Assembling cellular networks of colloids via emulsions of partially miscible liquids: a compositional approach†
Niek
Hijnen
and
Paul S.
Clegg
*
School of Physics & Astronomy, The University of Edinburgh, Edinburgh, EH9 3JZ, UK. E-mail: n.hijnen@ed.ac.uk; paul.clegg@ed.ac.uk
First published on 3rd March 2014
Abstract
Colloidal particles are often regarded as building blocks for creating new materials, so guiding them to form specific structures is crucial. Here we present a simple method to assemble colloids into a cellular network. It relies on compositional changes in a colloid-stabilized emulsion of partially miscible liquids, and is induced by the disappearance of the continuous phase via evaporation. The particles are hereby forced into a cellular network as the droplets are squeezed together. Re-mixing of the liquid phases eventually occurs, leaving a stable structure surrounded by a single fluid phase. We demonstrate in detail the formation of the networks and discuss some general features of this approach. Finally, we show the macroscopic volume change and ultimate stability of the structure.
Porosity is essential for a great variety of materials applications1–3 although it can be demanding to incorporate. Hence, much effort is put into making and designing porous materials, while they are also common in nature. A class of macroporous materials (pores > 50 nm) are cellular solids which contain a space-filling packing of polyhedral voids, separated by solid cell walls; these therefore are a type of foam. The main characteristic of a cellular material is that its density, relative to that of the material from which the cell walls are made, is very low. Additionally, properties like the mechanical response, surface area, and thermal conductivity are very different compared to the non-porous counterpart.4 A cellular structure could also act as backbone, or underlying scaffold, in a composite material, where it might for instance offer mechanical strength or a conducting pathway.
It is clear that these structures have great potential for developing new materials, and one way to create them is through the directed assembly5 of colloidal particles. An example is a liquid crystal–colloid composite, where colloids are in a thermotropic nematic liquid crystal.6 The particles form a network when the suspending liquid is quenched from the isotropic phase into the nematic phase, due to the particles becoming trapped in between growing nematic regions. Freeze-drying an aqueous colloidal suspension is another example.7,8 Similar to the liquid crystal case this involves a transition of the dispersing liquid into a more ordered state, since here the particles become stuck in between growing ice crystals.
Colloid-stabilized emulsions (“Pickering” emulsions) of immiscible liquids can also act as a template for macroporous, foam-like solids. In this type of emulsion, coalescence of droplets is prevented by solid colloidal particles trapped at the liquid–liquid interfaces. In particular, high internal phase emulsions (HIPEs) serve this purpose well with droplets packed densely inside a minority fluid. Processing of these emulsions to obtain the final materials can involve polymerization of the continuous minority phase, or drying and sintering of the emulsion.9–11 Starting from colloid-stabilized emulsions with approximately equal volumes of the 2 liquids is also an option. Such an emulsion can lead to the formation of macroporous ceramics upon slow evaporation of both liquids at ambient temperature.12–14 Typically, the cells of material resulting from these emulsion-based methods lack the polyhedral geometry characteristic for a foam. Deformation of emulsion droplets to overcome this could potentially be achieved via centrifugation.15
A different route to assembling colloids into a more foam-like cellular network by using Pickering emulsions involves swapping immiscible liquids for partially miscible liquids, as was recently reported.17 It entails varying the temperature towards re-mixing of the liquid phases. As this happens, buoyancy, phase behaviour, and a decreasing interfacial tension all play roles in the formation of the network. Approaching the temperature where the liquids re-mix, droplets grow and coalesce more-easily, while they also deform as the volume fraction of droplets rises. In a narrow temperature interval, close to complete mixing of the liquid components, the particles end up trapped in a polyhedral cellular network. Held together by residual continuous phase, there is only temporary stabilization of the structure. Over time it coarsens as film thinning and rupture occur, similar to liquid foams in the absence of solid particles. The networks also fall apart when the liquid phases mix completely.
