An organogelator design without solubilizing side chains by backbone contortion of a perylene bisimide pigment†
Zengqi
Xie‡
,
Vladimir
Stepanenko
,
Benjamin
Fimmel
and
Frank
Würthner
*
Universität Würzburg, Institut für Organische Chemie & Center for Nanosystems Chemistry, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@chemie.uni-wuerzburg.de
First published on 17th February 2014
Abstract
Here we report a perylene bisimide (PBI) based gelator molecule that contains neither a long alkyl chain nor any other solubilizing group which are considered to be essential moieties of organogelators. Instead, the solubility of the newly designed PBI gelator 4a is imparted by a contorted aromatic core. Its self-assembly into highly fluorescent one-dimensional nanostructures is directed by H-bonding, affording extremely low critical gelation concentrations (CGCs) below 0.1 wt%.
Whilst solution-processing is considered as a major advantage of organic materials for electronic, photovoltaic or photonic applications,1–3 the need for solubilizing side chains is often a severe penalty. Solubilizing groups are electronically inactive and often even isolating, prohibiting the desired percolation of exciton, electron or hole carriers within the material.4 Nevertheless, in the research field of conjugated polymers significant weight fractions of alkyl side chains are typically attached to the conjugated backbone to ensure sufficient solubility for the processing of the functional conjugated structures. For supramolecular polymers that are formed through non-covalent bonds, such as hydrogen bonding and π–π interactions between small molecules, the situation appears more promising for entropic reasons.1 Albeit, also in this research field alkyl chains are widely applied to increase the solubility of the molecular building blocks to reach the concentration required for the respective intermolecular interaction, e.g. hydrogen bonding, that directs a one-dimensional fiber growth instead of the undesired early precipitation of the material. Accordingly, many functional dyes bearing solubilizing long alkyl chains were employed as low molecular-mass organic gelators (LMOGs) in the past decades because of their strong tendency to self-assemble into organic networks, which may possess desirable charge transport, fluorescence, and sensing abilities for various (opto)electronic applications.5,6 Previously reported LMOGs such as oligophenylenevinylene derivatives,7–9 perylene bisimide (PBI) derivatives,10–13 and others14–17 all possess important features including, on the one hand, strong and directional intermolecular interactions (e.g. π–π interaction, hydrogen bonding) that promote self-assembly and, on the other hand, factors preventing neat crystallization (alkyl chains, steroidal unit, etc.) and thus to form intertwined one-dimensional nanostructures.
