From supramolecular polymers to hydrogel materials†
Christianus M. A.
Leenders
,
Tristan
Mes
,
Matthew B.
Baker
,
Marcel. M. E.
Koenigs
,
Pol
Besenius
,
Anja R. A.
Palmans
* and
E. W.
Meijer
*
Institute for Complex Molecular Systems, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands. E-mail: e.w.meijer@tue.nl; a.palmans@tue.nl; Fax: +31 (0)40 2451036; Tel: +31 (0)40 2473101
First published on 10th October 2013
Abstract
Supramolecular hydrogels formed by decorating benzene-1,3,5-tricarboxamide (BTA) units with amphiphilic ethylene glycol-based side chains are presented; careful selection of the substituents of the BTAs allows for the tuning of the self-assembly behaviour and hence the mechanical properties of the resultant hydrogel.
Hydrogels are an intriguing class of soft material with a wide scope of applications ranging from regenerative medicine1 to the preparation of soft machines.2 Over the past decade, systems have been explored in which non-covalent interactions, such as hydrogen bonding, π–π stacking, metal complexation, ionic, and solvophobic interactions drive the hydrogel formation. In contrast to covalent bonds, non-covalent interactions can make these systems dynamic, stimuli responsive, and potentially self-healing. The two main strategies followed to form a non-covalent transient network employ either the self-assembly of small molecules, or macromolecules decorated with associating groups. In low molecular weight (LMW) hydrogelators, small molecules self-assemble into fibrillar aggregates which subsequently form a transient network.3 Initially, serendipity played an important role in the identification of LMW hydrogelators, but the development of structure–property relationships has enabled a more rational, design-based approach towards functional and responsive hydrogelators.3d,4 Alternatively, the decoration of polymer chains with self-assembling moieties results in network formation where the polymer backbone is crosslinked via non-covalent interactions.5
In contrast to most synthetic hydrogels, biological hydrogels display unique nonlinear mechanical properties as a result of finely tuned filament stiffness and crosslink density.6 Only recently, rigid, synthetic polymers were reported that display unique strain stiffening by controlled bundle formation.7 Hydrogels based on non-covalent interactions are, in principle, uniquely suited to tune bundle stiffness via hierarchical self-assembly principles.8 Additionally, the crosslink density can be readily controlled by balancing the number of associating groups per polymer chain.9 Because of the necessary balance between multiple interactions, the relationship between molecular structure and mechanical properties of supramolecular hydrogels remains a challenge.
In this communication, we report a set of hydrogels utilizing the N,N′,N′′-trialkyl-benzene-1,3,5-tricarboxamide (BTA) unit to go from supramolecular polymers in water to tailored hydrogel materials with properties dictated by the BTA self-assembly.10 With a bottom-up approach, we clearly show how small changes in the molecular structure of a self-assembling monomer allows one to tailor the properties of the resulting supramolecular hydrogel materials. In organic solvents, BTAs self-assemble into columnar helical stacks driven by threefold intermolecular hydrogen bonding by a cooperative mechanism.11 In aqueous solution, the BTA amides need to be shielded from the surrounding polar media; introducing a hydrophobic spacer effectively allows self-assembly to be driven by a combination of hydrogen bonding and hydrophobic interactions.12 Here, we systematically studied a small library of BTA derivatives, both in dilute solution and in the gel, with the aim to relate the molecular structure to the gel morphology and mechanical properties (Scheme 1, see ESI† for synthetic details). To establish structure–property relationships, the substitution of LMW BTA derivatives 1–3 varies in the number of hydrophobic side chains. In addition, telechelic polymers 4a–c and 5 are obtained by connecting two BTAs via a poly(ethyleneglycol) (PEG) based linker with aliphatic spacers which are varied in length (4a–c).