In this Communication, we present a method where the starting point for assembling colloids into cellular networks is also a colloid-stabilized emulsion. We have taken emulsions of the partially miscible liquids nitromethane and ethylene glycol (phase diagram in Fig. 1a, preparation details in ESI†) stabilized by silica particles (Fig. S1†), and use solvent evaporation to drive network formation. Here, nitromethane will be by far the fastest evaporating component, since its vapour pressure (4.79 kPa at 25 °C) is orders of magnitude higher than that of ethylene glycol (0.01 kPa at 25 °C).18 Samples were studied with confocal laser scanning microscopy (CLSM). Fluorescent labeling of both the particles (FITC, λex = 488 nm) and liquids (Nile Red, λex = 555 nm) conveniently allowed the simultaneous observation of structure formation and the underlying behaviour of the liquid phases.
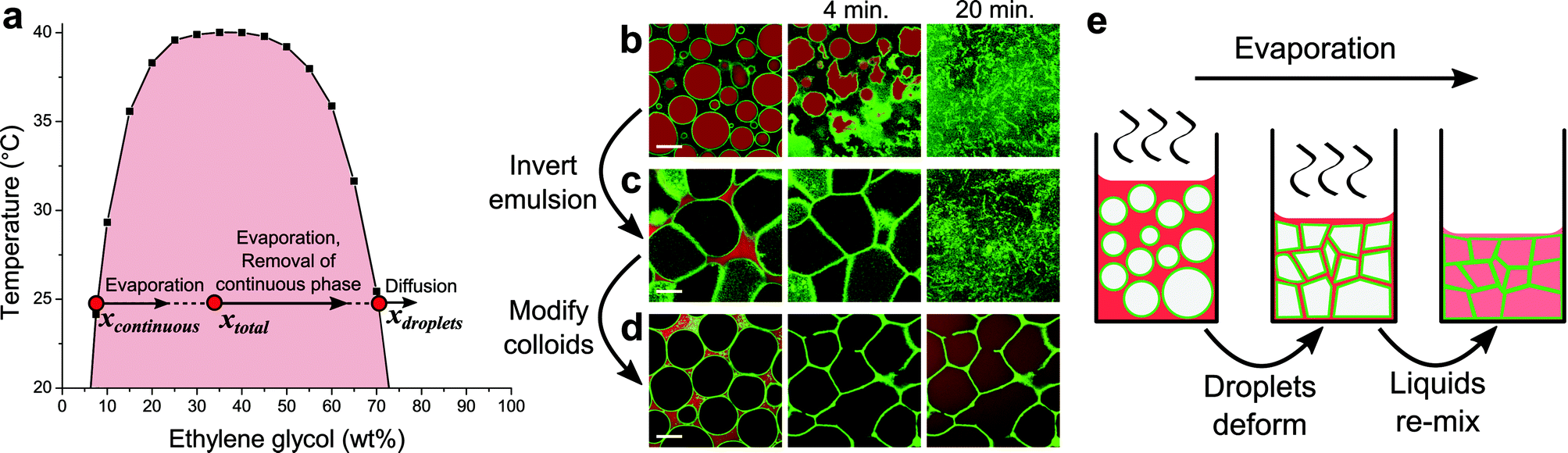 | ||
| Fig. 1 Evaporation in colloid-stabilized emulsions of nitromethane and ethylene glycol. (a) The phase diagram of the liquid pair, where black squares indicate data points found in literature.16 Initial compositions x for the emulsions used for images in (c) and (d), directly after preparation, are indicated by red dots. (b–d) CLSM snapshots demonstrate the structural evolution of emulsions (4 vol% silica particles) with varying initial compositions, as solvent evaporation proceeds from left to right: (b) hydrophilic particles and 45/55 (w/w) nitromethane/ethylene glycol, (c) hydrophilic particles and 65/35 (w/w) nitromethane/ethylene glycol, (d) hydrophobic particles and 65/35 (w/w) nitromethane/ethylene glycol. In these images, green is fluorescence from the particles and red is fluorescence from Nile Red initially located mainly in the nitromethane-rich phase. Scale bars are 20 μm. (e) Schematic sketch of the formation of a cellular network. | ||
Solvent evaporation has a dramatic effect on structuring in these emulsions. When composed of nitromethane-rich droplets in an ethylene glycol-rich continuous phase, as evaporation proceeds unhindered, nitromethane being removed from the continuous phase will be replenished from within the droplets. Consequently, droplets shrink, crumple and eventually disappear as the liquids re-mix, leaving the colloids to re-disperse in a single fluid phase (Fig. 1b). The particles used in this case are relatively hydrophilic (likely contact angle θ ≈ 120°, see ESI†), and changing the liquid composition inverts the emulsion, completely changing the structural evolution of the sample (Fig. 1c). Now the droplets deform as they become squeezed together due to the decreasing volume of the continuous phase. A cellular structure forms as a result, which falls apart upon re-mixing of the liquid phases (more detail in Fig. S2†). Thus, again the ultimate fate of the colloids is to be re-dispersed.