One intriguing gelator molecule, namely 1-cyano-trans-1,2-bis(3′,5′-bis-trifluoromethyl-biphenyl)ethylene,18 reported by Park and co-workers in 2004, as well as some further structurally related derivatives19 constitute an exception since they lack alkyl chains but, nevertheless, self-assemble into one-dimensional nanowires by CN⋯F and π–π interactions. Recently, Hisaki et al. reported another example, i.e. octadehydrodibenzo[12]annulene-based organogels using methyl ester groups to prevent crystallization and promote gelation.20 Such gelators may form highly condensed networks in the solid state and are particularly promising for electronic devices owing to the lack of inactive and isolating alkyl chains. These few examples raise now the questions whether an alternative design concept is conceivable for the formation of self-assembled functional materials that gets along without solubilizing alkyl chains and, more importantly, whether such a concept would be applicable even to large aromatic π-scaffolds like those present in perylene bisimides (PBIs) whose outstanding n-type semiconducting and fluorescence properties are of interest for many applications.21–23
Unfortunately, PBIs are among the most insoluble organic pigments24 and their processing into nanostructures by self-assembly protocols in solution has so far only been accomplished for derivatives bearing alkyl side chains.10–13,25–28 Herein, we report a new PBI derivative that lacks any flexible solubilizing groups in the molecular structure and demonstrate its self-assembly into three-dimensional networks composed of nanofibers in solution and even in polymer matrices. The concept for our molecular design is based on π-scaffold contortion,29–32 which has the potential to evolve as a key design element to accomplish similar organogelators and nanofibers for other functional dyes and semiconductor molecules. For PBIs we could demonstrate in our previous work that substitution of the perylene core at 1,6,7,12 positions (bay area) with phenoxy groups affords an about 25° rotational twist between the two naphthalene imide subunits, concomitant with strongly improved solubility and slipped stack J-aggregation properties.33 Thus, by core-contortion the archetype PBI pigments were shown to convert into soluble dyes or dye aggregates.34 Our newly designed target compound PBI 4a, which contains hydrogen atoms at the imide positions and ortho-methylphenoxy groups at bay areas, was synthesized according to the route outlined in Scheme 1. The starting compound tetrachloro-substituted PBI 1, bearing α-methylbenzyl groups at the imide positions, was synthesized according to our previously reported method.33 The nucleophilic substitution of all four chlorine atoms of PBI 1 by ortho-cresol afforded PBI 2a in 72% yield. The saponification of bisimide 2a with potassium hydroxide in t-BuOH at 80 °C provided perylene bisanhydride 3a. The latter was converted to the target ditopic PBI 4a by reaction with ammonium acetate in 86% yield. The reference compound 4b (ref. 34) was synthesized according to the same method as applied for PBI 4a. The only structural difference between 4a and 4b is the substituent at the ortho-position of the bay substituents (CH3 in 4a and H in 4b). Appreciably, the solubility of 4a is significantly higher than that of reference 4b in organic solvents, e.g. ∼5 mg mL−1 in chloroform for 4a but only around 1 μg mL−1 in chloroform for 4b at room temperature. For this reason no organogels were obtained for 4b in the absence of further additives such as melamines whose addition, however, modifies the electronic structure of the self-assembled material in an undesirable way (i.e. from J- to H-aggregation, leading to a quenching of the fluorescence).34 This difference in solubility and self-assembly capability of 4a and 4b underlines the important role of the methyl groups in the fixation of the contorted conformation of PBI 4a and the associated possibilities of intermolecular contacts to neighbour molecules in a three-dimensional solid state.
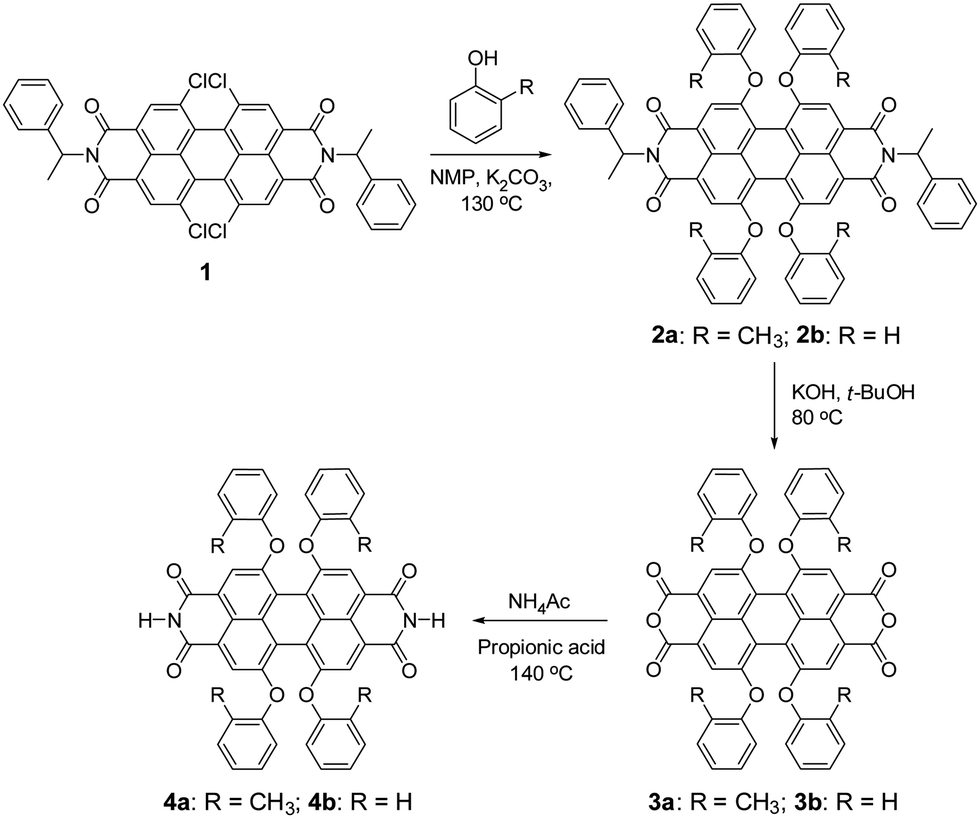 | ||
| Scheme 1 Synthesis of PBI dyes 4a,b. NMP = N-methyl-2-pyrrolidone. | ||
PBI 4a is well soluble in polar solvents like dichloromethane, but it is insoluble in nonpolar methylcyclohexane (MCH). Thus, aggregates of 4a were readily formed when a concentrated solution in dichloromethane was dropped to the “bad” solvent MCH. These aggregates can be easily dispersed in MCH by supersonic treatment and the mixture is stable enough for spectroscopic measurements. Fig. 1 displays the absorption and emission spectra of 4a in dichloromethane (molecularly dissolved solution) and in MCH (aggregate dispersed by supersonic treatment), which show large red-shifts for both absorption and emission of the aggregate compared to those of monomers as typically observed upon formation of hydrogen-bonded PBI J-aggregates.33 Indeed, the narrowed absorption and emission spectra in MCH with full-width-at-half-maximum (fwhm) values of 920 cm−1 (absorption) and 865 cm−1 (emission) compared with those of monomer solution in dichloromethane with fwhm values of 2190 cm−1 (absorption) and 1343 cm−1 (emission), respectively, clearly confirm the formation of J-aggregates with ordered molecular stacking in MCH. The fluorescence quantum yields of 4a in dichloromethane and in MCH were determined to be as high as 0.93 ± 0.01 and 0.77 ± 0.03, respectively, applying the conventional method for the determination of the relative fluorescence quantum yield.35
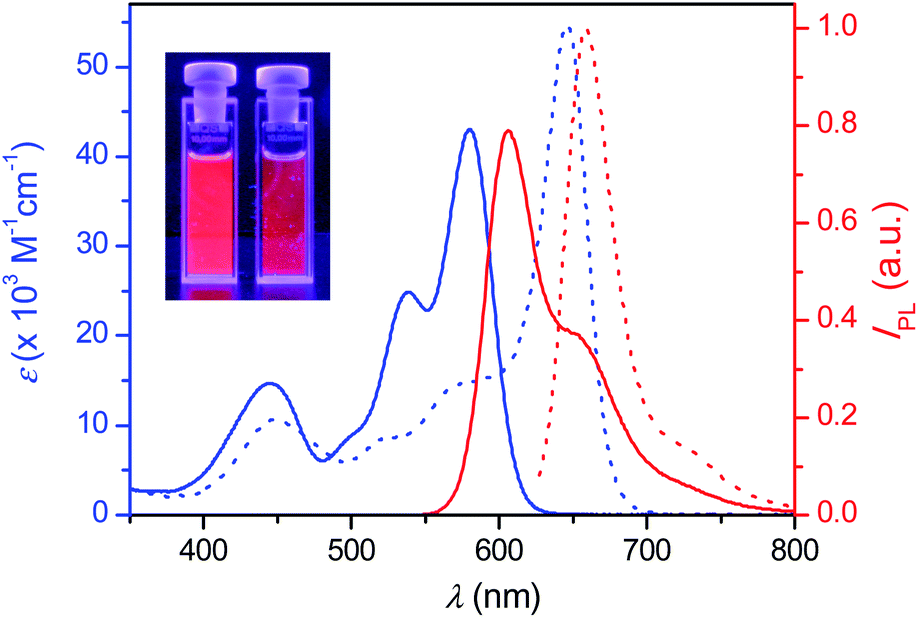 | ||
| Fig. 1 Absorption (blue) and emission (red) spectra of 4a in dichloromethane (solid lines) and MCH (dotted lines) at a concentration of 2.0 × 10−6 M at room temperature (λexc = 530 and 620 nm for monomer and aggregates, respectively). The inset shows a photograph of 4a solution in dichloromethane (left) and 4a aggregates dispersed in MCH by supersonic treatment (right) under a UV lamp. | ||
PBI 4a could be well dissolved in toluene by gentle heating. More interestingly, a highly fluorescent black gel was formed after the solution was cooled down to room temperature. The gelation of 4a in toluene occurs rapidly even at a very low concentration of 0.1 wt% (1.2 mM) and the “stable-to-inversion” test succeeded just after the gelation process as shown in Fig. 2. We found that the critical gelation concentration (CGC) of 4a in toluene is as low as 0.05 wt% and the gelation process is completely thermoreversible by heating and cooling the mixture. Temperature- and concentration-dependent studies corroborate the notion that the growth process of gel fibers follows a nucleation–elongation growth mechanism by cooperative hydrogen-bonding and π–π-interactions between PBI building blocks (see ESI, Fig. S1 and S2†).33 Furthermore, we have explored the gelation ability of 4a at a concentration of ∼0.1 wt% (1.0 mg mL−1) in various solvents. Our studies showed that gelation takes place in chlorobenzene, 1,2-dichloroethane, and ethanol as it is the case in toluene; however, no gelation was observed in chloroform, ethyl acetate, tetrahydrofuran, or acetone. As the CGC of 4a in different solvents is much lower than 1 wt%, this new PBI derivative can be classified as a super gelator.36
 | ||
| Fig. 2 Different states of the 4a gelation process in toluene at a very low concentration of 0.1 wt% (1.2 mM). (A) Homogeneous warm solution; (B and C) start and finish of the gelation process that takes ∼2 min at room temperature; (D) the “stable-to-inversion test” succeeds after gelation; and (E) the gel under a UV lamp. | ||
In order to get insight into the morphology of 4a aggregates in the gel, a suspension of a shake-destroyed gel in 1,2-dichloroethane was transferred onto a carbon film by drop-casting and subjected to transmission electron microscopy (TEM) observation. The TEM image shown in Fig. 3a suggests that gelator 4a creates an interlaced three-dimensional (3D) network consisting of bundles of nanofibrous aggregates with a width of 10–20 nm. It was evaluated that the length to width ratio of the nanofibres is up to 1000. The network of 4a nanofibres was also formed readily on drop-casting from a very dilute solution. Fig. 3b shows the scanning electron microscopy (SEM) image of a drop-cast film from a diluted solution of 1,2-dichloroethane (0.1 mM), where 3D networks composing of nanofibres with a width of ∼35 nm could be found. Here the width of the nanofibres is larger than that formed in the gel observed by TEM, which might be an outcome of a longer preparation time and co-aggregation of the nanofibres with each other during the drop-casting process. Atomic force microscopy (AFM) observations of gels of 4a in 1,2-dichloroethane (1.2 mM) on highly oriented pyrolytic graphite (HOPG) also confirmed the 3D network structure as shown in Fig. 3c. After tenfold dilution of the gel to 0.12 mM, the aggregated structure could still be observed as the image in Fig. 3d shows. Many rigid nanofibres with a uniform height of 1.35 ± 0.10 nm were found, which corresponds to the single PBI 4a fibre according to the molecular size. With regard to potential applications it is particularly noteworthy that these PBI 4a nanofibres can be easily embedded into polymeric matrices by addition of the respective polymer to the PBI 4a solution (ESI, Fig. S2 and S3†).
