Co/CoO/CoFe2O4/G nanocomposites derived from layered double hydroxides towards mass production of efficient Pt-free electrocatalysts for oxygen reduction reaction†
Ruijie
Huo‡
a,
Wen-Jie
Jiang‡
b,
Sailong
Xu
*a,
Fazhi
Zhang
a and
Jin-Song
Hu
*b
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: xusl@mail.buct.edu.cn
bCAS Key Laboratory of Molecular Nanostructure and Nanotechnology, Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100090, China. E-mail: hujs@iccas.ac.cn
First published on 28th October 2013
Abstract
Development of a simple, reproducible and cost-effective protocol for mass production of non-precious-metal electrocatalysts for oxygen reduction reaction (ORR) is still challenging but highly desirable for their practical applications in industry. Herein, we developed a facile and scalable method to directly produce graphene (G) supported CoFe-LDHs and successfully used them as a precursor for mass production of Co/CoO/CoFe2O4/G as a low-cost and Pt-free efficient electrocatalyst, which exhibits comparable electrocatalytic activity and much better durability for ORR in comparison with commercial Pt/C catalysts. The result may provide a way for cost-effective production of ORR electrocatalysts on a large scale for practical applications.
Fuel cells have generated diverse scientific and technological interest as clean and high-efficiency devices although their performance is still limited by the sluggish oxygen reduction reaction on the cathode. Platinum-based materials are well-known to be one of the most active catalysts for ORR,1,2 but suffer from the issues of high cost, limited supply, and weak durability, which hinder them from practical applications in large amounts. Alternatively, some non-precious-metal catalysts have been demonstrated to be effective in either reducing the usage of Pt or replacing it.3–6 For example, transition-metal chalcogenides,7 oxides, carbides, and nitrides have shown reasonable ORR activity and practical durability.8–11 Among these catalysts, graphene supported transition metal oxides have been considered as one of the most promising non-precious-metal catalysts thanks to their large surface area, excellent conductivity and chemical stability, and potential synergetic effects of two-dimensional honeycomb-type graphene sheets.8,12,13 Several kinds of nanoparticles supported on graphene sheets, such as Co/CoO/G,8 Co3O4/G,11 Fe3O4/G,10 and MnCo2O4/G,9 are able to show enhanced performances for ORR compared with pristine nanoparticles. Given the complex procedures or necessary control of elaborate-designed nanostructures involved, however, it still remains a great challenge to develop a simple, reproducible and cost-effective protocol for mass production of electrocatalysts towards practical applications in industry.
Layered double hydroxides (LDHs), known as hydrotalcites and expressed by the general formula [M1−x2+Mx3+(OH)2]x+[An−]x/n·yH2O, where M2+ and M3+ are typically divalent and trivalent metal cations in a brucite (Mg(OH)2)-like layer and An− is the anion between the hydrated interlayer galleries, are widely used in industry due to their versatility, low-cost and ease of mass production.14–16 The diversity of LDHs in metal cations, the MII/MIII molar ratio, and the interlayer anion has endowed the LDH-based materials with extensive applications, including heterogeneous catalysts,17 superconductive and magnetic devices,18 photocatalysts,19,20 and antimicrobial support.21 Recently, we have developed a general and scalable synthesis method involving separate nucleation and aging steps (SNAS) to successfully mass produce pristine CaAl-LDH.22 In this communication, we extended this facile and scalable method to directly produce composite precursors of graphene supported CoFe-LDH for mass production of the Co/CoO/CoFe2O4/G nanocomposite, which has demonstrated the efficient electrocatalytic performance with superior durability for ORR. The result may provide a way for cost-effective production of electrocatalysts on a large scale for industrial applications.
