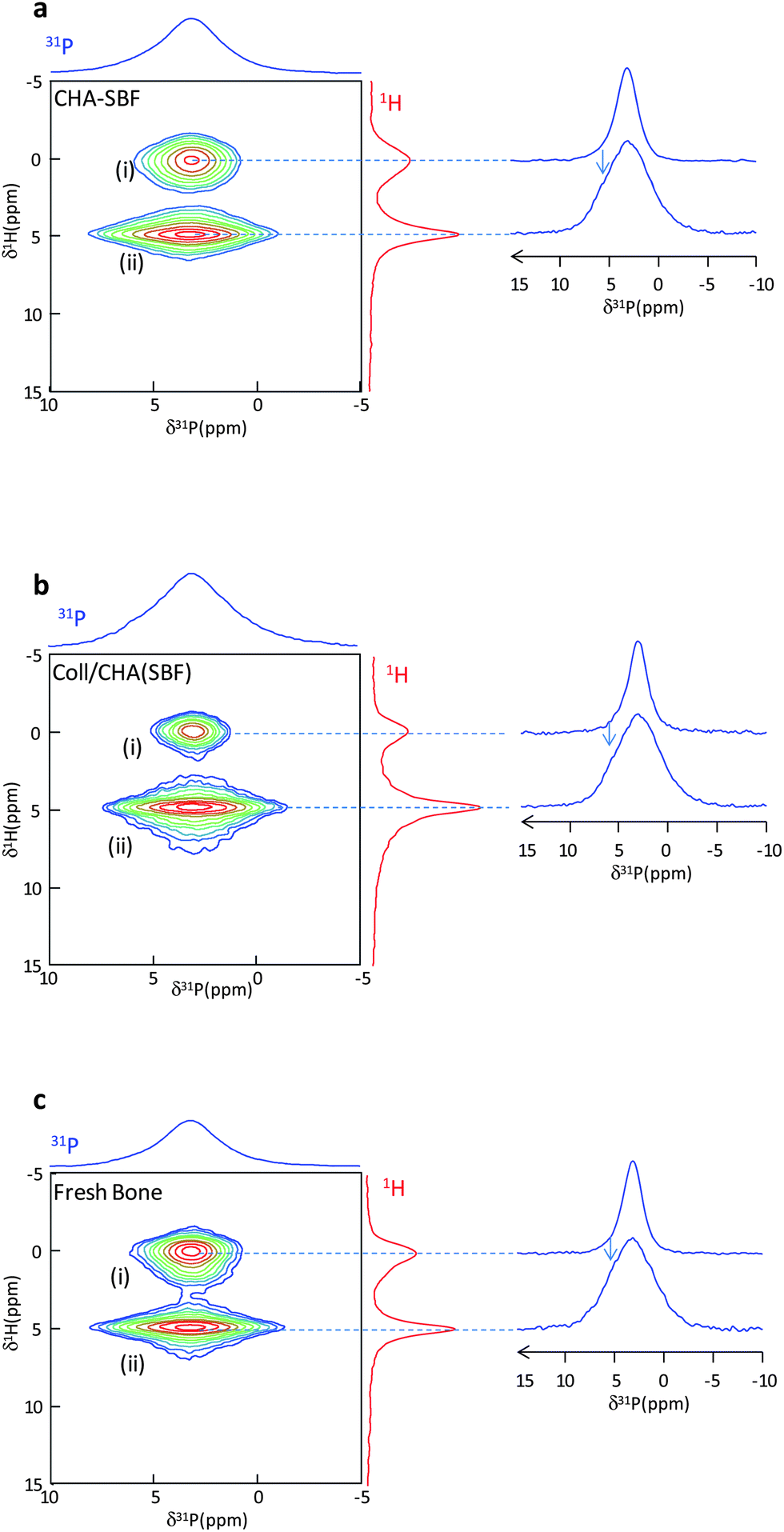 | ||
| Fig. 3 (a, b and c) 2D {1H}31P HetCor spectra of wet CHA–SBF, Coll/CHA(SBF) and fresh bone displaying two distinct domains corresponding to (i) the apatitic core and (ii) the hydrated disordered layer. The arrows on the 31P projections indicate the characteristic shoulder of the bone mineral 31P spectrum. | ||
Hence, as previously demonstrated,22 there is an impact of the collagen confinement on apatite formation at the atomic scale. However, this effect appears to occur only at the initial formation steps of the bone apatite mineral since the collagen confinement does not affect the size of the Hap particles and the proportion of the amorphous layer vs. the apatitic domain. Here, we show that this latter characteristic may actually be determined by the ionic composition of the mother solution.