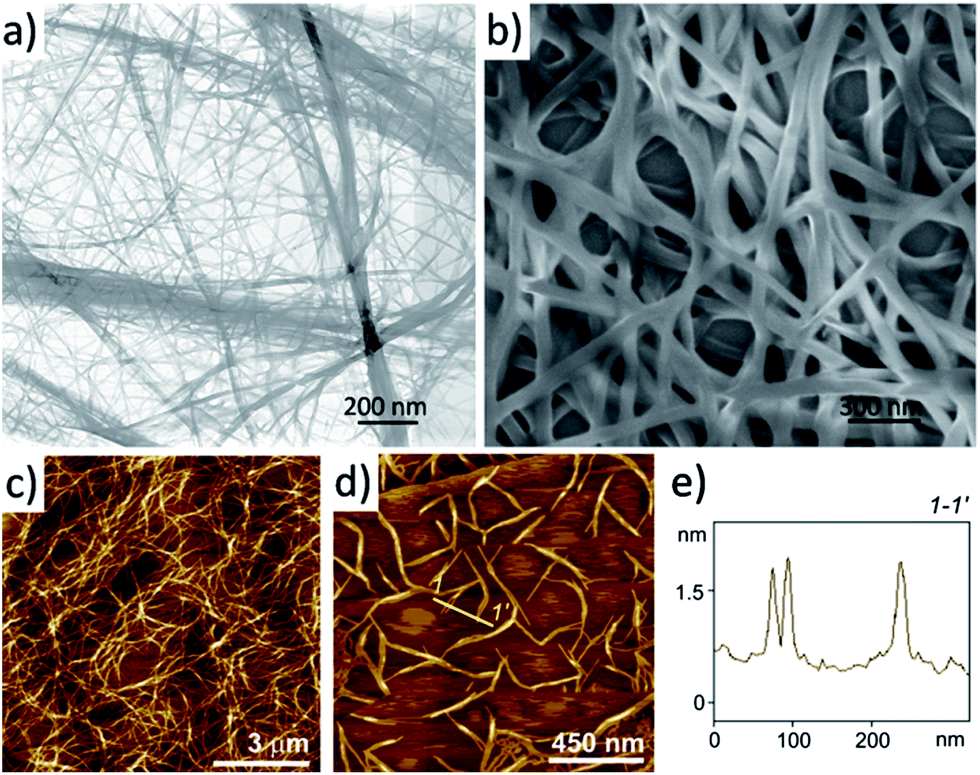 | ||
| Fig. 3 (a) TEM image of a suspension of 4a in 1,2-dichloroethane (1.2 mM) after drop-casting onto a carbon film. (b) SEM image of a film prepared by drop-casting a solution of 4a in 1,2-dichloroethane (0.1 mM) onto a silicon substrate. (c and d) Height AFM images of a thin film prepared by spin-coating the solution of 4a in 1,2-dichloroethane onto HOPG. The concentration for (c) is 1.2 mM and for (d) is 0.12 mM. The Z scale is 50 nm (c) and 5 nm (d). (e) Cross-section analysis along the yellow line is indicated as 1–1′ in (d). | ||
In contrast to the well soluble bisanhydride 3a, which does not contain any NH group for hydrogen bonding, PBI 4a possesses strong gelation ability. It indicates that the intermolecular hydrogen bonds play an important role in the formation of the nanofibres, which favour the gelation process. The FT-IR spectroscopy of solid 4a (Fig. S4†) shows different stretching frequencies of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups at 1682 and 1702 cm−1 that correspond to the hydrogen-bonded carbonyl groups and non-hydrogen-bonded carbonyl groups, respectively, as assigned before,33 confirming the existence of hydrogen bonds (CO⋯H–N). Considering the observed large bathochromic shift of the absorption band and the morphology of PBI 4a gels, a ‘brick-wall’ motif of the molecular stacking in nanofibres is proposed (Fig. 4). The molecules are aligned in a ‘head-to-tail’ fashion directed by hydrogen bonds and the adjacent molecular strings slip about a half molecular length distance along the molecule long axis. In this packing arrangement one molecule interacts with four neighboring molecules through π–π interactions, leading to strong J-type excitonic coupling (dipole transition is along the N–N axis).
O groups at 1682 and 1702 cm−1 that correspond to the hydrogen-bonded carbonyl groups and non-hydrogen-bonded carbonyl groups, respectively, as assigned before,33 confirming the existence of hydrogen bonds (CO⋯H–N). Considering the observed large bathochromic shift of the absorption band and the morphology of PBI 4a gels, a ‘brick-wall’ motif of the molecular stacking in nanofibres is proposed (Fig. 4). The molecules are aligned in a ‘head-to-tail’ fashion directed by hydrogen bonds and the adjacent molecular strings slip about a half molecular length distance along the molecule long axis. In this packing arrangement one molecule interacts with four neighboring molecules through π–π interactions, leading to strong J-type excitonic coupling (dipole transition is along the N–N axis).