 | ||
| Scheme 1 Chemical structures of BTA derivatives 1–5. | ||
First, the self-assembly of BTA derivatives 1–5 in solution was assessed by cryogenic transmission electron microscopy (cryo-TEM) measurements on samples prepared at 1 mg mL−1 in water (Fig. 1). Gratifyingly, 1 forms high aspect-ratio supramolecular polymers with a diameter of approximately 5 nm and of micrometer lengths, and no bundling or clustering is observed. At this concentration, 2 and 3 are insoluble, highlighting the importance of the hydrophilic and hydrophobic balance. Telechelic 4c assembles into larger fibers with diameters up to 100 nm, but close inspection reveals that these fibers contain small fibrils, with a diameter comparable to the fibers observed in samples of 1. This observation suggests that the hydrophobic substituents on the BTA moieties in 4c induce the clustering of individual BTA stacks. While the substitution on the BTA moiety in 5 is comparable to 1, no fibrillar aggregates are visible in the cryo-TEM images; instead, small spherical objects are observed. The lower molar concentration of BTA assembling units (ten times lower in the cryo-TEM of 5 compared to 1) and the balance of hydrophobic to hydrophilic regions (when compared to 1 and 4) significantly influence the self-assembly of the BTA units in molecule 5, consistent with Israelachvili's theory of surfactants.13
 | ||
| Fig. 1 Cryo-TEM images of 1, 4c and 5. (A) Network structure of 1 at 1 mg mL−1 (cBTA = 7.5 × 10−4 M). The network consists of micrometer long fibrils with a width of 5 nm (scale bar 100 nm). (B) Bundle of fibrils of 4c at 1 mg mL−1 (cBTA = 5 × 10−5 M). Fibrils are bundled into micrometer long fibers and have a width of 80 nm (scale bar 100 nm). (C) Micellar structure of 5 at 1 mg mL−1 (cBTA = 5 × 10−5 M). The micelles have an approximate diameter of 10 nm (scale bar 100 nm). | ||
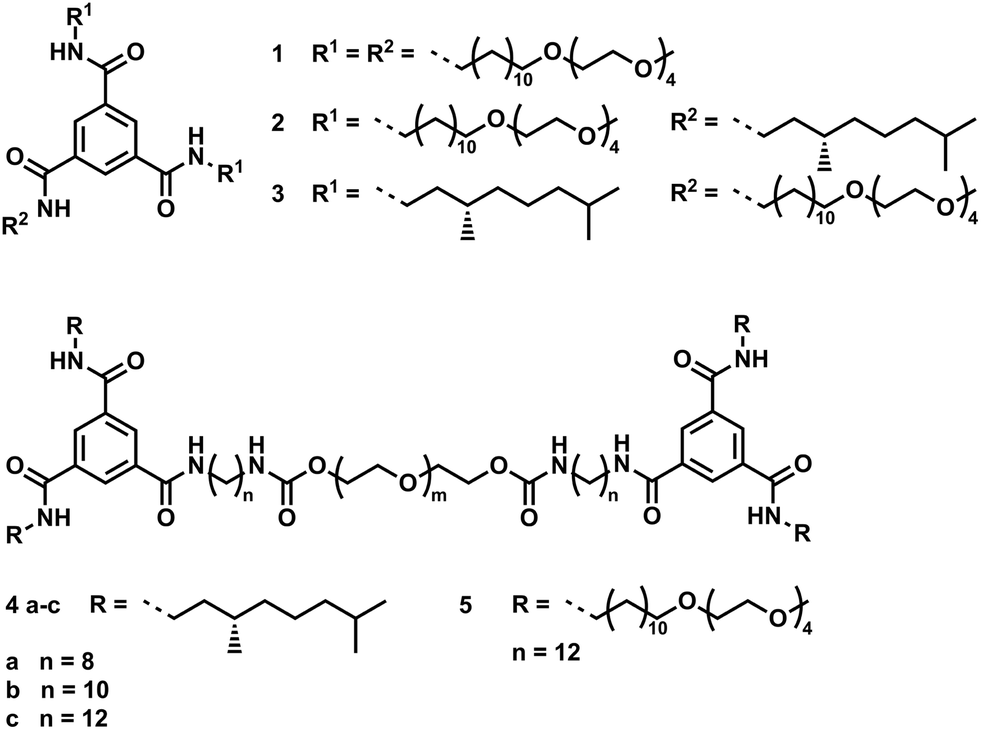
Additional spectroscopic measurements on solutions of 1, 4a–c and 5 in water at similar concentrations indicate that the BTA units are aggregated under dilute conditions. The UV-vis absorption spectrum of 1 (c = 5 × 10−5 M) in water displays maxima at 209 and 225 nm, indicating BTA aggregation (Fig. S5†).12a The PEG linker in 4a–c and 5 dominates the absorption spectrum in the region of interest (Fig. S6†), preventing analysis. Circular dichroism (CD) spectroscopy on optically active derivatives 4a–c shows negative Cotton effects at 223 nm, indicating the self-assembly of BTAs into left handed helical columnar stacks (Fig. 2A).11,12a Interestingly, the molar circular dichroism Δε strongly depends on the length of the hydrophobic spacer between the BTA moiety and the PEG linker and varies from −16, to −24, to −36 L mol−1 cm−1 for 4a, 4b and 4c, respectively. The Δε of 4c is close to the maximum value for small BTA derivatives in water (|Δε| = 43 L mol−1 cm−1),12a suggesting a high degree of aggregation of the BTA end groups into helical columnar stacks. The lower intensities recorded for 4a and 4b imply only partial aggregation into helical stacks. Apparently, the shorter spacers insufficiently shield the BTA amides from the surrounding polar media, limiting the self-assembly into helical aggregates.