Comparison of the 31P apatitic core signal of the pure apatite powders, CHA vs. CHA–SBF, shows that the line width is broader for CHA-SBF (Table 1). Thus, the local disorder around the phosphate ions in the apatitic lattice is higher. This could be due to an effect of maturation that would increase the crystallinity as soon as the time of maturation of the particles in solution is increased.10 However, this assumption appears unlikely since the time of maturation is nearly the same for both samples. Thus, it suggests that the local disorder is due to the ionic substitutions in the apatitic lattice. As the SBF is composed of many ions that are known to be present as ionic substituents in bone minerals, various substitutions can be expected such as Na+, CO32−, Mg2+, K+ and Cl−. They might be responsible for the structural disorder of the apatitic lattice as already reported for carbonate ions.39 Furthermore, the insertion of cations can play a role in the apparent depletion of OH− ions in the Hap lattice of bone minerals.29,40 The fact that the OH− resonance is barely visible in the 1H MAS spectrum of CHA–SBF (that is similar to bone minerals) strengthens this assumption (Fig. S2†).
As carbonate ions are the most important ionic substitutions in bone (5–8 wt%), we pursued the study to clarify their role in the related NMR observations.
Influence of the CO32− content on apatite formation
Different syntheses of carbonated Hap were achieved by incorporating various amounts of CO32− (cHA-x) (x = 3.6, 5.2 and 8.1 wt%) and compared to stoichiometric Hap (HA).27 As mentioned previously, 5–8 wt% is the CO32− amount reported for bone minerals. To only assess the influence of the CO32− substitutions (i.e. excluding the occurrence of confinement effects), the apatites were precipitated in the absence of a 3D collagen matrix. The different XRD diffractograms and TEM observations of HA, cHA-3.6, cHA-5.2, cHA-8.1, powders are displayed in Fig S3† and Fig. 4a, respectively. The line width of the diffraction peaks increases with the carbonate ion content (Fig. S3†). The estimation of the lattice parameters and crystallite size (in the ab plane and along the c axis) was carried out through a Rietveld refinement (FullProf software)41 (Table S3†). In agreement with previous data reported on carbonated apatites,42 the c/a ratio increases with the carbonate ion content. Moreover, the crystallites are elongated in the c-axis direction with a decrease in length when the CO32− content increases; HA is characterized by the largest crystallite size (∼25 nm × 25 nm × 62 nm) and cHA-8.1 by the smallest (∼7 nm × 7 nm × 10 nm). According to TEM observations (Fig. 4a), it appears that the particles are actually single crystals. As described earlier,43 the size of the nanorod particles decreases and tends to a spherical morphology as soon as the carbonate content increases. This observation confirms the inhibitor role of carbonate ions in the growth of Hap particles.44 | ||
| Fig. 4 (a) TEM micrographs of HA, cHA-3.6, cHA-5.2, cHA-8.1 (scale bar = 100 nm). (b) 31P MAS quantitative spectra of HA, cHA-3.6, cHA-5.2, cHA-8.1. (c) 1H MAS spectra of HA, cHA-3.6, cHA-5.2, cHA-8.1 (* residual ethanol) and (d–g) 2D {1H}31P HetCor spectra of HA, cHA-3.6, cHA-5.2, cHA-8.1 (the samples are studied in the dry state). | ||
The structural modifications induced by the carbonate ions at the atomic level were assessed by 1H and 31P MAS NMR spectroscopy on dry powders. The 31P NMR MAS spectra of HA, cHA-3.6, cHA-5.2 and cHA-8.1 display one single resonance centered around 2.8–3.1 ppm (Fig. 4b). The 31P spectrum of HA sample displays a sharper signal (LW = 80 Hz) compared to carbonated samples. This is characteristic of a long-range order that is associated with a higher degree of crystallinity. The LW of the 31P MAS spectra increases with the carbonate ion content. In particular, a noticeable increase in line width is observed for cHA-8.1 (210 Hz) compared to cHA-5.2 (116 Hz) and cHA-3.6 (113 Hz). This indicates a higher local disorder reflected by an increase of the 31P chemical shift distribution. It is worth mentioning that the δ(31P) shifts to higher chemical shift values with the carbonate ion content, which is in agreement with previously reported observations.45 Interestingly, cHA-8.1 also displays a shoulder on the left side of the resonance (arrow) similar to that observed for bone minerals, CHA–SBF and Coll/CHA(SBF). This observation suggests that this characteristic shoulder may be a consequence of the carbonate ion incorporation above 5.2 wt%.