The key features for producing graphene supported CoFe-LDH involve rapid mixing of an aqueous metal source solution and basic graphene oxide (GO) dispersion, a nucleation process in a microreactor for several minutes, and a subsequent aging process (see Experimental in the ESI†). This procedure is distinctly different from the traditional coprecipitation process for LDH preparation. The resulting CoFe-LDH/G was then calcinated at 650 °C for 2 h to obtain the final nanocomposite. The powder X-ray diffraction (XRD) pattern of the calcinated nanocomposite presents typical features of metallic cobalt, cubic CoO and spinel CoFe2O4, as shown in Fig. 1a. The peaks centered at 44.2° and 51.5° can be ascribed to the diffraction from (111) and (200) planes of metallic cobalt (JCPDS 15-0806), respectively. The peaks at 36.5°, 42.4° and 61.5° can be well-indexed to (111), (200) and (220) planes of cubic CoO (JCPDS 75-0533), respectively. All other peaks at 18.2°, 30.1°, 35.5°, 43.5°, 57.2° and 62.7° are the typical diffractions from spinel CoFe2O4 (JCPDS 22-1086). This result indicates that the product should be the composite of metallic cobalt, cubic CoO and spinel CoFe2O4 (designated as Co/CoO/CoFe2O4/G herein after). The co-existence of Co with CoO and CoFe2O4 could be attributed to the partial reduction of the precursor by graphene at a moderate temperature of 650 °C, i.e., the conversion from CoO to Co. This is consistent with the findings in a previous study concerning the conversion from the precursor of Co-containing compounds to the metallic nanoparticles at a high temperature of no less than 700 °C.23
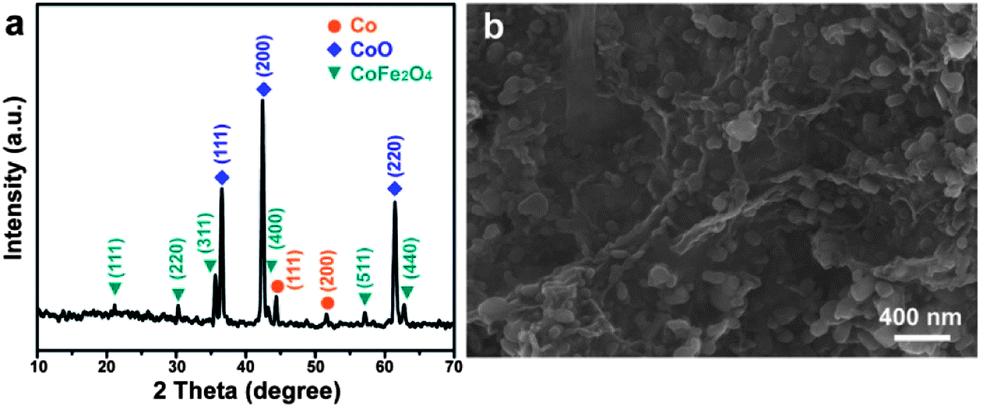 | ||
| Fig. 1 (a) XRD pattern and (b) SEM image of the Co/CoO/CoFe2O4/G nanocomposite. | ||
The morphology of the Co/CoO/CoFe2O4/G nanocomposite was investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM image clearly shows that various nanoplatelets are anchored to the underlying graphene substrate (Fig. 1b). TEM visualization confirms the co-existence of the nanoparticles and the G support (Fig. 2a). The wrinkled folds shown in Fig. 1b and dark lines in Fig. 2a are typical features of graphene layers. Elemental mapping results reveal that except for metallic Co domains (as marked by a yellow dotted line), CoO and CoFe2O4 are well dispersed in the graphene support (Fig. 2b–f). This uniform dispersion of CoO/CoFe2O4 could originate from the thermal decomposition of single-source CoFe-LDH precursors with a fully ordered arrangement of metallic cations.24 The XPS spectrum was further recorded to reveal the chemical state of Co species and the relative content of each component in the composite (Fig. S1†). The XPS signals in the Co 2p region exhibit complex shake-up satellite structures arising from multiple interactions between the core-hole generated on photoemission and the unpaired 3d valence electrons, which are characteristic of high spin Co2+ centers.25 The complex XPS signals can be well deconvoluted into several components. Two peaks at 779.8 and 781.3 eV can be identified to the characteristic peaks of Co2+ from both CoFe2O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and CoO,27 and the peak at 778.2 eV can be ascribed to Co0 species. Elemental analysis on the basis of the area ratio of the corresponding XPS peaks gives a Co/Fe molar ratio of 1.85, quantitatively in agreement with that of 2.0 in the starting materials. This yields that the mass ratio of each component in Co/CoO/CoFe2O4 is 10.8:32.8:53.7.
26 and CoO,27 and the peak at 778.2 eV can be ascribed to Co0 species. Elemental analysis on the basis of the area ratio of the corresponding XPS peaks gives a Co/Fe molar ratio of 1.85, quantitatively in agreement with that of 2.0 in the starting materials. This yields that the mass ratio of each component in Co/CoO/CoFe2O4 is 10.8:32.8:53.7.
