Preparation of porous nanocarbons with tunable morphology and pore size from copolymer templated precursors†
Mingjiang
Zhong
a,
Chuanbing
Tang
*b,
Eun Kyung
Kim
ac,
Michal
Kruk‡
a,
Ewa B.
Celer
d,
Mietek
Jaroniec
d,
Krzysztof
Matyjaszewski
*a and
Tomasz
Kowalewski
*a
aDepartment of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, Pennsylvania 15213, USA. E-mail: km3b@andrew.cmu.edu; tomek@andrew.cmu.edu; Fax: +1-412-929-8102; Tel: +1-412-268-5927
bDepartment of Chemistry and Biochemistry, University of South Carolina, 631 Sumter Street, Columbia, South Carolina 29208, USA. E-mail: tang4@mailbox.sc.edu
cCurrent address: Battery R&D, AMD PJT, LG Chem Research Park, LG Chem, 104-1, Moonji-dong, Yuseong-gu, Daejeon 305-738, Korea
dDepartment of Chemistry, Kent State University, Kent, Ohio 44242, USA
First published on 19th September 2013
Abstract
Porous nanocarbons with tunable morphology and pore size were prepared via the carbonization of polyacrylonitrile containing block copolymers. Retention of the nanostructure of the precursor upon pyrolysis was studied for both thin-film and bulk materials. Morphology-dependent preservation of the resulting nanostructure was observed in the latter case.
The chemical diversity of carbon is behind a variety of industrial scale carbon materials with diverse physical forms, such as powders, fibers, films, monoliths, and well-defined nanostructures including fullerenes, nanotubes and graphenes.1 Among these, porous nanocarbon materials are of particular interest, with applications including filters, catalysts, absorbents, and energy conversion and storage devices.2 Some of the desirable features of nanoporous materials include high specific surface area, large pore volume, adjustable pore shape and pore diameter, and tailorable surface properties.3 Nanoporous materials are typically synthesized through templating methods, often based on micelle and hard templating strategies.3 Another elegant approach to the synthesis of nanoporous materials is based on the phase behavior of block copolymers (BCP).4 Provided that the structure of domains, formed by one of the blocks, can be “locked” and the domains of the other sacrificial block can be selectively removed, a remarkable variety of nanoporous structures of tailorable dimensions becomes accessible.4
Previously, we have reported the preparation of copolymer templated nitrogen-enriched porous nanocarbon (CTNC) materials by the carbonization of well-defined BCP precursors containing polyacrylonitrile (PAN) segments as the nitrogen containing carbonaceous precursors, and poly(n-butyl acrylate) (PBA) as the sacrificial block, which can be thermally degraded to generate mesopores.5 The concurrent introduction of nitrogen heteroatoms facilitated by this approach has been shown to lead to the modification of the electronic properties, catalytic activity, and chemical functionality, opening the way to a broad spectrum of applications ranging from electrode materials for supercapacitors, CO2 sorbents, and electrocatalysts for the oxygen reduction reaction.5a–c For instance, the CO2 adsorption capacity and selectivity over N2 of CTNCs exceeded values reported for general activated carbons in spite of lower specific surface areas.5b,c Performance of CTNC's in these kinds of applications is critically dependent on the accessibility of surface carbon and nitrogen functionalities, which, in turn, depend on the nanostructure. Herein, we report the results of studies demonstrating the dependence of thin-film and bulk carbon nanostructures on the morphology and block size of phase-separated PAN-b-PBA copolymer precursors. Scheme 1 illustrates the synthetic route to CTNCs starting from the PAN-b-PBA, with PAN representing a minority phase. As shown in the scheme, BCPs with this range of compositions provide the resulting CTNCs of diverse nanostructures with highly accessible interfaces.