In contrast, changing the surface chemistry of the particles to make them more hydrophobic (likely contact angle θ ≈ 40°, see ESI†) gives structurally stable cellular networks (Fig. 1d). In this case, trimethylsilyl groups were covalently attached to silanol groups on the particles, reducing the amount of ionizable surface groups, and therefore surface charge. It is likely that the corresponding decrease in interparticle repulsions ultimately allows particles to be bound together more strongly through van der Waals interactions. This is similar to monogel formation from bijels, where particle structures remain intact after removal of the interfacial scaffold that has assembled them.19 To rule out the possibility that residual continuous phase is responsible for structural stability, first samples were heated to far above the upper critical solution temperature (60 °C) to no effect. Additionally, diffusion of ethanol into samples, which forces re-mixing of the liquid phases,20 left the networks intact (Fig. S3†).
During evaporation the liquid composition changes, while the particles remain trapped at the droplet interfaces. The overall liquid composition (xtotal) becomes more ethylene glycol-rich, as indicated by an arrow in Fig. 1a.21 This is mainly mediated by the composition change of the continuous phase associated with nitromethane evaporating more quickly than ethylene glycol (arrow in Fig. 1a). At the same time a little diffusion takes place between the 2 liquid phases, which would act towards again reaching the equilibrium compositions and volumes of the individual phases. However, the lack of a clear change in the size of droplets suggests that it is insignificant compared to the effect of evaporation. This is not surprising considering the time scale of structure formation and the relatively small area of exposed liquid–liquid interface. Ultimately, after xtotal crosses the binodal, the changing liquid composition results in re-mixing of the liquid phases, as evidenced from the diffusion of Nile Red throughout the sample (final 2 images in Fig. 1d). A cellular network of colloids within a single fluid phase is what remains hereafter. Overall structure formation is sketched schematically in Fig. 1e.
Images from a time series of CLSM snapshots recorded during solvent evaporation (movie in the ESI†) reveal several detailed aspects of the process. First of all, fluorescence from the particles demonstrates that despite the clear rigidity of the colloid-stabilized interfaces, droplets can coalesce when pushed into one another (Fig. 2a, first 2 images). However, the occurrence of coalescence is fairly limited, and therefore does not much influence the final cell size on a large scale (Fig. S4†). Later, as the continuous phase disappears, particles attached to the droplet interfaces are quite suddenly jammed into the final cellular network (Fig. 2a, last 2 images). At this point the walls and nodes of the particle network are formed, and fixed into place. Surplus particles in the continuous phase get pushed mainly into the gaps where 3 or more droplets meet, fortifying the nodes of the network.