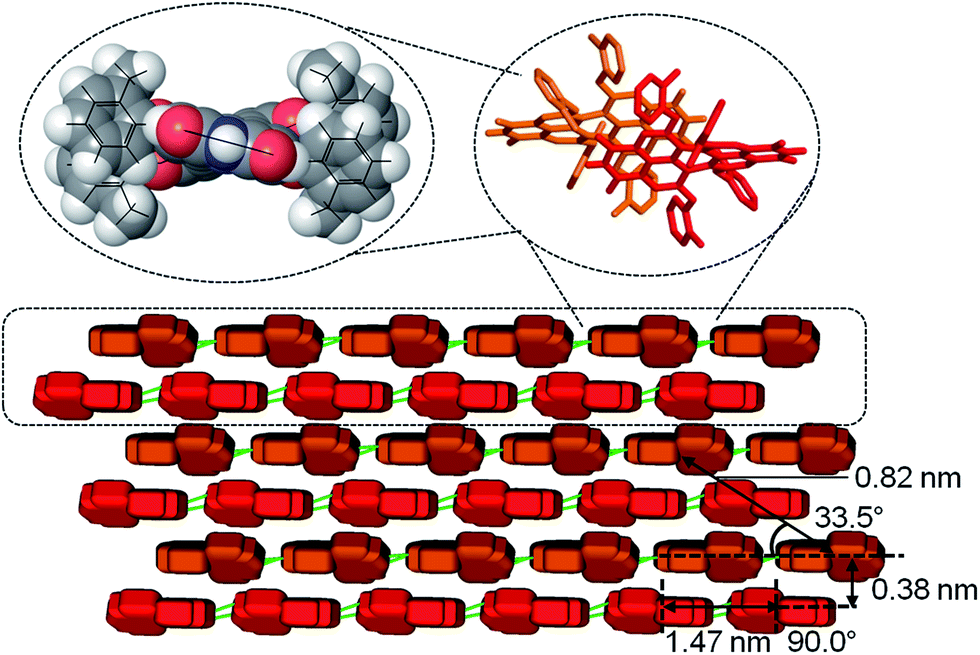 | ||
| Fig. 4 Schematic illustration of the molecular stacking in the nanofibres from monomer to dimer (top), extended M-/P-double string and brick-wall layers (bottom) including values of spacing determined by SAED. The orange and red ‘bricks’ represent the two enantiomers of 4a whose molecular structure is based on the lowest energy conformation according to B3LYP/STO-3G calculations (see ESI†). Intermolecular hydrogen bonds (depicted as green sticks) direct the elongation of the fibre along the PBI long molecular axis and π–π-stacking interactions the orthogonal growth of the brick wall. | ||
By using a solvent exchange method (see ESI†) we were also able to prepare needle-like single crystals of 4a with a width of about ten micrometres. This width is not sufficient for single crystal X-ray analysis but can be used for powder X-ray (see Fig. S7†) and selected area electron diffraction (SAED, see Fig. 5A and S5†). Both techniques support the layered arrangement of the dyes as suggested in Fig. 4. In particular the SAED pattern exhibits sharp diffraction spots that allow us to determine intermolecular distances in the single crystal. The triangle, circle, square and rectangle sets of spots with d-spacings of 0.46 nm, 0.38 nm, 0.82 nm and 1.47 nm, respectively, are indeed in excellent agreement with the molecular packing proposed in Fig. 4. Furthermore we could determine the optical axis by taking photographs of consecutive rotating single crystalline needles under a cross-polarized microscope taken in dark field (Fig. S5B–D and S6†). Fig. 5B–D show that the anisotropic birefringence was maximized only when the micro-needle was aligned at 45° to the direction of the polarizer, which supports our proposed uniaxial molecular stacking scheme as shown in Fig. 4.
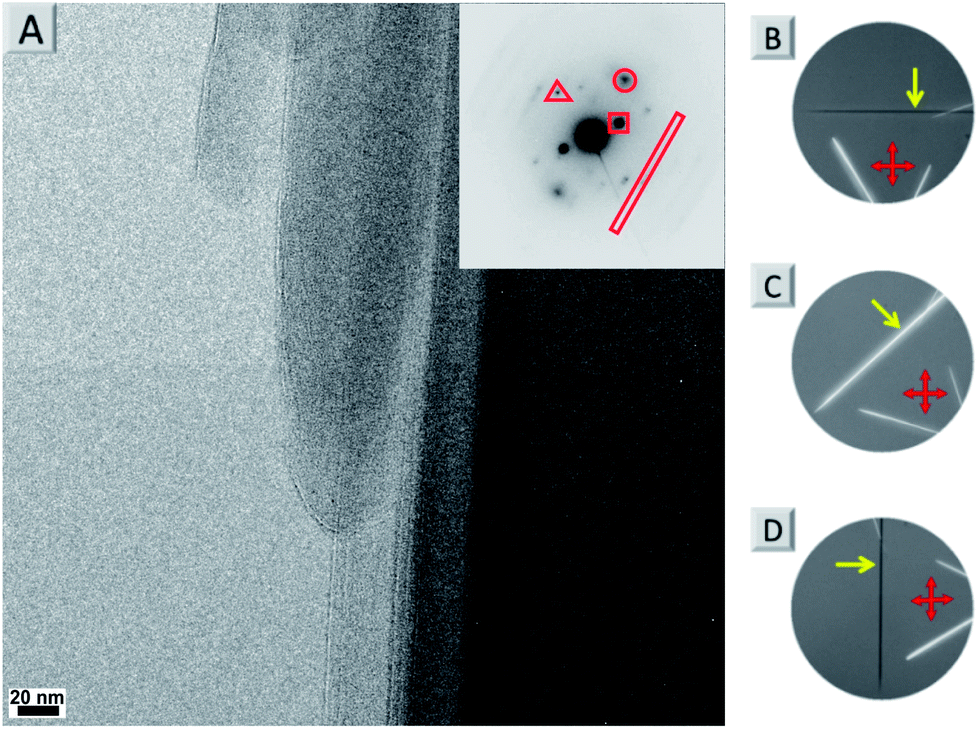 | ||
| Fig. 5 (A) TEM image of 4a nanoneedles with the SAED pattern (inset) showing distances of 0.46 nm (red triangle), 0.38 nm (red circle), 0.82 nm (red square) and 1.47 nm (red rectangle). (B–D) Photographs of consecutive rotating single crystalline needles under a cross-polarized microscope taken in dark field. The red double arrows indicate the directions of the polarizers and the yellow arrows indicate the same crystal in all figures. | ||
Conclusions
Our present study demonstrates that contorted aromatic cores may lead to organogelator molecules even in the absence of long alkyl chains or any other solubilizing substituents. Accordingly, for one of the most insoluble pigment colorants, i.e. PBIs bearing hydrogen-bonding functionalities, self-assembly by directional intermolecular H-bonding and π–π interaction could be applied to direct the formation of one-dimensional nanofibres, which cross-link to create three-dimensional networks that gelate organic solvents. The alkyl-chainless molecular structure of 4a, which is unique in comparison to all previously reported PBI-based organogelators, makes it prone to form nanofibres with desirable absorption and fluorescence features of J-aggregates. Such low dimensional fluorescent nanofibres might enable important applications, e.g. as nanocrystal transistors,37 laser materials,38 optical waveguides,39 or as non-fullerene exciton and n-channel transport components in heterojunction solar cells.40 In our most recent work we could already demonstrate the highly successful application of 4a nanofibres as cathode interlayer materials in polymer solar cells.41 The general concept of π-scaffold contortion might prove useful for the development of other alkyl-chainless organogelator molecules as well.Acknowledgements
This project was financially supported by the Alexander-von-Humboldt foundation (postdoctoral stipend for ZX). We thank Dr Nadezda V. Tarakina (Experimental Physics, Würzburg) for her help with the SAED measurement and data analysis.Notes and references
- D. Gonzólez-Rodríguez and A. P. H. J. Schenning, Chem. Mater., 2011, 23, 310–325 CrossRef.
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23, 397–415 CrossRef CAS.
- W. Pisula, X. L. Feng and K. Müllen, Chem. Mater., 2011, 23, 554–567 CrossRef CAS.
- F. C. Grozema and L. D. A. Siebbeles, Int. Rev. Phys. Chem., 2008, 27, 87–138 CrossRef CAS.
- S. S. Babu, S. Prasanthkumar and A. Ajayaghosh, Angew. Chem., Int. Ed., 2012, 51, 1766–1776 CrossRef CAS PubMed.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed.
- S. J. George and A. Ajayaghosh, Chem. – Eur. J., 2005, 11, 3217–3227 CrossRef CAS PubMed.
- S. Yagai, S. Kubota, T. Iwashima, K. Kishikawa, T. Nakanishi, T. Karatsu and A. Kitamura, Chem. – Eur. J., 2008, 14, 5246–5257 CrossRef CAS PubMed.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS PubMed.
- K. Sugiyasu, N. Fujita and S. Shinkai, Angew. Chem., Int. Ed., 2004, 43, 1229–1233 CrossRef CAS PubMed.
- S. Yagai, Y. Monma, N. Kawauchi and T. Karatsu, Org. Lett., 2007, 9, 1137–1140 CrossRef CAS PubMed.
- F. Würthner, C. Bauer, V. Stepanenko and S. Yagai, Adv. Mater., 2008, 20, 1695–1698 CrossRef.
- X.-Q. Li, X. Zhang, S. Ghosh and F. Würthner, Chem. – Eur. J., 2008, 14, 8074–8078 CrossRef CAS PubMed.