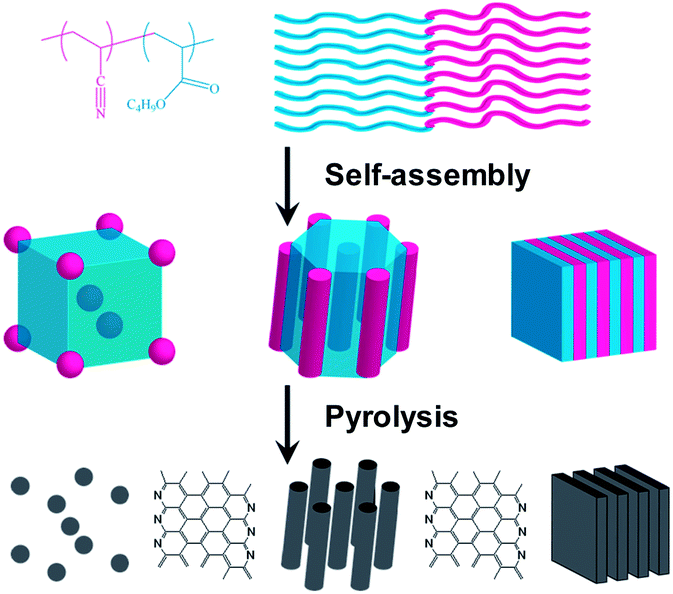 | ||
| Scheme 1 Preparation of porous CTNCs from PAN-b-PBA (adapted from ref. 5a). | ||
The BCP precursors were synthesized via atom transfer radical polymerization (ATRP) as reported before.6 Table S1 (ESI†) lists all samples discussed in this work. In our past work we have demonstrated that in order to preserve the nanostructure and prevent mixing of the two phases during thermal treatment, PAN has to be stabilized by heating to 280 °C under air.5 This stabilization step allows the morphology of the PAN phase to be fixed through side chain cross-linking and cyclization, after which the nanostructure can be maintained during carbonization between 500 and 1000 °C under an inert atmosphere.5d,7 In what follows, all CTNCs were pre-stabilized for 1 hour under the above conditions prior to carbonization.
The evolution of morphology of thin films upon thermal treatment was followed with the aid of tapping mode atomic force microscopy (AFM). As shown in Fig. 1, the carbonization of thin films of PAN-b-PBA drop-cast on Si substrates at 600 °C under N2 resulted in materials with morphology closely resembling the original morphology of PAN domains. For instance, the copolymer containing 5.8 wt% PAN had PAN domains of spherical morphology (Fig. 1a), which was preserved after the stabilization and subsequent pyrolysis (Fig. 1b). These results corroborate the findings reported earlier for the triblock copolymers of PAN.5d The copolymer BA240AN104, where the subscripts stand for the degree of polymerization (DP) of corresponding blocks, containing 15.2 wt% PAN resulted in the formation of short cylindrical domains of PAN (Fig. 1c). The AFM images indicate that after carbonization, the short PAN cylinders were all converted into short carbon filaments (Fig. 1d). A copolymer of 17.6 wt% PAN, but higher DP, i.e., BA240AN124, formed PAN domains in the form of long cylinders (Fig. 1e). The resulting CTNC also maintained to a large extent its original structure (Fig. 1f). A BA78AN114 copolymer containing 37.7 wt% PAN exhibited a complicated branched morphology (Fig. 1g), which was also well retained upon thermal treatment (Fig. 1h).
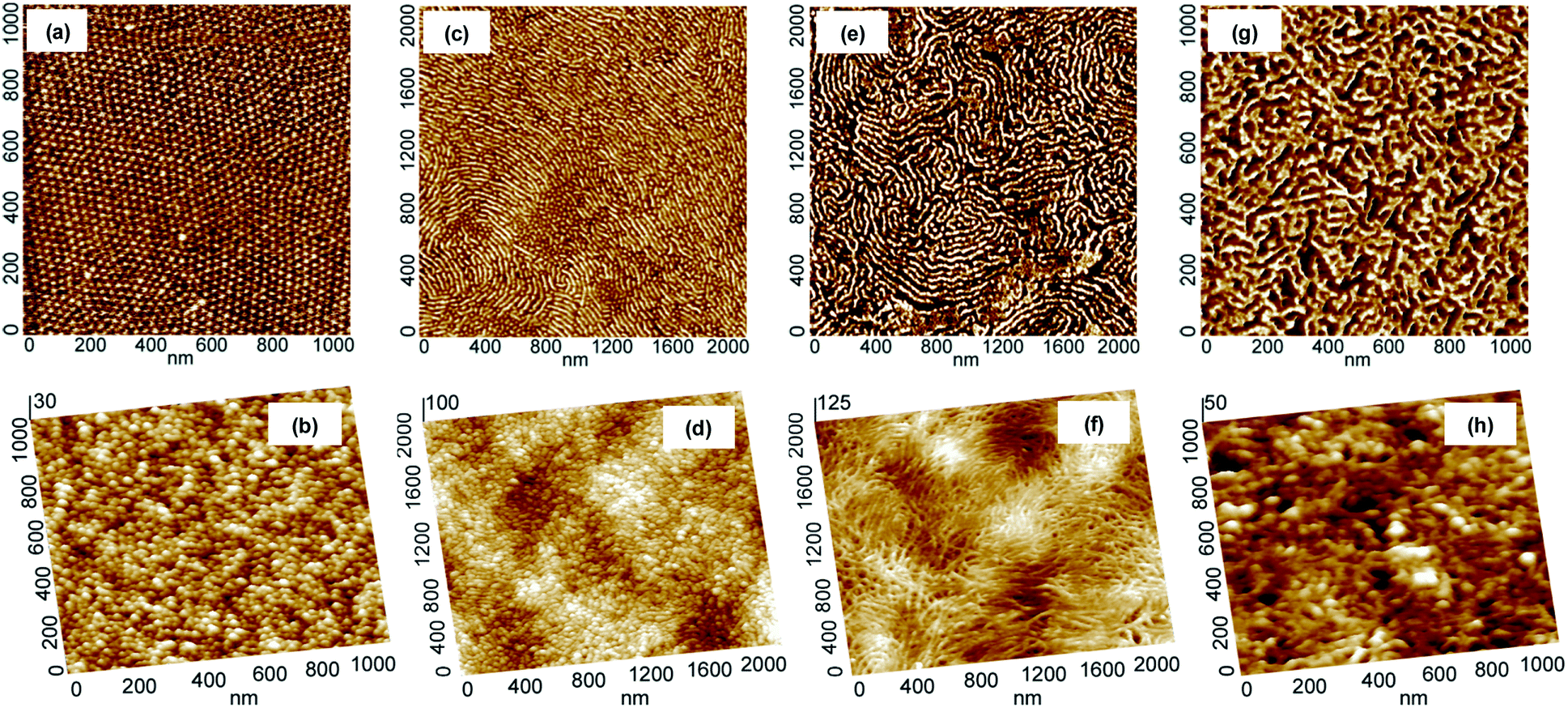 | ||
| Fig. 1 Tapping mode AFM phase images (top) of thin film of PAN-b-PBA block copolymers prepared by drop-casting and height images (bottom) of the resulting CTNCs after pyrolysis under N2 at 600 °C. (a) BA240AN36 (5.8 wt% PAN); (b) CTNC from BA240AN36; (c) BA240AN104 (15.2 wt% PAN); (d) CTNC from BA240AN104; (e) BA240AN124 (17.6 wt% PAN); (f) CTNC from BA240AN124; (g) BA78AN114 (37.7 wt% PAN); (h) CTNC from BA78AN114. | ||
In general, preservation of original morphology and formation of nanoporosity may be more readily achieved in thin films, owing to the ease of removal of the sacrificial block and to the support provided by the underlying substrate.8 However, in practical applications, bulk carbon materials are often more desirable than thin films. Therefore, preservation of the original nanostructure in the bulk without the external support presents an important challenge.