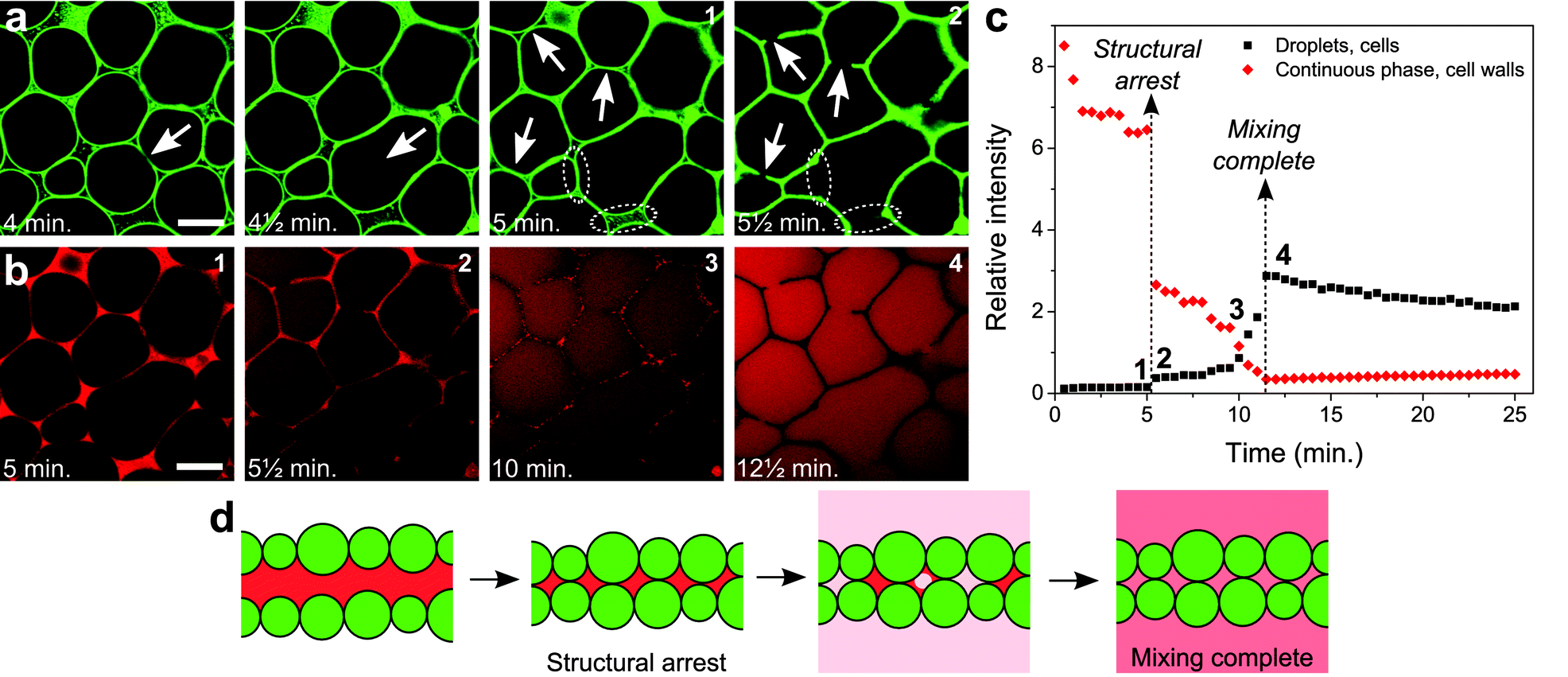 | ||
| Fig. 2 Detailed structure formation. (a) Structuring of the colloids at different times, during the process of structure formation. (b) Nile Red fluorescence at different times, initially locating the presence of the nitromethane-rich continuous phase, and at a later stage present equally throughout the entire sample as the liquid phases have re-mixed. Scale bars are 20 μm. A movie showing the full time series of CLSM images is provided as part of the ESI.† (c) The relative intensities of Nile Red fluorescence from inside the droplets/cells (black squares) and from inside the continuous phase/cell walls (red diamonds) plotted as a function of time (Fig. S5† and its caption explain how this graph was generated). (d) Schematic drawing indicating the formation of the cell walls and the re-mixing of the liquids. | ||
The current process appears to be only initially analogous to the coarsening of a liquid foam, in contrast to previous observations in a similar system.17 While film thinning and rupture are clearly involved as evidenced by coalescence events, these processes are halted, preventing a slow coarsening of the structure. When the volume of the continuous phase is reduced past a critical value, the particles are jammed into place as they are stuck at the interfaces. Film rupture occurring at the very last moments can therefore be arrested by the presence of the particles. In this case the film cannot pull back completely, resulting in holes in the cell walls (indicated by arrows in the last 2 images in Fig. 2a). Also, fortified sections of the wall can result from film rupture just before the structure locks in, when particles are dragged along by the interface of the film (dotted ellipses in the last 2 images in Fig. 2a). The involvement of liquid–liquid interfaces in the formation of the particle network appears from some walls separating a small and a large cell, where the curvature is inwards into the large cell as a result of the balance of the Laplace pressures prior to structural arrest.
The removal of the continuous phase, which drives structure formation, can be followed in detail through the fluorescence of Nile Red. As the volume of the continuous phase decreases, large thin sections of continuous phase result from the deformation of droplets when they are being pushed together (1st image in Fig. 2b). Droplet deformation is probably facilitated by a decreasing interfacial tension as in this non-equilibrium situation the compositions of the liquid phases become more alike. During this early stage, a decreasing relative intensity of Nile Red fluorescence is observed from the continuous phase (Fig. 2c), despite the fact that here the Nile Red concentration most likely increases. Since Nile Red fluorescence is highly solvent dependent, this could well be due to changes in the composition of the continuous phase.22 Bleaching and an increased concentration of free silica particles could contribute as well. Just after structural arrest, clearly Nile Red remains in the cell walls (2nd image in Fig. 2b). A sudden drop in the relative fluorescence intensity from the cell walls is observed here (Fig. 2c), however, it is not eliminated completely. Nitromethane-rich phase thus appears to be confined within the cell walls for some time. The fluorescence intensity in the droplets/cells makes a slight jump upon structural arrest, however this is merely caused by the decrease in fluorescence intensity from the cell walls.
After structural arrest, fluorescence from the cell walls is observed to slowly disappear in a heterogeneous fashion, and Nile Red clearly diffuses into the cells (3rd image in Fig. 2b). This is marked by the relative fluorescence intensity from the cell walls slowly falling off further, while there also is a corresponding increase of the relative fluorescence intensity from within the cells (Fig. 2c, in between the dashed lines). Here, mixing of the 2 phases goes towards completion. Judging from heterogeneities in fluorescence from the cell walls, this probably involves distinct pockets of residual nitromethane-rich phase trapped between the closely packed particles. When the liquids have mixed completely and the Nile Red has diffused throughout the cells, the cell walls appear dark in the images due to the presence of the particles (4th image in Fig. 2b). What happens to the liquid phases just before and after structure lock-in, can now be summarized by schematic drawings (Fig. 2d).
Macroscopically, evaporation and network formation are manifested by a volume decrease and gelation (Fig. 3a). While clearly exhibiting a yield stress, the networks are quite fragile and easily destroyed by shear. Slight deformations at the edges of the sample, induced by a bit of surplus liquid, indicate that the structure is quite soft. Holding such a sample upside down, the structure remains self-supporting, from which the order of magnitude of the yield stress is estimated to be ∼50 Pa (Fig. S6†). That these self-supporting structures, although soft and fragile, are able to form, most likely owes mainly to the partial miscibility of the liquid components. As a result of the low interfacial tension associated with this, and in combination with the eventual disappearance of the interfaces through re-mixing, film rupture can be prevented and halted by the jamming together of interfacial particles. This is illustrated nicely by the gaps in cell walls (Fig. 2a). In the case of immiscible liquids, where interfacial tension is high, the complete disappearance of the continuous phase would, most likely, result in film rupture. This conjecture is supported by studies of the complete drying of Pickering emulsions with a volatile continuous phase: a surplus of particles in the continuous phase is required in order to preserve the droplets.12
 | ||
| Fig. 3 Cellular networks on a larger scale. (a) An emulsion (4 vol% silica) left to evaporate overnight demonstrates both a decrease in sample volume and a yield stress after network formation. (b) Varying cell size after changing initial droplet size by using different initial colloid concentrations (vol%), where the inset shows a higher resolution image of the 4 vol% sample. The scale bars are 250 μm for the main images and 10 μm for the inset. | ||
To observe the large scale structure of networks, CLSM enabled imaging up to several layers of cells (Fig. S7†). Near the bottom glass surface slightly larger and more open cells are observed compared to deeper into the sample. Coalescence and film rupture might be more common here, while also more rapid sedimentation of larger droplets can be involved. By increasing the particle concentration, emulsions with smaller droplets could be created. This proved to be a straightforward way to adjust the final cell size (Fig. 3b).
In conclusion, a relatively straightforward approach to assembling colloids into cellular networks has been presented, using colloid-stabilized emulsions of partially miscible liquids. Besides a fast evaporating continuous phase, sufficient interparticle attractions appeared to be required to create a stable structure. The complete disappearance of the continuous phase through mixing of the initial liquid phases could be followed in detail and is a unique feature, giving a soft cellular network in a single fluid phase. Controlling the final cell size through the initial droplet size is easy, demonstrating some of the method's flexibility. Structure formation with another pair of liquids demonstrates the feasibility albeit for shorter times; evidently the key challenge lies in achieving particle bonding after mixing of the liquid phases (Fig. S8†). Additionally, a recent study based on this approach aims at battery applications by employing another different pair of partially miscible liquids, while also including polymer in the cell walls.23 Thus, changing starting materials and introducing additional components, for instance in the continuous phase, might offer straightforward ways to functionalize and tune the structures for applications.
Acknowledgements
We thank Dr Andy Schofield for providing the bare fluorescent silica particles that were used for further modification. This work was supported by the Marie Curie Initial Training Network COMPLOIDS no. 234810 and EPSRC grant EP/J007404/1.References
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- T. M. Freyman, I. V. Yannas and L. J. Gibson, Prog. Mater. Sci., 2001, 273–282 CrossRef CAS.
- L. Borchardt, M. Oschatza and S. Kaskel, Mater. Horiz., 2014, 1, 157–168 RSC.
- L. J. Gibson and M. F. Ashby, Cellular solids: Structure and properties, Cambridge University Press, 2nd edn, 1999 Search PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- S. P. Meeker, W. C. K. Poon, J. Crain and E. M. Terentjev, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 61, R6083–R6086 CrossRef CAS.
- C. A. L. Colard, R. A. Cave, N. Grossiord, J. A. Covington and S. A. F. Bon, Adv. Mater., 2009, 21, 2894–2898 CrossRef CAS.
- L. Qiu, J. Z. Liu, S. L. Y. Chang, Y. Wu and D. Li, Nat. Commun., 2012, 3, 1241 CrossRef PubMed.
- M. Destribats, B. Faure, M. Birot, B. Odile, V. Schmitt and R. Backov, Adv. Funct. Mater., 2012, 22, 2642–2654 CrossRef CAS.
- I. Akartuna, A. R. Studart, E. Tervoort and L. J. Gauckler, Adv. Mater., 2008, 20, 4714–4718 CrossRef CAS.
- V. O. Ikem, A. Menner, T. S. Horozov and A. Bismarck, Adv. Mater., 2010, 22, 3588–3592 CrossRef CAS PubMed.
- B. P. Binks, Adv. Mater., 2002, 14, 1824–1827 CrossRef CAS.
- I. Aranberri, B. P. Binks, J. H. Clint and P. D. I. Fletcher, J. Porous Mater., 2008, 16, 429–437 CrossRef.
- S. Barg, B. P. Binks, H. Wang, D. Koch and G. Grathwohl, J. Porous Mater., 2011, 19, 859–867 CrossRef.
- L. Maurice, R. A. Maguire, A. B. Schofield, M. E. Cates, P. S. Clegg and J. H. J. Thijssen, Soft Matter, 2013, 9, 7757–7765 RSC.
- J. Sørenson and W. Arlt, Liquid–liquid Equilibrium Data Collection, Dechema, 1979 Search PubMed.
- J. H. J. Thijssen and P. S. Clegg, Soft Matter, 2010, 6, 1182–1190 RSC.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, 82nd edn, 2001 Search PubMed.
- E. Sanz, K. A. White, P. S. Clegg and M. E. Cates, Phys. Rev. Lett., 2009, 103, 255502 CrossRef.
- J. W. Tavacoli, J. H. J. Thijssen, A. B. Schofield and P. S. Clegg, Adv. Funct. Mater., 2011, 21, 2020–2027 CrossRef CAS.
- In most of the experiments some of the continuous phase was also removed when transferring small portions of the sample onto a glass slide for solvent evaporation.
- A. Hawe, M. Sutter and W. Jiskoot, Pharm. Res., 2008, 25, 1487–1499 CrossRef CAS PubMed.
- D. Cai, J. H. J. Thijssen and P. S. Clegg, 2014, in preparation.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional experimental results. See DOI: 10.1039/c3mh00165b |
| This journal is © The Royal Society of Chemistry 2014 |