- A. Del Guerzo, A. G. L. Olive, J. Reichwagen and H. Hopf, J. Am. Chem. Soc., 2005, 127, 17984–17985 CrossRef CAS PubMed.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 1092–1097 CrossRef CAS PubMed.
- A. Das and S. Ghosh, Chem. Commun., 2011, 8922–8924 RSC.
- S. Prasanthkumar, A. Saeki, S. Seki and A. Ajayaghosh, J. Am. Chem. Soc., 2010, 132, 8866–8867 CrossRef CAS PubMed.
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS PubMed.
- B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS PubMed.
- I. Hisaki, H. Shigemitsu, Y. Sakamoto, Y. Hasegawa, Y. Okajima, K. Nakano, N. Tohnai and M. Miyata, Angew. Chem., Int. Ed., 2009, 48, 5465–5469 CrossRef CAS PubMed.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- Perylene and Perinone Pigments in Industrial Organic Pigments, ed. W. Herbst and K. Hunger, Wiley-VCH, 3rd edn, Weinheim, 2004, pp. 473–484 Search PubMed.
- K. Balakrishnan, A. Datar, T. Naddo, J. L. Huang, R. Oitker, M. Yen, J. C. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS PubMed.
- Y. K. Che, A. Datar, K. Balakrishnan and L. Zang, J. Am. Chem. Soc., 2007, 129, 7234–7235 CrossRef CAS PubMed.
- L. T. Kang, Z. C. Wang, Z. W. Cao, Y. Ma, H. B. Fu and J. N. Yao, J. Am. Chem. Soc., 2007, 129, 7305–7312 CrossRef CAS PubMed.
- Y. L. Lei, Q. Liao, H. B. Fu and J. N. Yao, J. Phys. Chem. C, 2009, 11, 10038–10043 Search PubMed.
- Z. Chen, M. G. Debije, T. Debaerdemaeker, P. Osswald and F. Würthner, ChemPhysChem, 2004, 5, 137–140 CrossRef CAS PubMed.
- S. X. Xiao, M. Myers, Q. Miao, S. Sanaur, K. L. Pang, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2005, 44, 7390–7394 CrossRef CAS PubMed.
- Y. Shi, H. Qian, Y. Li, W. Yue and Z. Wang, Org. Lett., 2008, 10, 2337–2340 CrossRef CAS PubMed.
- K. Mouri, S. Saito and S. Yamaguchi, Angew. Chem., Int. Ed., 2012, 51, 5971–5975 CrossRef CAS PubMed.
- T. E. Kaiser, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6719–6732 CrossRef CAS PubMed.
- F. Würthner, C. Thalacker, A. Sautter, W. Schärtl, W. Ibach and O. Hollricher, Chem. – Eur. J., 2000, 6, 3871–3886 CrossRef.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- H. Bouas-Laurent and J.-P. Desvergne, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, The Netherlands, 2006, pp. 363–430 Search PubMed.
- R. Li, W. P. Hu, Y. Q. Liu and D. B. Zhu, Acc. Chem. Res., 2010, 43, 529–540 CrossRef CAS PubMed.
- E. M. Calzado, J. M. Villalvilla, P. G. Boj, J. A. Quintana, R. Gomez, J. L. Segura and M. A. Diaz-Garcia, J. Phys. Chem. C, 2007, 111, 13595–13605 CAS.
- Y. S. Zhao, J. J. Xu, A. D. Peng, H. B. Fu, Y. Ma, L. Jiang and J. N. Yao, Angew. Chem., Int. Ed., 2008, 47, 7301–7305 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Mater., 2011, 23, 583–590 CrossRef CAS.
- Z. Xie, B. Xiao, Z. He, W. Zhang, Y. Zhang, C. Wang, H. Wu, F. Xie, L. Liu, Y. Ma, W.-Y. Wong, F. Würthner and Y. Cao, submitted.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of new PBI dyes, evaluation of temperature and concentration dependent self-assembly processes by a nucleation–elongation model, UV/vis, FT-IR, OPM, DSC, XRD, SAED studies, theoretical studies, and NMR and MS spectra. See DOI: 10.1039/c3mh00159h |
| ‡ Current address: Institute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, P. R. China. |
| This journal is © The Royal Society of Chemistry 2014 |