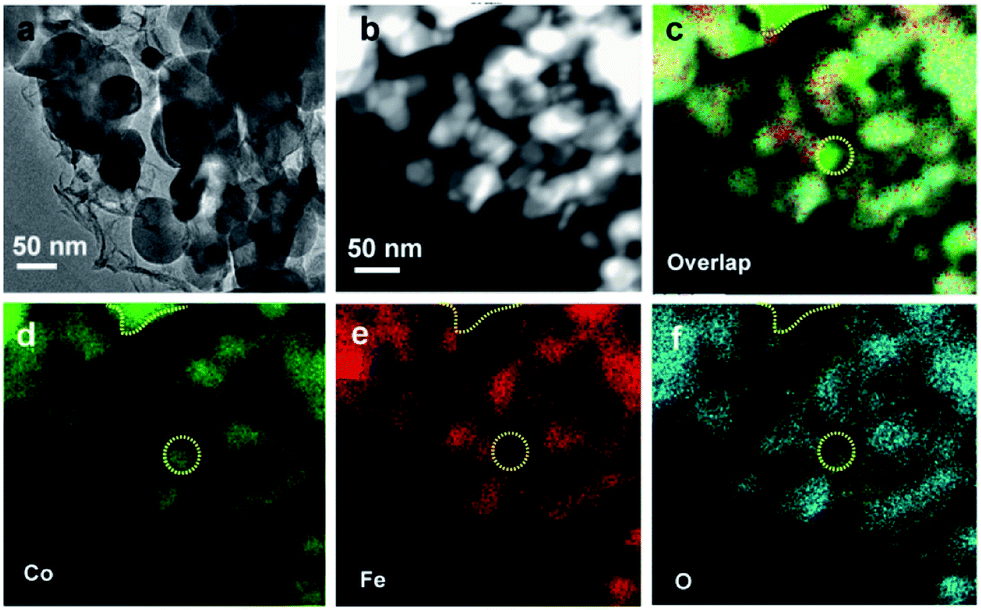 | ||
| Fig. 2 TEM and elemental mapping images of the Co/CoO/CoFe2O4/G nanocomposite. (a) TEM image, (b) STEM image, (c), (d), (e), and (f) overlap cobalt, iron, and oxygen elemental mapping images, respectively. | ||
To investigate the electrocatalytic activity of the Co/CoO/CoFe2O4/G nanocomposite for ORR, we first performed three-electrode cyclic voltammetry (CV) experiments in O2-free and O2-saturated solutions with 0.1 M KOH at a scan rate of 50 mV s−1. The corresponding voltammograms (Fig. 3a) show that a distinct ORR peak at −0.163 V (marked by a dotted ellipse) is observed in O2-saturated solution in contrast to the featureless CV in O2-free solution, indicating the electrocatalytic activity of Co/CoO/CoFe2O4/G for oxygen reduction. The electrocatalytic activity for ORR of Co/CoO/CoFe2O4/G was further investigated by linear sweep voltammetry (LSV) using a rotating disk electrode (RDE) and compared to commercial Johnson Matthey (JM) Pt/C catalysts (20 wt% Pt loading). As shown in Fig. 3b, Co/CoO/CoFe2O4/G exhibits a similar onset potential and a slightly lower limiting current density. The half-wave potential (E1/2), viz. a potential at which the current is half of the limiting current in the LSV curve, was also obtained to assess both catalysts.28 The E1/2 for Co/CoO/CoFe2O4/G is −0.25 V vs. Ag/AgCl, only 50 mV negative than that of JM Pt/C, indicating that the electrocatalytic activity comparable to the JM Pt/C catalyst can be achieved by the present Pt-free composite. The electrocatalytic activity of pure graphene was also measured under the same conditions for comparison. The result (Fig. S2†) shows that pure graphene has very poor ORR activity, which reveals that the electrocatalytic active sites for ORR of the nanocomposite should mainly come from cobalt-based particles. To get further insight into electron transfer parameters of the Co/CoO/CoFe2O4/G catalyst, a series of RDE measurements were carried out at varying rotating speeds (Fig. 3c). The corresponding Koutecky–Levich (K–L) plots (Fig. 3d) were then calculated based on the Koutecky–Levich equation (see Electrochemical test in the ESI†). For comparison, K–L plots