The same conclusions can be drawn from the 1H ssNMR analysis (Fig. 4c). The LW of the apatitic OH− resonance (δ(1H) = 0 ppm) is increasing with the carbonate ion content due to the increase of the local disorder around hydroxyl ions. Moreover, a drastic modification of the LW is evidence for cHA-8.1 (compared to cHA-3.6 and cHA-5.2 that exhibit rather similar 1H spectra) where the signal becomes asymmetric with a prominent shoulder on the left side highlighting the presence of distinct environments for the hydroxyl ions in the apatitic lattice. The broad proton resonance at 5 ppm corresponding to adsorbed water is also increasing with the carbonate content probably due to the increase of the specific surface area of the particles exposed to atmospheric moisture.
Aside from the effect of carbonate ions on the local disorder of the apatitic core, we investigate their impact on the amorphous layer. The series of 2D {1H}31 P HetCor experiments for HA, cHA-3.6, cHA-5.2 and cHA-8.1 (Fig. 4d–g) show a strong correlation at δ31P ∼ 3 ppm and δ1H = 0 ppm characteristic of apatitic PO43− ions correlating with hydroxyl ions. A broader correlation centered at δ1H ∼ 5 ppm arises with the increase of the carbonate ion content that corresponds to the non-apatitic surface species. In agreement with measurements on the dry state of the samples, the corresponding 1H signal is spread over a large range of chemical shifts (from 5 to 15 ppm for cHA-8.1). This is in contrast to previous hydrated samples for which the whole signal merges at 4.85 ppm due to a fast proton chemical exchange process involving water molecules. These data strongly suggest that the incorporation of carbonate ions is directly responsible for the formation of the amorphous surface layer. Furthermore, the line width of the resonance corresponding to apatitic phosphate increases with the carbonate ion content (Table 1). This increase of the chemical shift distribution could result from the substitution of carbonate ions in the apatitic lattice.45 A similar behavior is observed for non-apatitic phosphates suggesting also the presence of carbonate ions in the outer layer8 (Table 1).
The simulations of the 31P MAS spectra confirm an increase of the relative amount of phosphate ions in the disordered domain compared to apatitic phosphates (up to 65% for cHA-8.1) (Table 1). If the thickness of the surface layer remains unchanged, this trend can be related to the decrease in particle size and thus in the relative amount of the core species compared to surface species.
The results show that carbonate ions are directly involved in the formation of the outer disordered domain proposed as an ACP layer in bone apatites.8 In addition to their effect on size,44 carbonate ions influence the ACP-to-crystalline ratio of the apatite particles. A related effect may occur for bone apatite where a high amount of carbonate ion substitutions is reported for both young and mature crystals (>6 wt%).
Keeping in mind the higher local disorder in the apatite lattice for Coll/CHA compared to CHA, we propose that a higher amount of CO32− may be incorporated in the apatitic lattice of Coll/CHA. Since the amount of carbonate ions in the mother solution is identical for both samples, this difference can result from a local increase of the carbonate ion concentration within the dense collagen matrix. This may occur through electrostatic attractive forces induced by positive net charges close to the C-terminal domain of the collagen molecules.46
The comparison between CHA–SBF and cHA-8.1 is particularly informative. The initial CO32− concentration in SBF is relatively low (300 times less than the mother solution for cHA-8.1), but the resulting particles possess a similar carbonate ion content (7 wt%). The amount of carbonate ions incorporated may be mediated by the carbonate to phosphate initial ratio (CO3/P = 4.2 vs. CO3/P = 1 for CHA–SBF and cHA-8.1, respectively). The fact that the CO3/P ratio is even more important in the human plasma (up to 27) strengthens this hypothesis. Other ions from SBF described as substitution ions in the Hap lattice (Na+, K+, Cl−) might also help for the insertion of carbonate ions.
Conclusion
In summary, we demonstrate that both, collagen confinement and CO32− substitutions influence the local disorder of the apatitic core and induce the formation of an hydrated amorphous layer. The impact of the 3D confinement in a dense collagen matrix may be of importance in the first stage of bone mineral formation. This 3D confinement impacts both the 31P and the 1H NMR response for Hap particles synthesized in an aqueous medium where O3/P = 1. In that case, we observe a higher proportion of the ACP layer as well as a higher structural disorder of the apatitic core in the presence of the collagen matrix. This may be explained by a local increase of carbonate ion concentration within the gap region due to a positive net charge close to the C-terminal domain of the collagen molecules. We also show that, in addition to a collagen confinement,22 the carbonate substitutions may also interfere in mediating the particle sizes in vivo. The proportion of the ACP layer relative to the crystalline core increases with the CO32− content and is concomitant with the decrease of the particle size. This latter characteristic appears to control the degree of crystallinity of newly formed bone apatite nanocrystals (while other parameters may play a role during the maturation). The incorporation of ∼7–8 wt% carbonate ions allows the setting of apatitic models with similar spectral characteristics to those found for bone minerals.Acknowledgements
We thank IMM Recherche especially Dr L. Behr for providing the fresh bone samples, A. Anglo and C. Illoul for preparation of bone thin sections for TEM observations, Dr Ö. Sel and Dr J. Seto for insightful discussions and critical suggestions. This work was supported by the Agence Nationale de la Recherche (ANR) through the ANR- 09-BLAN-0120-01 “NanoShap” program.References
- M. J. Glimcher, in Medical Mineralogy and Geochemistry, ed. N. Sahai and M. A. A. Schoonen, 2006, vol. 64, pp. 223–282 Search PubMed.
- S. Fitton-Jackson, Proc. R. Soc. London, Ser. B, 1957, 146, 270–280 CrossRef.
- S. Weiner and W. Traub, FEBS Lett., 1986, 206, 262–266 CrossRef CAS.
- W. J. Landis, K. J. Hodgens, J. Arena, M. J. Song and B. F. McEwen, Microsc. Res. Tech., 1996, 33, 192–202 CrossRef CAS.
- H. Frenkel-Mullerad and D. Avnir, J. Am. Chem. Soc., 2005, 127, 8077–8081 CrossRef CAS PubMed.
- G. K. Hunter and H. A. Goldberg, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8562–8565 CrossRef CAS.
- C. Rey, C. Combes, C. Drouet and M. J. Glimcher, Osteoporosis Int., 2009, 20, 1013–1021 CrossRef CAS PubMed.
- Y. Wang, S. Von Euw, F. M. Fernandes, S. Cassaignon, M. Selmane, G. Laurent, G. Pehau-Arnaudet, C. Coelho, L. Bonhomme-Coury, M.-M. Giraud-Guille, F. Babonneau, T. Azaïs and N. Nassif, Nat. Mater., 2013 DOI:10.1038/NMAT3787.
- R. Legros, N. Balmain and G. Bonel, J. Chem. Res., 1986, 8–9 CAS.
- C. Rey, C. Combes, C. Drouet, H. Sfihi and A. Barroug, Mater. Sci. Eng., C, 2007, 27, 198–205 CrossRef CAS PubMed.
- A. K. Bembey, A. J. Bushby, A. Boyde, V. L. Ferguson and M. L. Oyen, J. Mater. Res., 2006, 21, 1962–1968 CrossRef CAS.
- S. C. Cowin, J. Biomech. Eng., 1999, 32, 217–238 CAS.
- W. Kolodziejski, in New Techniques in Solid-State NMR, ed. J. Klinowski, Springer, Berlin/Heidelberg, 2004, vol. 246, pp. 235–270 Search PubMed.
- J. Xu, P. Zhu, Z. Gan, N. Sahar, M. Tecklenburg, M. D. Morris, D. H. Kohn and A. Ramamoorthy, J. Am. Chem. Soc., 2010, 132, 11504–11509 CrossRef CAS PubMed.
- P. Zhu, J. Xu, N. Sahar, M. D. Morris, D. H. Kohn and A. Ramamoorthy, J. Am. Chem. Soc., 2009, 131, 17064–17065 CrossRef CAS PubMed.
- Y.-Y. Hu, A. Rawal and K. Schmidt-Rohr, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22425–22429 CrossRef CAS PubMed.
- O. Nikel, D. Laurencin, C. Bonhomme, G. E. Sroga, S. Besdo, A. Lorenz and D. Vashishth, J. Phys. Chem. C, 2012, 116, 6320–6331 CAS.
- R. K. Rai and N. Sinha, J. Phys. Chem. C, 2011, 115, 14219–14227 CAS.
- E. R. Wise, S. Maltsev, M. E. Davies, M. J. Duer, C. Jaeger, N. Loveridge, R. C. Murray and D. G. Reid, Chem. Mater., 2007, 19, 5055–5057 CrossRef CAS.
- A. H. Roufosse, W. P. Aue, J. E. Roberts, M. J. Glimcher and R. G. Griffin, Biochemistry, 1984, 23, 6115–6120 CrossRef CAS.
- J. P. Yesinowski and H. Eckert, J. Am. Chem. Soc., 1987, 109, 6274–6282 CrossRef CAS.