 | ||
| Fig. 2 Molar circular dichroism Δε of 4a–c: (A) in solution (cBTA for 4a = cBTA for 4b = 9.6 × 10−5 M and cBTA for 4c = 6.6 × 10−5 M in water, l = 1.0 cm, T = 20 °C); (B) in the gel state (cBTA for 4a–c = 9.4 × 10−3 M in water, l = 0.2 mm, T = 20 °C). | ||
A qualitative assessment of the ability of compounds 1–5 to form hydrogels was obtained from vial inversion tests. The critical gelation concentration (CGC)—corresponding to the minimum concentration necessary for the solution to not flow upon vial inversion—was determined and the results are presented in Table 1. BTA 1 forms clear, viscous solutions at concentrations up to 2.5 × 10−3 M (0.33% w/w). At higher concentrations the solution starts to become slightly opaque, and above roughly 5% w/w a slightly opaque gel is formed. Presumably, at this high concentration enough physical entanglements between the aggregates occur to form a transient network. Telechelic 4a–c form stable (self-supporting for several months), clear, and fully transparent gels with CGCs between 7 and 11 wt% (Table 1); interestingly, the CGC depends on the hydrophobic spacer length. The CGC of 4a, with the shortest spacer length of eight carbon atoms, is the highest at 11% w/w. With increasing spacer length, the observed CGC decreases to 7% w/w for 4c. This suggests weaker association of the endgroups, possibly due to less effective shielding of the BTA core from the surrounding polar medium by the shorter spacers, hampering the self-assembly and resulting in a weaker gel. In fact, the CD spectra of the gels of 4a–c (Fig. 2B) show a similar trend. The Δε values vary from −5, to −7.5, to −16 L mol−1 cm−1 for 4a, 4b and 4c, respectively, suggesting a decrease in BTA aggregation upon shortening the spacer length. Interestingly, the recorded Δε values are all lower than those found in dilute solution (Fig. 2A), indicating the formation of less well-defined aggregates by the BTA moieties in the network responsible for gelation. In addition, the shape of the Cotton effect is slightly different, indicating slight changes in the packing between the BTAs.14 Telechelic 5 also forms a stable and clear gel with a CGC of 8% w/w. Thus, while the cryo-TEM revealed large differences in the aggregation of 4c and 5 at 0.1% w/w, 5 does form stable gels with a CGC close to that of 4c.

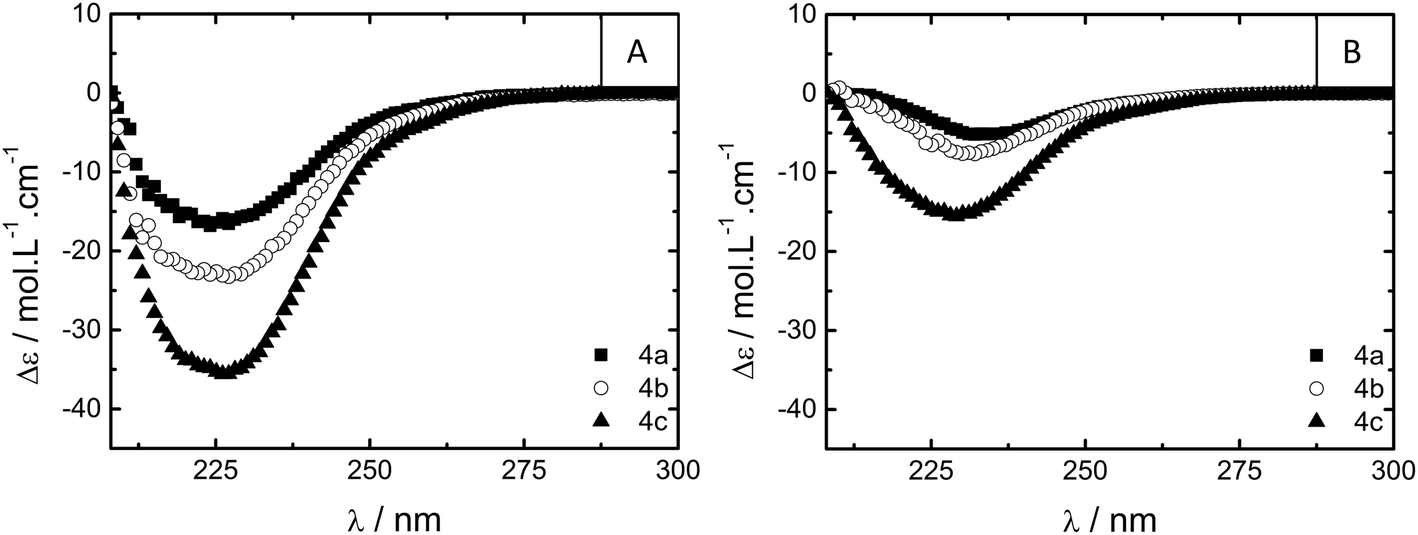
After this qualitative analysis, oscillatory rheology measurements on samples of 1 (c = 5% w/w), 4a–c (c = 10% w/w) and 5 (c = 10% w/w) were performed to study the gels in more detail (Table 2, see ESI† for details). All compounds showed visco-elastic character during the timescales investigated. Compounds 4a–c, containing the hydrophobic (S)-3,7-dimethyloctyl substituents, showed similarly high plateau storage moduli (G0, ∼104 Pa) while polymer 5 and small-molecule 1, containing tetraethylene glycol substituents, showed lower G0 values (1730 Pa and ∼80 Pa, respectively).
| Compound | G 0 | λ (s) | η 0 (Pa s) | γ Y (%) |
|---|---|---|---|---|
| a Relaxation times were estimated from the crossover points of frequency sweep measurements. b The intrinsic viscosity was determined from plateau values during frequency sweeps. c The yield point was defined as the deviation from linearity for shear stress during strain sweep. d ND = not determined. | ||||
| 1 | ∼80 | NDd | NDd | 11 |
| 4a | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
2.1 | 3500 | 110 |
| 4b | 12500 |
16 | 29000 |
72 |
| 4c | 15400 |
57 | 70000 |
55 |
| 5 | 1730 | 0.42 | 740 | 160 |
During frequency sweep measurements (Fig. 3), the telechelic polymers 4a–c and 5 all show flow on longer timescales (G′′ > G′ as angular frequency, ω, is smaller) while for 1, G′ and G′′ remain more frequency-independent within the measured range, suggesting the formation of significantly different gel networks. Within the telechelic polymers, the spectra of 4a–c are nearly superimposable upon a frequency shift, all culminating in very similar G0 values; this similarity suggests similar morphology and crosslink density within the series. Thus, expanding the spacer between BTA and PEG from eight carbons in 4a to twelve carbons in 4c results in more than an order of magnitude increase in the relaxation time of the gel network, ultimately forming a less dynamic gel (Table 2). This increase in relaxation time (λ) concomitant with an increase in intrinsic viscosity (η0) has also been observed in PEG polymers capped with hydrocarbons and fluorocarbons.15 These trends have been connected to an increase in the energy required to disrupt non-covalent crosslinks due to persistent self-assembly; in conjunction with the CD data (vide supra), the BTAs in the polymers 4a–c appear to interact more strongly upon increasing the hydrophobic spacer length. Telechelic compound 5 forms both a weaker (low G0) and more dynamic (low λ) gel when compared to series 4. While this would suggest a gel with a lower number of more dynamic crosslinks, direct comparisons are difficult to make due to potential differences in morphology (see cryo-TEM).
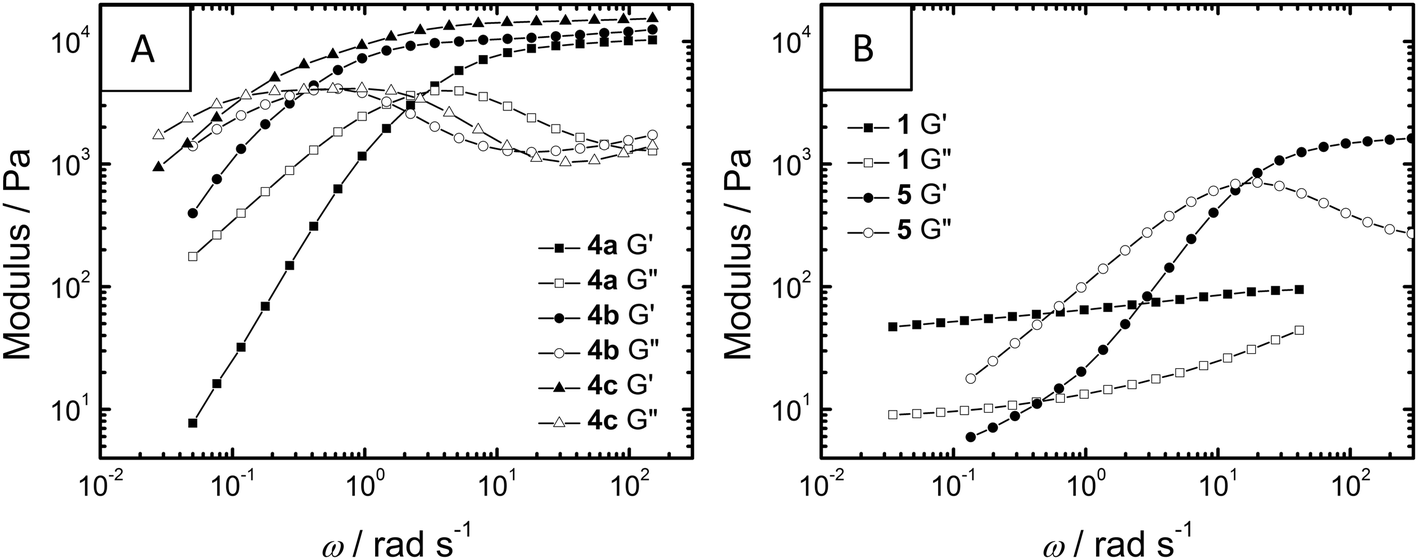 | ||
| Fig. 3 Oscillatory rheology frequency sweeps at 20 °C of storage modulus G′ and loss modulus G′′ of 1 (5% w/w), 4a–c (10% w/w) and 5 (10% w/w). | ||
When the gel moduli were measured as a function of strain (Fig. S1 and S2†), small molecule 1 took the lowest amount of strain (γ) until loss of gel integrity (11%) while telechelic compound 5 took the highest amount of strain (160%). Interestingly, for the telechelic hydrogelators 4a–c and 5, the molecules showing the shortest relaxation times also tolerate the most strain.
For the series 4a–c a linear increase of the aliphatic spacer length leads to a non-linear response in the self-assembly as observed in the CD measurements; moreover, the rheological data mirrors this non-linear trend. This suggests that the macroscopic properties of the hydrogel materials are influenced by the molecular structure but, more accurately, are dictated by the resultant supramolecular structure.
In conclusion, we have successfully prepared hydrogelators based on the self-assembly of BTAs with amphiphilic substituents. By carefully choosing the molecular structure of the BTA unit, we can tune the self-assembly behaviour in solution and hence modulate the mechanical properties of the resulting hydrogels. The BTA unit as a LMW hydrogelator forms only weak gels (G0 ∼ 102 Pa). By coupling two BTA units together via a PEG-based linker, stronger (G0 ∼ 103–104 Pa), stable, clear gels are formed, and by modifying the substituents on these BTA units, the mechanical properties can be tuned. The length of the hydrophobic spacer between the PEG linker and the BTA units has a direct effect on the mechanical properties of the gel. Furthermore, the induction of bundle formation in 4a–c compared to 5 affects the mechanical properties, presumably through the formation of a different type of network. Finally, the BTA end-groups still adopt helical packing in the gel-state as seen in CD measurements, suggesting that BTA self-assembly in combination with hydrophobic effects provides the non-covalent network. Therefore, small BTA derivatives may be incorporated into these one-dimensional BTA motifs, allowing a facile method for scaffold functionalization, toward modular and rationally designed biomaterials.
Notes and references
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- P. Calvert, Adv. Mater., 2009, 21, 743–756 CrossRef CAS.
- (a) R. G. Weiss and P. Terech, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, Springer London, Limited, 2006 Search PubMed; (b) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS PubMed; (c) M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS; (d) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed; (e) J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC.
- (a) J. H. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS PubMed; (b) M. Zelzer and R. V. Ulijn, Chem. Soc. Rev., 2010, 39, 3351–3357 RSC.
- (a) E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC; (b) C. Tsitsilianis, Soft Matter, 2010, 6, 2372–2388 RSC; (c) A. Noro, M. Hayashi and Y. Matsushita, Soft Matter, 2012, 8, 6416–6429 RSC; (d) S. Seiffert and J. Sprakel, Chem. Soc. Rev., 2012, 41, 909–930 RSC.
- (a) C. Storm, J. J. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nature, 2005, 435, 191–194 CrossRef CAS PubMed; (b) M. L. Gardel, J. H. Shin, F. C. MacKintosh, L. Mahadevan, P. Matsudaira and D. A. Weitz, Science, 2004, 304, 1301–1305 CrossRef CAS PubMed.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, P. van Rijn, M. C. A. Stuart, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2011, 50, 12285–12289 CrossRef CAS PubMed.
- (a) P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed; (b) P. Y. W. Dankers, M. J. A. van Luyn, A. Huizinga-van der Vlag, G. M. L. van Gemert, A. H. Petersen, E. W. Meijer, H. M. Janssen, A. W. Bosman and E. R. Popa, Biomaterials, 2012, 33, 5144–5155 CrossRef CAS PubMed.
- (a) A. Bernet, R. Q. Albuquerque, M. Behr, S. T. Hoffmann and H.-W. Schmidt, Soft Matter, 2012, 8, 66–69 RSC; (b) R. C. T. Howe, A. P. Smalley, A. P. M. Guttenplan, M. W. R. Doggett, M. D. Eddleston, J. C. Tan and G. O. Lloyd, Chem. Commun., 2013, 49, 4268–4270 RSC.
- (a) M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606–611 CrossRef CAS PubMed; (b) S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC; (c) P. J. M. Stals, M. M. J. Smulders, R. Martin-Rapun, A. R. A. Palmans and E. W. Meijer, Chem.–Eur. J., 2009, 15, 2071–2080 CrossRef CAS PubMed; (d) T. Mes, M. M. J. Smulders, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2010, 43, 1981–1991 CrossRef CAS; (e) P. J. M. Stals, J. F. Haveman, R. Martin-Rapun, C. F. C. Fitie, A. R. A. Palmans and E. W. Meijer, J. Mater. Chem., 2009, 19, 124–130 RSC.
- (a) C. M. A. Leenders, L. Albertazzi, T. Mes, M. M. E. Koenigs, A. R. A. Palmans and E. W. Meijer, Chem. Commun., 2013, 49, 1963–1965 RSC; (b) P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef CAS PubMed; (c) P. Besenius, K. P. van den Hout, H. M. H. G. Albers, T. F. A. de Greef, L. L. C. Olijve, T. M. Hermans, B. F. M. de Waal, P. H. H. Bomans, N. A. J. M. Sommerdijk, G. Portale, A. R. A. Palmans, M. H. P. van Genderen, J. A. J. M. Vekemans and E. W. Meijer, Chem.–Eur. J., 2011, 17, 5193–5203 CrossRef CAS PubMed; (d) M. von Groning, I. de Feijter, M. C. A. Stuart, I. K. Voets and P. Besenius, J. Mater. Chem. B, 2013, 1, 2008–2012 RSC.
- J. B. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Perkin Trans. 2, 1976, 1525–1568 Search PubMed.
- Y. Nakano, T. Hirose, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Chem. Sci., 2012, 3, 148–155 RSC.
- (a) T. Annable, R. Buscall, R. Ettelaie and D. Whittlestone, J. Rheol., 1993, 37, 695–726 CrossRef CAS; (b) B. Xu, L. Li, A. Yekta, Z. Masoumi, S. Kanagalingam, M. A. Winnik, K. Zhang, P. M. Macdonald and S. Menchen, Langmuir, 1997, 13, 2447–2456 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization by UV and CD spectroscopy and oscillatory rheology. See DOI: 10.1039/c3mh00103b |
| This journal is © The Royal Society of Chemistry 2014 |