obtained by theoretical calculations and representing the 2 or 4 electron transfer process are also plotted in Fig. 3d (orange dashed line). It can be seen that all K–L plots from Co/CoO/CoFe2O4/G at different potentials are perfectly parallel to the dashed line for the 4 electron transfer process, suggesting that ORR on Co/CoO/CoFe2O4/G was dominated by a more efficient and beneficial four-electron reaction pathway. The electron transfer numbers (n) at different potentials are listed in Fig. S3.†
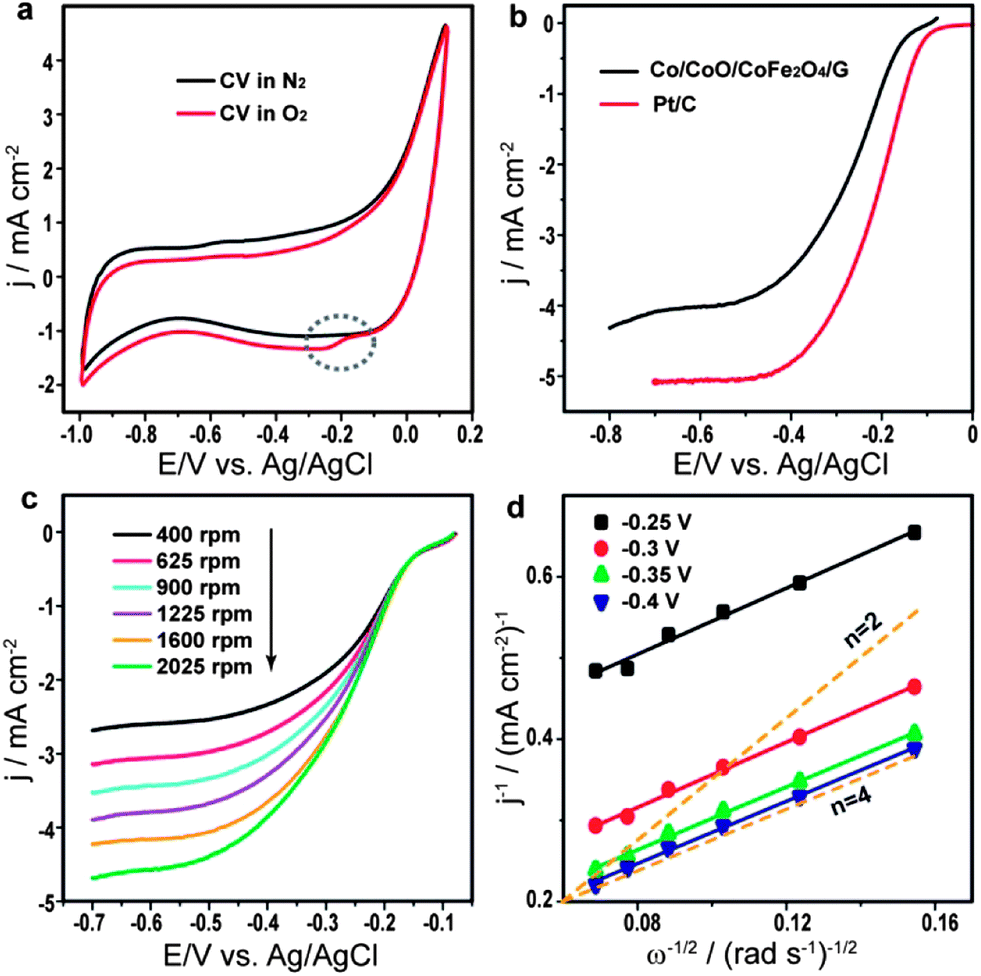 | ||
| Fig. 3 (a) CVs in O2-free or O2-saturated 0.1 M KOH at a scan rate of 50 mV s−1 for Co/CoO/CoFe2O4/G, (b) LSV curves for ORR recorded at a scan rate of 10 mV s−1 and a rotation speed of 1600 rpm, (c) LSV curves for Co/CoO/CoFe2O4/G at different rotation speeds, and (d) K–L plots of Co/CoO/CoFe2O4/G derived from data in (c) at different electrode potentials. | ||
Furthermore, it is generally known that methanol can penetrate through proton exchange membranes and deteriorate cathode catalysts, which is a major concern for the performance of direct methanol fuel cells.29 To investigate the capability of Co/CoO/CoFe2O4/G against methanol poisoning, LSVs in O2-saturated 0.1 M KOH in the presence of 0.5 M methanol were also measured for both Co/CoO/CoFe2O4/G and JM Pt/C. As shown in Fig. 4a, almost no loss in ORR activity in the presence of methanol was found for Co/CoO/CoFe2O4/G, but the ORR activity of JM Pt/C was observed to decrease drastically in terms of the 146 mV negative shift of E1/2 (Fig. 4b), indicative of its excellent tolerance to methanol poisoning effects much better than that of commercial JM Pt/C. In addition, the durability of the Co/CoO/CoFe2O4/G catalyst was examined by running current–time chronoamperometric experiments in O2-saturated solution at −0.2 V vs. Ag/AgCl and at a rotation speed of 1600 rpm. Fig. 4b shows that JM Pt/C suffered from a decrease of 39% in the current density after a 20000 s test, while only a 20% loss of current density was observed on the Co/CoO/CoFe2O4/G catalyst (Fig. 4c). The result clearly suggests that the Co/CoO/CoFe2O4/G catalyst exhibited much better durability in comparison with JM Pt/C.