- Y. Wang, T. Azaïs, M. Robin, A. Vallée, C. Catania, P. Legriel, G. Pehau-Arnaudet, F. Babonneau, M.-M. Giraud-Guille and n. Nassif, Nat. Mater., 2012, 11, 724–733 CrossRef CAS PubMed.
- L. Wang and G. H. Nancollas, Chem. Rev., 2008, 108, 4628–4669 CrossRef CAS PubMed.
- M. M. G. Guille, C. Helary, S. Vigier and N. Nassif, Soft Matter, 2010, 6, 4963–4967 RSC.
- J. Silvent, N. Nassif, C. Helary, T. Azais, J. Y. Sire and M. M. G. Guille, PLoS One, 2013, 8, e57344 CAS.
- N. Nassif, F. Martineau, O. Syzgantseva, F. Gobeaux, M. Willinger, T. Coradin, S. Cassaignon, T. Azais and M. M. Giraud-Guille, Chem. Mater., 2010, 22, 3653–3663 CrossRef CAS.
- S. Takemoto, Y. Kusudo, K. Tsuru, S. Hayakawa, A. Osaka and S. Takashima, J. Biomed. Mater. Res., Part A, 2004, 69, 544–551 Search PubMed.
- C. Bonhomme, C. Coelho, N. Baccile, C. Gervais, T. Azais and F. Babonneau, Acc. Chem. Res., 2007, 40, 738–746 CrossRef CAS PubMed.
- G. Cho, Y. Wu and J. L. Ackerman, Science, 2003, 300, 1123–1127 CrossRef CAS PubMed.
- S. Maltsev, M. J. Duer, R. C. Murray and C. Jaeger, J. Mater. Sci., 2007, 42, 8804–8810 CrossRef CAS.
- R. A. Santos, R. A. Wind and C. E. Bronnimann, J. Magn. Reson., Ser. B, 1994, 105, 183–187 CrossRef CAS.
- C. Jäger, T. Welzel, W. Meyer-Zaika and M. Epple, Magn. Reson. Chem., 2006, 44, 573–580 CrossRef PubMed.
- Y. Wang, J. Silvent, M. Robin, F. Babonneau, A. Meddahi-Pelle, N. Nassif and M. M. Giraud Guille, Soft Matter, 2011, 7, 9659–9664 RSC.
- A. Hodge and J. Petruska, in Aspects of Protein Structure, ed. G. Ramachandran, Academic Press, 1963, pp. 289–300 Search PubMed.
- N. Folliet, C. Roiland, S. Begu, A. Aubert, T. Mineva, A. Goursot, K. Selvaraj, L. Duma, F. Tielens, F. Mauri, G. Laurent, C. Bonhomme, C. Gervais, F. Babonneau and T. Azais, J. Am. Chem. Soc., 2011, 133, 16815–16827 CrossRef CAS PubMed.
- S. H. Rhee and J. Tanaka, J. Mater. Sci.: Mater. Med., 2000, 11, 449–452 CrossRef CAS.
- A. Kaflak-Hachulska, A. Samoson and W. Kolodziejski, Calcif. Tissue Int., 2003, 73, 476–486 CrossRef CAS PubMed.
- E. Davies, M. J. Duer, S. E. Ashbrook and J. M. Griffin, J. Am. Chem. Soc., 2012, 134, 12508–12515 CrossRef CAS PubMed.
- C. F. Nawrot, D. J. Campbell, J. K. Schroeder and M. Van Valkenburg, Biochemistry, 1976, 15, 3445–3449 CrossRef CAS.
- C. K. Loong, C. Rey, L. T. Kuhn, C. Combes, Y. Wu, S. H. Chen and M. J. Glimcher, Bone, 2000, 26, 599–602 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- D. G. Nelson and J. D. Featherstone, Calcif. Tissue Int., 1982, 34, S69–S81 Search PubMed.
- D. G. A. Nelson, J. Dent. Res., 1981, 60, 1621–1629 CrossRef CAS PubMed.
- R. Z. Legeros, O. R. Trautz, J. P. Legeros, E. Klein and W. P. Shirra, Science, 1967, 155, 1409–1411 CAS.
- W. P. Aue, A. H. Roufosse, M. J. Glimcher and R. G. Griffin, Biochemistry, 1984, 23, 6110–6114 CrossRef CAS.
- F. Nudelman, K. Pieterse, A. George, P. H. H. Bomans, H. Friedrich, L. J. Brylka, P. A. J. Hilbers, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1004–1009 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of the sample synthesis and characterization techniques used. See DOI: 10.1039/c3mh00071k |
| This journal is © The Royal Society of Chemistry 2014 |