Herein, the porous structures of CTNCs from thin films and bulk precursors were compared by examination of nitrogen adsorption isotherms. In comparison with the bulk CTNC, thin-film CTNC from copolymer BA202AN106 exhibited higher Brunauer–Emmett–Teller (BET) specific surface area (SBET) (300 m2 g−1vs. 170 m2 g−1) and lower pore diameters (Fig. 2a). As mentioned above, higher surface area of thin-film samples can be explained by better pore accessibility and perhaps by the physical support of the substrate preventing partial collapse of the corresponding cylindrical morphology and pore closure upon pyrolysis. Lower pore diameter, on the other hand, can be explained by partial alignment of copolymer domains in the thin films prior to stabilization, facilitating higher packing density, and thus smaller pore size in the resulting CTNC. In contrast, pyrolysis of the copolymer with branched morphology resulted in negligible collapse of nanoporous structure. For instance, Fig. 2b shows similar mesopore size distribution in the thin-film (SBET = 490 m2 g−1) and bulk CTNC (SBET = 460 m2 g−1) from BA78AN114. The preservation of nanostructure in bulk pyrolysis was also evaluated by Porod analysis of small angle X-ray scattering patterns, as described in our earlier work.5aFig. 2c presents the dependence of SBET of bulk CTNCs from precursors with different PAN content, where all of the precursors contained the same length of PBA segment (DPPBA = 90). The sharp increase of SBET with PAN content indicates that the preservation of nanoporous structure in bulk pyrolysis is morphologically dependent. It turns out that the branched structure, with roughly ca. 40 wt% PAN, provides an ideal precursor for bulk pyrolysis, facilitating good retention of nanostructure after removal of the soft template. Transmission electron microscopy (TEM) further confirmed the nanoporous structure in the bulk CTNC at this copolymer composition (Fig. 2d).
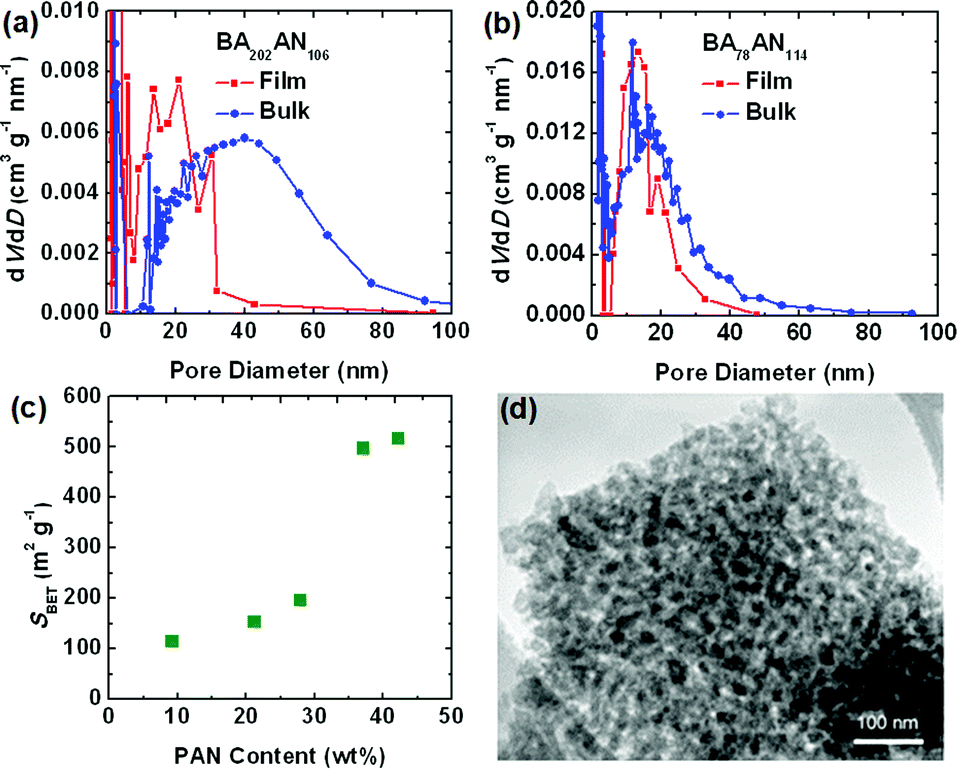 | ||
| Fig. 2 Pore size distributions of thin film and bulk CTNCs from PAN-b-PBA (a) BA202AN106; (b) BA78AN114; (c) plot of the BET specific surface area (SBET) vs. PAN content (DPPBA = 90); (d) TEM image of CTNC from BA78AN114. | ||
Since the mesopores in the CTNCs originate from the burn-off of the PBA domains, one can envision controlling the mesopore size by adjusting the DPPBA while retaining similar average compositions. Results presented in Fig. 3 show the dependence of mesopore size and SBET on DPPBA for copolymers containing ∼40 wt% of PAN, exhibiting branched phase-separated morphology and presumably yielding CTNCs with well-preserved nanoporous structure. Indeed, as shown by blue hollow bars, the size of mesopores consistently increased with the increase of DPPBA. Likewise, SBET (red solid bars) increased with the decrease of DPPBA down to DPPBA = 90. The subsequent drop of SBET with further decrease of DPPBA can be attributed to the loss of phase-separated structure with the decrease of DP of both blocks.
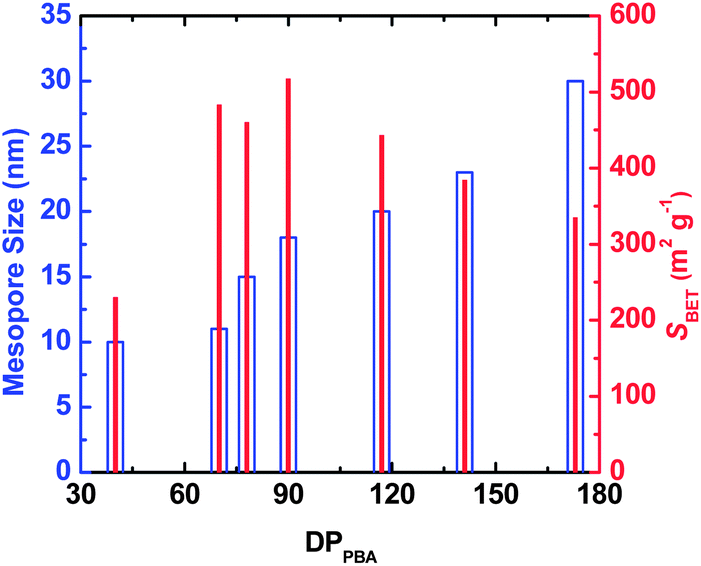 | ||
| Fig. 3 Effect of DPPBA on the mesopore size and the BET specific surface area (SBET) of bulk CTNCs from PAN-b-PBA with branched morphology. | ||
In conclusion, the study of porous CTNCs prepared by pyrolysis of thin films of PAN-b-PBA copolymers demonstrated the achievement of diverse nanostructures by tuning the ratio of the block lengths. Moreover, those nanostructures were retained upon pyrolysis, presumably at least partially owing to the physical support provided by the substrate of thin film. This demonstration of the ability to tailor the nanostructure of carbons by controlling the composition of the copolymer precursor opens new avenues for the controlled synthesis of this important class of materials. This procedure was then extended to bulk materials and it was discovered that a 3-dimensional branched morphology retains its shape during pyrolysis, yielding carbon nanostructures with mesopore size and surface controlled simply by tuning the length of the sacrificial block. One remaining challenge in this system would be to overcome the loss of the degree of preservation of nanoscale morphology upon thermal treatment caused by the decrease of order–disorder transition temperature with the decrease of the block lengths. Limits of the thermal stability of nanoscale morphology of BCP precursor establish the limits for maximizing the surface area and minimizing the mesopore size. It can be envisioned that this challenge could be addressed either by developing the low temperature PAN stabilization chemistry or by identifying the sacrificial block exhibiting a stronger immiscibility with PAN.
Financial support was provided by the National Science Foundation (DMR-0304508 and DMR-0969301), U.S. Department of Energy's National Energy Technology Laboratory (DE-FE0004000) and CRP Consortium at Carnegie Mellon University. M.Z. thanks for the financial support from Astrid and Bruce McWilliams Fellowship.
Notes and references
- (a) L. H. Peebles, Carbon fibers: formation, structure, and properties, CRC Press, Boca Raton, 1995 Search PubMed; (b) A. Krüger, Carbon Materials and Nanotechnology, Wiley-VCH, 2010 Search PubMed.
- (a) J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073 CrossRef CAS; (b) E. Frackowiak and F. Beguin, Carbon, 2001, 39, 937 CrossRef CAS; (c) F. Rodriguez-Reinoso, Carbon, 1998, 36, 159 CrossRef CAS; (d) R. L. McCreery, Chem. Rev., 2008, 108, 2646 CrossRef CAS PubMed; (e) Y. Liang, R. Fu and D. Wu, ACS Nano, 2013, 7, 1748 CrossRef CAS PubMed; (f) Y. Liang, D. Wu and R. Fu, Sci. Rep., 2013, 3, 1119 CAS; (g) Y. Ouyang, H. Shi, R. Fu and D. Wu, Sci. Rep., 2013, 3, 1430 CrossRef PubMed.
- (a) D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959 CrossRef CAS PubMed; (b) Nanoporous Materials: Science and Engineering, ed. G. Q. Lu and X. S. Zhao, Imperial College Press, London, 2004 Search PubMed.
- (a) M. Seo and M. A. Hillmyer, Science, 2012, 336, 1422 CrossRef CAS PubMed; (b) A. S. Zalusky, R. Olayo-Valles, J. H. Wolf and M. A. Hillmyer, J. Am. Chem. Soc., 2002, 124, 12761 CrossRef CAS PubMed; (c) E. A. Jackson, Y. Lee and M. A. Hillmyer, Macromolecules, 2013, 46, 1484 CrossRef CAS; (d) V. Z. H. Chan, J. Hoffman, V. Y. Lee, H. Latrou, A. Avgeropoulos, N. Hadjichristidis, R. D. Miller and E. L. Thomas, Science, 1999, 286, 1716 CrossRef CAS; (e) H. Sai, K. W. Tan, K. Hur, E. Asenath-Smith, R. Hovden, Y. Jiang, M. Riccio, D. A. Muller, V. Elser, L. A. Estroff, S. M. Gruner and U. Wiesner, Science, 2013, 341, 530 CrossRef CAS PubMed; (f) D. H. Lee, S. Park, W. Gu and T. P. Russell, ACS Nano, 2011, 5, 1207 CrossRef CAS PubMed.
- (a) M. Zhong, E. K. Kim, J. P. McGann, S.-E. Chun, J. F. Whitacre, M. Jaroniec, K. Matyjaszewski and T. Kowalewski, J. Am. Chem. Soc., 2012, 134, 14846 CrossRef CAS PubMed; (b) J. P. McGann, M. Zhong, E. K. Kim, S. Natesakhawat, M. Jaroniec, J. F. Whitacre, K. Matyjaszewski and T. Kowalewski, Macromol. Chem. Phys., 2012, 213, 1078 CrossRef CAS; (c) M. Zhong, S. Natesakhawat, J. P. Baltrus, D. Luebke, H. Nulwala, K. Matyjaszewski and T. Kowalewski, Chem. Commun., 2012, 48, 11516 RSC; (d) T. Kowalewski, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2002, 124, 10632 CrossRef CAS PubMed; (e) C. Tang, A. Tracz, M. Kruk, R. Zhang, D. M. Smilgies, K. Matyjaszewski and T. Kowalewski, J. Am. Chem. Soc., 2005, 127, 6918 CrossRef CAS PubMed.
- (a) C. Tang, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2003, 36, 1465 CrossRef CAS; (b) K. Matyjaszewski, Macromolecules, 2012, 45, 4015 CrossRef CAS; (c) K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed.
- E. Fitzer, Carbon, 1989, 27, 621 CrossRef.
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of Experimental details and relevant characterization. See DOI: 10.1039/c3mh00084b |
| ‡ Current address: Department of Chemistry, College of Staten Island and Graduate Center, City University of New York, 2800 Victory Boulevard, Building 6S-241, Staten Island, New York, 10314, USA. |
| This journal is © The Royal Society of Chemistry 2014 |