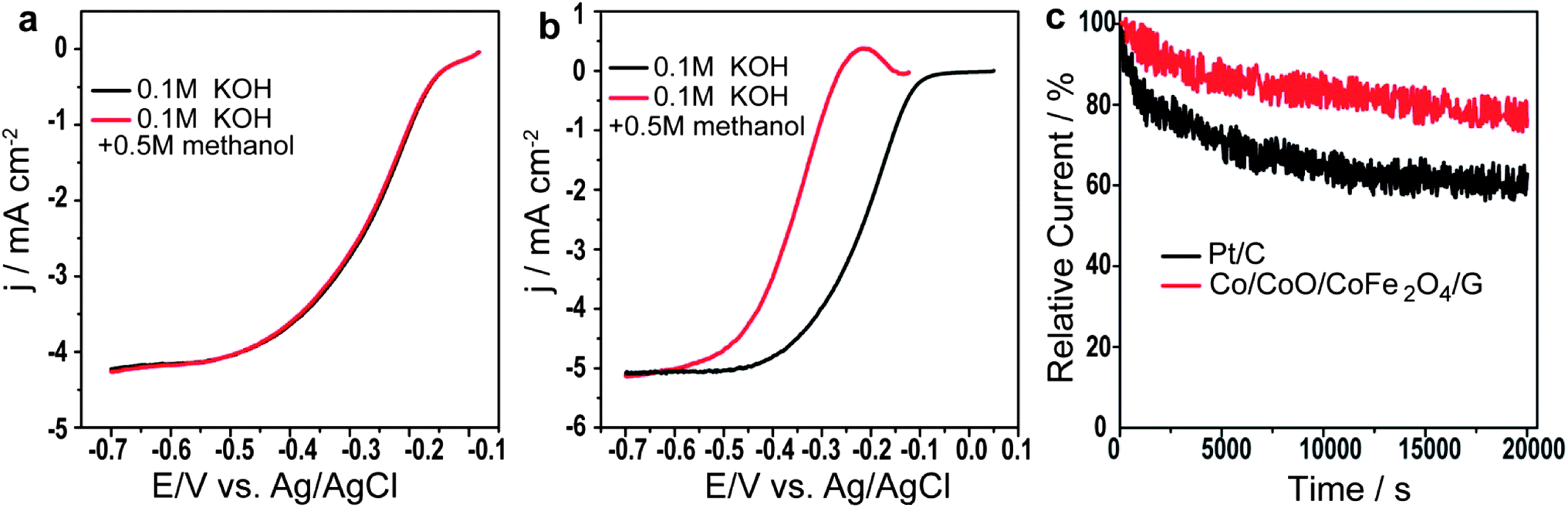 | ||
| Fig. 4 (a) RDE polarization curves of Co/CoO/CoFe2O4/G in O2-saturated 0.1 M KOH in the presence or absence of 0.5 M methanol. (b) RDE polarization curves of JM Pt/C in O2-saturated 0.1 M KOH in the presence or absence of 0.5 M methanol. (c) Current–time (i–t) chronoamperometric responses of Co/CoO/CoFe2O4/G and Pt/C at −0.2 V vs. Ag/AgCl in O2-saturated 0.1 M KOH at a rotation rate of 1600 rpm. | ||
In summary, we have explored a facile method for mass production of low-cost and Pt-free Co/CoO/CoFe2O4/G electrocatalysts for ORR by using graphene supported CoFe-LDH as a precursor. The catalyst exhibits comparable electrocatalytic activity and much better durability in comparison with commercial Pt/C catalysts. The reported route could be extended to design and produce various nanostructured catalysts in a cost-effective and scalable way in view of the versatility of LDH precursors.
Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program 2014CB932102 and 2011CB808700) and National Natural Science Foundation of China (no. 21071012, 21121063 and 91127044).Notes and references
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- Y.-P. Xiao, S. Wan, X. Zhang, J.-S. Hu, Z.-D. Wei and L.-J. Wan, Chem. Commun., 2012, 48, 10331–10333 RSC.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526–8544 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS.
- Y.-P. Xiao, W.-J. Jiang, S. Wan, X. Zhang, J.-S. Hu, Z.-D. Wei and L.-J. Wan, J. Mater. Chem. A, 2013, 1, 7463–7468 CAS.
- M.-R. Gao, J. Jiang and S.-H. Yu, Small, 2012, 8, 13–27 CrossRef CAS PubMed.
- S. Guo, S. Zhang, L. Wu and S.-H. Sun, Angew. Chem., Int. Ed., 2012, 51, 11770–11773 CrossRef CAS PubMed.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Z. Regier and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- C. Zhu and S. Dong, Nanoscale, 2013, 5, 1753–1767 RSC.
- J. Zhang, C. Guo, L. Zhang and C. M. Li, Chem. Commun., 2013, 49, 6334–6336 RSC.
- X. Guo, F. Zhang, D. G. Evans and X. Duan, Chem. Commun., 2010, 46, 5197–5210 RSC.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- M.-Q. Zhao, Q. Zhang, J.-Q. Huang and F. Wei, Adv. Funct. Mater., 2012, 22, 675–694 CrossRef CAS.
- M. B. J. Roeffaers, B. F. Sels, H. Uji-i, F. C. D. Schryver, P. A. Jacobs, D. E. D. Vos and J. Hofkens, Nature, 2006, 439, 572–575 CrossRef CAS PubMed.
- E. Coronado, C. Martí-Gastaldo, E. Navarro-Moratalla, A. Ribera, S. J. Blundell and P. J. Baker, Nat. Chem., 2010, 2, 1031–1036 CrossRef CAS PubMed.
- K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., Int. Ed., 2012, 51, 8008–8011 CrossRef CAS PubMed.
- J. L. Gunjakar, T. W. Kim, H. N. Kim, I. Y. Kim and S.-J. Hwang, J. Am. Chem. Soc., 2011, 133, 14998–15007 CrossRef CAS PubMed.
- C. Chen, P. Gunawan, X. W. Lou and R. Xu, Adv. Funct. Mater., 2012, 22, 780–787 CrossRef CAS.
- S. Xu, B. Zhang, Z. Chen, J. Yu, D. G. Evans and F. Zhang, Ind. Eng. Chem. Res., 2011, 50, 6567–6572 CrossRef CAS.
- S. Yang, G. Cui, S. Pang, Q. Cao, U. Kolb, X. Feng, J. Maier and K. Müllen, ChemSusChem, 2010, 3, 236–239 CrossRef CAS PubMed.
- M. Li, Y.-X. Yin, C. Li, F. Zhang, L.-J. Wan, S. Xu and D. G. Evans, Chem. Commun., 2012, 48, 410–412 RSC.
- D. Briggs and M. P. Seah, Practical Surface Analysis: Auger and X-Ray Photoelectron Spectroscopy, Wiley, 1996 Search PubMed.
- N. Ballarini, F. Cavani, S. Passeri, L. Pesaresi, A. F. Lee and K. Wilson, Appl. Catal., A, 2009, 366, 184–192 CrossRef CAS PubMed.
- S. S.-Y. Lin, D. H. Kim, M. H. Engelhard and S. Y. Ha, J. Catal., 2010, 273, 229–235 CrossRef CAS PubMed.
- S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J.-B. Baek and L. M. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209–4212 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. L. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nr05352k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |