Tailoring photocatalytic nanostructures for sustainable hydrogen production
Matteo
Cargnello
* and
Benjamin T.
Diroll
Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, PA 19104, USA. E-mail: mcargn@sas.upenn.edu
First published on 8th November 2013
Abstract
Photocatalysis is an important component for achieving sustainability in chemical transformations. It requires light absorption by a semiconductor and efficient extraction of the photogenerated electron–hole pairs to chemically active sites. One of the main problems in photocatalytic materials is to avoid electron–hole recombination following excitation. Tailored nanostructures offer a new way for achieving this goal by facilitating electron–hole separation. Nanoscaling of materials offers additional opportunities to generate unique photocatalysts that demonstrated novel light absorption, thermodynamic and kinetic properties. In this feature article we highlight some recent approaches towards the preparation of materials and nanostructures that showed improved activity for the sustainable production of hydrogen. This reaction has received much attention for the supply of future demand both for chemical industry and energy-related applications.
 Matteo Cargnello | Matteo Cargnello obtained his Ph.D. in Nanotechnology in 2012 at the University of Trieste (Italy). For his thesis he was awarded the Italian Chemical Society – Inorganic Chemistry Division Award in 2012, the ENI Award 2013 “Debut in Research” and the European Federation of Catalysis Societies Award in 2013. He is currently a post-doctoral scholar at the University of Pennsylvania (Philadelphia, USA) in the Department of Chemistry. His interests are in uniform and tailored materials and nanostructures for catalysis and photocatalysis and optical metamaterials. |
 Benjamin T. Diroll | Benjamin T. Diroll obtained his B.S. in Chemistry from the University of Chicago in 2009 and is currently a Ph.D. student in the Department of Chemistry at the University of Pennsylvania. His interests are in the optoelectronic properties of nanocrystals and their thin-films. |
1. Introduction
The Sun is the primary source of energy for the life on our planet. Solar energy is safe, abundant and carbon neutral. Alongside nuclear energy and a mix of other renewables, it is among the best options to replace fossil fuels, which have caused environmental problems due to their extensive use.1 However, solar energy is diffuse, intermittent and its harvesting, concentration and storage need to be improved for the full exploitation of its potential. Plants and organisms have learnt how to use it to convert abundant compounds such as water and CO2 into useful chemicals for their growth. One solution for storing solar energy is to use chemical bonds similarly to what nature has done with fossil fuels. “Solar-to-fuel” photocatalysis aims to transform solar energy into compounds that can be used as fuels. Hydrogen is one of the target molecules for this strategy as it would allow a carbon-free energy solution, but its handling and storage are not easy tasks to solve as hydrogen is a gas under standard temperature and pressure. Producing hydrogen from an abundant, non-toxic and easily transportable source such as water is an ideal sustainable solution.In general, a photocatalytic reaction is based on the formation of charges (electrons and holes) following absorption of light by a semiconductor.2 Many events can occur after light is absorbed by the semiconductor (Fig. 1): (a) electrons are excited from the valence band (VB) to the conduction band (CB) of the semiconductor, and a positive hole is left behind in the valence band (given that the light carries an energy equal or greater to the semiconductor band gap); (b) photoexcited holes that reached the surface of the semiconductor can oxidize adsorbed donor species Dads; (c) photoexcited electrons that reached the surface of the semiconductor can reduce adsorbed acceptor species Aads; (d) and (e) charges can recombine either on the surface or in the bulk.
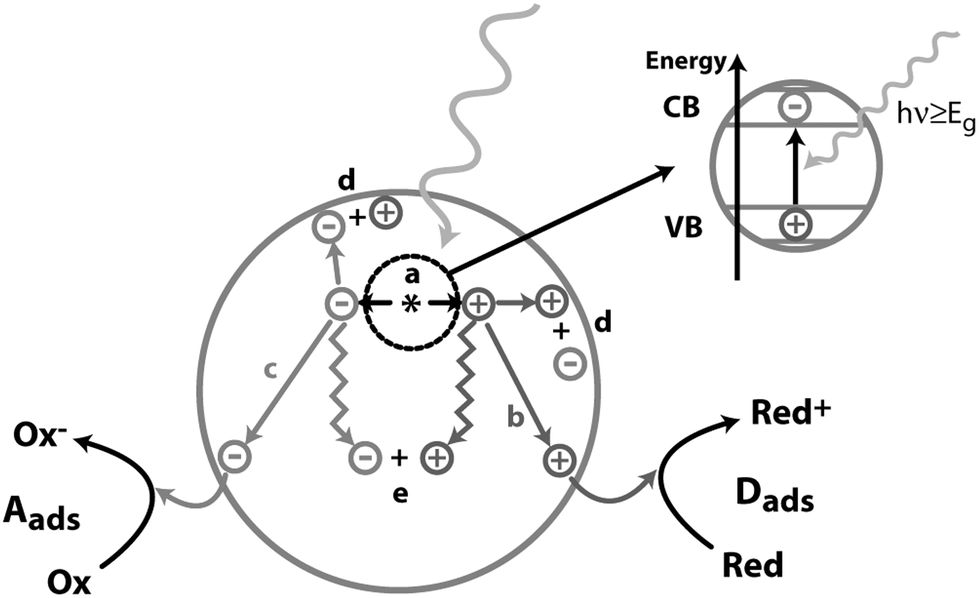 | ||
| Fig. 1 Schematic representation of the events following light absorption by a semiconductor. (a) Electron–hole pair formation; (b) donor oxidation by a photoexcited hole; (c) acceptor reduction by a photoexcited electron; (d and e) electron–hole recombination on the surface or in the bulk. Adapted with permission from ref. 2. Copyright 1995 American Chemical Society. | ||
Photoexcited species that reach the surface can be used to extract chemical work. Water splitting can be indeed realized in this way: water is the donor species that is oxidized to oxygen, while protons are the acceptor species that are reduced to hydrogen. However, water splitting is an energy-intensive reaction. This fact is evident not only by the 1.23 V potential requirement but also by the fact that it is a four electron process: four electrons must be removed from water to produce oxygen and protons, and protons must be reduced to hydrogen (eqn (1)).
| 2H2O + 4hν → 2H2 + O2 | (1) |
Fujishima and Honda first demonstrated the possibility to split water into H2 and O2 using titania and Pt electrodes under illumination of UV light.3 Their pioneering work sparked a huge interest in the field. One of the main issues is that UV light accounts for about 4–5% of the total incoming radiation and therefore there has been a tremendous effort to find better materials suitable for the process that could perform under visible light irradiation.
As an alternative to water splitting, compounds that are more easily oxidized than water can be used to promote the reaction by making the electron–hole recombination process less favorable. Biomass-derived compounds are the most attractive species in this class of sacrificial agents and the byproducts of their transformation can also have useful impact.4 In this way, organic species are oxidized to eventually CO2, and protons are still reduced to hydrogen in a process termed photoreforming.5
The control of the charges is pivotal for determining the efficiency of the final transformation. Careful design and preparation of structures where the charges are managed in such a way to maintain them separated and use them to carry out useful transformations are key elements for the development of photocatalytic materials with improved characteristics compared to the present ones. Nanotechnology can provide the tools needed to face this challenge. The fine manipulation of nanostructures is key for determining absorption, charge separation and reactivity. Also, nanostructuring a photocatalyst has many advantages. Active exposed surface area is increased by reducing the particle size. Charge recombination is also drastically reduced because charges have to travel less to reach the surface (and react) in small nanocrystals. Furthermore, quantum size effects can emerge and determine important changes in the electronic structure of the photocatalyst such that the nanostructure can show a different behaviour than the relative bulk counterpart.
This Feature Article highlights some of the recent works that focus on nanostructures as photocatalysts for sustainable hydrogen production. The goal is to show that manipulation of the structure has tremendous effect onto its final activity with the aim of contributing to the sustainability of hydrogen production from photocatalysis.
2. Oxides
Oxide semiconductors have several advantages over other photocatalytic materials in that they are stable under reaction conditions, non-toxic and abundant.Titania is one of the most studied materials in this class because of all the above cited elements. But as mentioned above, its wide band-gap results in little activity under solar illumination. However the UV contributes by 4–5% to the solar spectrum, so being able to maximize the quantum yield (ratio of H2 molecules produced per number of photons absorbed) in this part of the spectrum is also essential. In this sense a recent study by Chen et al. showed great potential for the improvement of solar absorption by titania.6 The hydrogenation of nanocrystalline TiO2 under 20 bar of H2 at 200 °C yielded a black titania sample that showed broad absorption in the visible range. The structure of the black titania contained crystalline cores with disordered shells and the authors calculated the presence of mid-gap states that contributed to the visible absorption. The lowered band gap allowed the black titania to be photocatalytically active under visible light for H2 production in the presence of Pt with high (10 mmol h−1 g−1 of photocatalysts) and sustained (22 days) activity under solar simulated light illumination. The engineering of defects in this case allowed the introduction of new electronic states which increased the activity of the system. In a previous publication, Zuo et al. similarly demonstrated that the presence of Ti(III) species self-doping the TiO2 host matrix beneficially influenced the rate of hydrogen production under visible light, thus highlighting the beneficial role of defect engineering onto the final activity.7
Specific facets can have a huge influence on photocatalytic reactions. Surface science techniques have been applied to understand the structure, reconstruction and activity of the most stable rutile and anatase facets8 or to engineer new phases with lowered bandgaps.9 However, knowledge gained from studies on single crystals can not always be straightforwardly applied to nanocrystalline samples exposing a variety of facets. Shape engineering in titania has seen a decisive contribution in the work by Yang et al. who demonstrated the preparation of anatase microcrystals exposing (001) facets.10 The work was inspired by theoretical calculations suggesting the higher reactivity of the (001) facet due to the presence of undercoordinated Ti centers.11 Unfortunately, the microcrystals prepared by Yang et al. had low surface area due to the relatively large size but the mechanism of shape control through the use of HF inspired a variety of approaches to prepare nanocrystalline samples more suited for catalysis.12,13
In a combined approach in which doping of titania and engineering of the exposed facets were combined, Liu et al. synthesized TiO2 sheets derived from TiN precursor.14 The synthesis used 1 M HF solution to increase the percentage of exposed (001) facets in a sheet-like morphology up to about 60% and the N-doping accounted for an extended absorption of the sample in the visible range, despite the band gap was not changed much (3.1 eV). The sample loaded with 1 wt% Pt showed H2 evolution from methanol–water under visible light. However, the activity of these high energy facets is still a debate and experimental comparisons on micron-size crystals15 might underestimate the effect of corners, edges or other sites which density is dramatically increased in the nanoscale regime.
An unprecedented control over the preparation of titania nanocrystals with defined size and shape was recently provided by Gordon et al.16 The use of TiF4 as precursor under non-hydrolytic conditions conveniently evolved limited but decisive amounts of HF in situ during the reaction thus avoiding the need to handle this very dangerous chemical. Additionally, by changing the ratio of TiF4 precursor in combination with TiCl4 and the type of surfactant (oleylamine or octadecanol), a full range of tunability in the shape of the titania nanocrystals based on a bipyramidal geometry was demonstrated (Fig. 2). The percentage of (101) and (001) facets was therefore well controlled and depended on the amount of HF released during the reaction, resulting in samples where F was still present on the surface as revealed by XPS. Furthermore, a ligand exchange procedure17 removed the shape-directing surfactants and allowed dispersibility in water. Samples were tested for H2 production in the presence of methanol as sacrificial agent and photodeposited Pt under simulated solar light. Samples with higher content of (101) facets were better than (001) for this particular reaction both with and without fluorine on the surface, clearly demonstrating that the higher energy (001) facets did not show improved activity for methanol photoreforming.
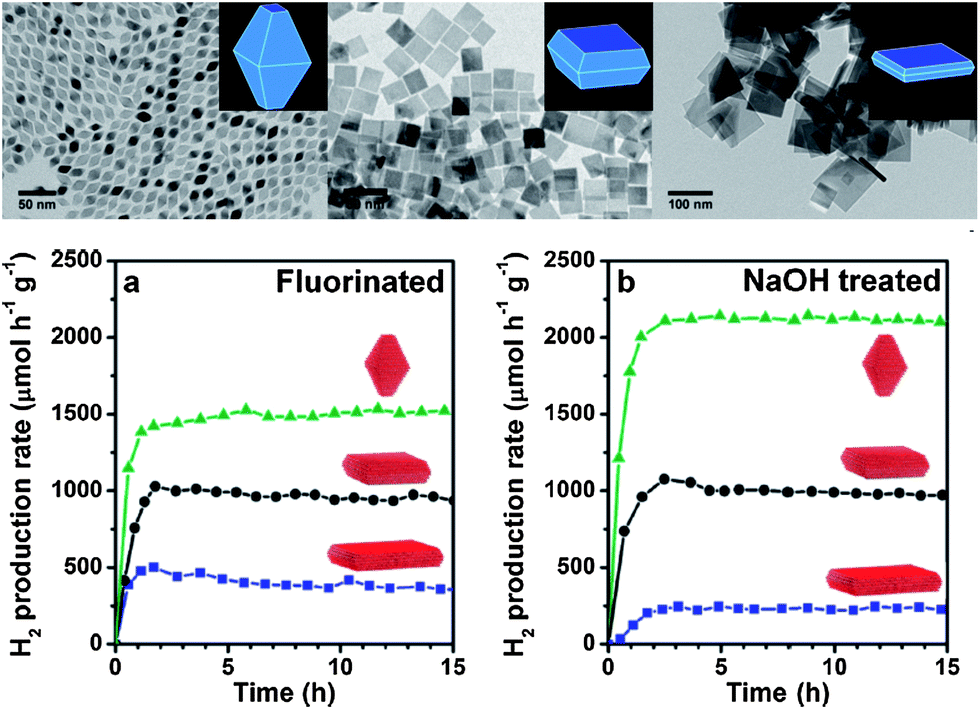 | ||
| Fig. 2 Top: TiO2 nanocrystals prepared from a TiF4 precursor with a tuneable increase in the concentration of specific facets, going from bipyramids (left) to truncated bipyramids (center) to nanoplates (right). Bottom: photoreforming of MeOH on Pt/TiO2 nanocrystals showed that {101} facets have higher H2 production rates than {001} facets both before (left) and after (right) removal of surface fluorine species. Adapted with permission from ref. 16. Copyright 2012 American Chemical Society. | ||
Core–shell structures are also attractive candidates as photocatalytic architectures. They allow manipulation of the exciton by tailoring the valence and conduction band of the core and shell materials to obtain type I or type II junctions. In this way the electron and holes can be more efficiently separated and used for chemical reactions at the photocatalyst surface. However, one or both charges may be isolated in the core material. This impedes turnover if reactants in solution are unable to access the surface of the core. However, if pores are introduced in the shell material, this issue can be avoided. An example of building such type of architecture was reported by Gombac et al. on CuOx/TiO2 core–shell type photocatalysts prepared by microemulsion.18 Cu particles were synthesized in the water droplets of the water-in-oil microemulsion and then embedded inside titania layers by hydrolysis of titanium butoxide followed by calcination of the resulting material. The core–shell sample showed higher photocatalytic H2 evolution rates from ethanol and glycerol aqueous solutions compared to conventional Cu/TiO2 samples prepared by deposition. HRTEM and EXAFS characterization suggested the presence of thin titania layers around the oxidized Cu particles (Fig. 3) and the possible dissolution of Cu ions inside the titania lattice, which might account for the improved activity. In this specific case, the core–shell structure increased the contact area between the metal particles and titania thus increasing the number of sites where electron–hole transfer can occur resulting in higher H2 production rates.
 | ||
| Fig. 3 (Top) HRTEM images showing the morphology of a conventional Cu/TiO2 photocatalysts prepared by sol–gel method (A) and a Cu@TiO2 photocatalyst prepared by microemulsion (B). EDS analysis revealed the presence of titania layers around the Cu particles in the embedded system (bottom). Adapted with permission from ref. 18. Copyright 2010 American Chemical Society. | ||
Exposed metal surfaces tend to favour hydrogen/oxygen recombination but coating Rh, Pt or Ir particles with Cr2O3 in a core–shell manner by photodeposition onto GaN–ZnO solid solution support allowed to reduce the back-reaction and to produce hydrogen under visible light illumination.19 The authors claim that the Cr2O3 shell is permeable to protons but not to oxygen, resulting in the high activity of the system. Improvement in the design consists in the deposition of another oxide (MnO) as oxygen evolving centre for complete water splitting.20
3. Quantum-dot based photocatalysts
Quantum dots (QDs), which demonstrate relatively narrow band gaps, are attractive class of materials for visible light photocatalysis. They have been employed both as sensitizing chromophores that transfer electrons to an optically-passive catalyst and as active hydrogen reduction catalyst. QDs offer distinctive advantages compared to other classes of materials and bulk phases. QD systems have a size-tunable band gap throughout the visible that allows size-dependent valence and conduction band energies.21 This makes QDs distinctive relative to the same bulk materials. The main drawback of QDs is their photo-oxidative instability. Tailoring the architecture and band-structure of QD-based materials offers an opportunity to overcome this issue.Although CdS is well-known to have an appropriate band-alignment for water splitting, it is unstable under photocatalytic conditions. Nanoscale cadmium chalcogenides are similar. A technical leap in the stability of hydrogen-producing chalcogenide photocatalysis was recently reported for a homogeneous QD/Ni photocatalyst.22 In this system of water-soluble CdSe QDs acting as chromophores and nickel salts, H2 production remained stable for 360 hours with turn-over numbers in excess of 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in the presence of ascorbic acid as electron donor, indicating that QDs have potential as much more stable chromophores than more commonly used organic dyes. This drastically-enhanced stability is specifically obtained without photodeposition of metal on to the QDs, as the authors claim that the active species is a soluble thiolate-Ni(II) complex formed in situ during the reaction. Although the precise reason for the increased stability is not clear, this study demonstrates that chalcogenide QDs can remain stable for prolonged periods under photocatalytic conditions. The long-term stability even without an electrical junction between the active catalyst and the QD sensitizer suggests a large space for optimization, potentially through formation of more sophisticated nanostructures that efficiently separate charges.
000 in the presence of ascorbic acid as electron donor, indicating that QDs have potential as much more stable chromophores than more commonly used organic dyes. This drastically-enhanced stability is specifically obtained without photodeposition of metal on to the QDs, as the authors claim that the active species is a soluble thiolate-Ni(II) complex formed in situ during the reaction. Although the precise reason for the increased stability is not clear, this study demonstrates that chalcogenide QDs can remain stable for prolonged periods under photocatalytic conditions. The long-term stability even without an electrical junction between the active catalyst and the QD sensitizer suggests a large space for optimization, potentially through formation of more sophisticated nanostructures that efficiently separate charges.
Careful tuning of the band-structure of quantum-confined nanostructures allows enhanced quantum efficiency and stability. Banin and co-workers have demonstrated that metal or metal oxide particles can be selectively grown on the tips or edges of anisotropic cadmium chalcogenide nanocrystals and that these structures demonstrate photocatalytic production of hydrogen.23–25 Building off of this work, Amirav and Alivisatos have studied in more detail the photocatalytic behaviour of Pt-tipped CdS nanorods.26,27 By comparing CdS nanorods with Pt-tipped CdSe/CdS “dot-in-rods”, the authors demonstrate that hole-localization to the CdSe dot core reduces recombination and slows photoxidation.27 By tuning the distance of the CdSe core from the Pt-tipped end of the rod, the authors tailor the distance between the electron and hole to prevent undesired recombination and separate the hole from the Pt/CdS interface. The result is that higher quantum efficiencies can be obtained by tailoring the length of the rods and the size of the CdSe core, which serves as the site of hole localization. (Fig. 4).
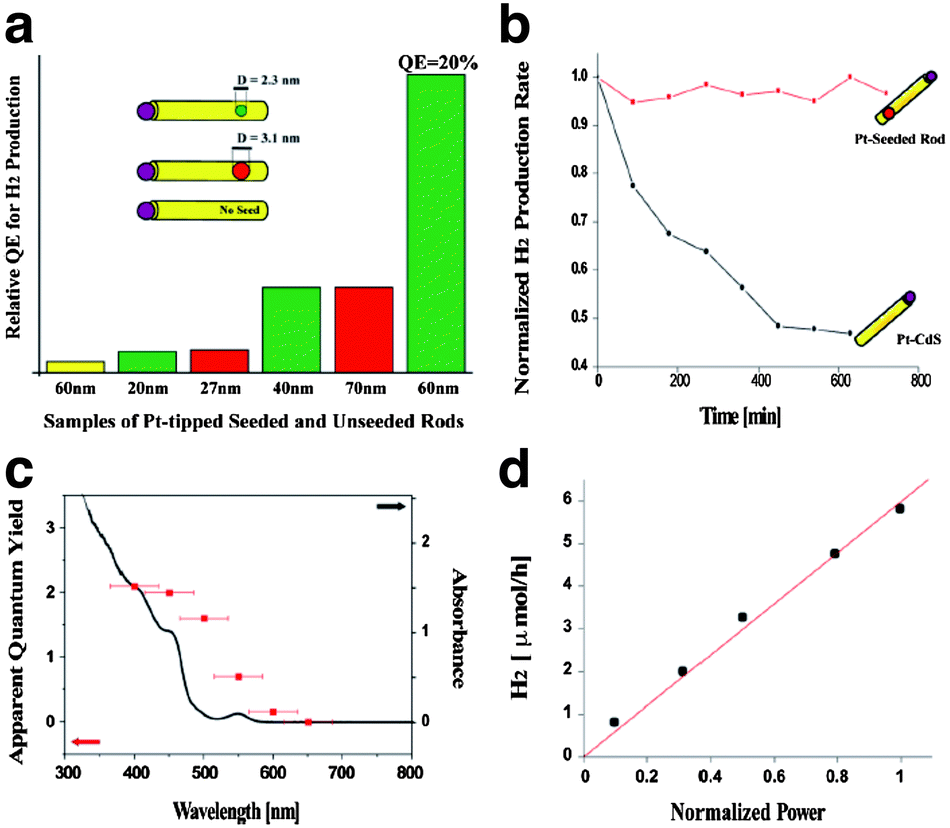 | ||
| Fig. 4 (a) Relative quantum efficiency for photocatalytic hydrogen production by Pt-tipped cadmium chalcogenide nanorods. (b) Time-dependent hydrogen production from Pt–CdS and Pt–CdSe/CdS nanorods. (c) Spectrally-dependent quantum yield of hydrogen production overlayed with the absorbance of the nanorods. (d) Power-dependence of the photocatalytic hydrogen production by CdSe/CdS nanorods. Reprinted with permission from ref. 27. Copyright 2010 American Chemical Society. | ||
A related photocatalytic H2-producing system using CdSe/CdS sensitizers coupled to platinum nanoparticles through methylviologen has been reported with efficiency close to 10%.28 Zhu et al. use transient absorption to show that the improvement in efficiency between the CdSe core and the CdSe/CdS heterostructure is due to the prolonged lifetime of the reduced methylviologen radical. This is presumably because the CdSe-localized hole remains spatially isolated and therefore unable to recombine with the excited electron that has been transferred to methylviologen. The two papers highlighted here show approaches using similar systems in distinctive configurations—metallurigical junctions versus solution-based redox couples. Continued emphasis on tailoring the size and structure of QD-based photocatalysts offers an opportunity to direct localization and energy of electrons and holes to optimize both activity and stability.
In addition to tuning the band-structure for stability, quantum confinement can make possible reactions such as proton reduction that are thermodynamically or kinetically unfavourable in the corresponding bulk semiconductors. As the size of nanocrystal approaches or goes beneath the Bohr exciton radius, the conduction and valence bands adopt a size-dependent energy related to the carrier effective masses. This effect is true to any semiconductor and at least one oxide system (WO3) has been reported to demonstrate enhanced catalytic activity in confined sizes.29 But the fact that Bohr exciton radii of oxides tend to be small (in many cases <3 nm), synthetic control relatively poorer than QDs, and the wide band-gap of water-splitting oxides makes confinement counterproductive for solar light absorption. In chalcogenide semiconductors with narrower band gaps, on the other hand, quantum confinement can be used to alter the energy level of the potential photocatalyst, tuning the kinetics and thermodynamics of proton reduction. Thus, quantum-confined CdSe may have a conduction band above the reduction potential of hydrogen, rather than below as in the bulk. Osterloh and co-workers reported hydrogen production from water using quantum-confined CdSe nanoribbons.30 Later, the same group more rigorously demonstrated that hydrogen reduction is possible because of the raised flatband potential of the confined CdSe.31
The synthesis of CdSe QDs can be tuned with near monolayer precision,32 which allows detailed study of the effect of size on the photocatalytic activity of CdSe. Quantum-confinement has two substantial and counterbalancing effects on the photocatalytic properties of QDs: quantum confinement increases the band-gap, reducing absorption, but increasing the energy of the conduction band. Holmes et al. show that quantum confinement controls photocatalytic hydrogen production from varying sizes of CdSe nanocrystals.33 They report the hydrogen production from CdSe nanocrystals in the range 1.75–4.81 nm in an aqueous electrolyte solution of Na2SO3 using a Xenon lamp. The authors find that detectable levels of hydrogen production are realized only with sample size below 3.5 nm, with the QDs showing no activity above this threshold. The authors show that the results of activity plotted versus the QD size can be understood quite well by assuming that the rate of electron transfer from the semiconductor to the acceptor follows eqn (2):
| kRed ∝ e−(ΔG−λ)2/4kTλ | (2) |
Although size-tunable reactivities of QD photocatalysts are currently proof-of-concept results, the exploitation of quantum confinement partially decouples reducing or oxidizing potentials from the materials choice. This observation, in connection with the development of increasingly better tools for tailoring particle size and shape in these systems and their integration in hierarchical structures can open new opportunities for improving the visible-light activity of photocatalysts based on QDs.
4. Plasmonic photocatalysts
Many promising new photocatalysts are made without a semiconductor absorber at all. Plasmonic photocatalysts function using the localized surface plasmon resonance (LSPR) of metallic nanoparticles. LSPRs arise from the oscillation of free electrons (or holes) on the surface of a nanometer-scale particle and the energy of the LSPR(s) supported on a particle depends on the number of free carriers in the material and the particle geometry. Plasmonic nanostructures have been shown to share important capabilities with semiconductors: electrons excited in LSPRs can be used in photodetection,34 photoreduction,35 “impossible” hot-electron splitting of hydrogen,36 and in combined photo-thermal catalysis.37 The latter two, in particular, demonstrate that excitation of the LSPR of noble metal nanoparticles can generate chemically-active electrons. Theoretical treatments of electron transfer from plasmonic nanostructures suggest that size and nanocrystal geometry dictate electron transfer from plasmonic systems to semiconductors or small molecules.38 Although gold nanostructures, with a visible plasmon resonance and exceptional chemical stability, are the most studied in this young field, other metals (Ag, Cu in particular) and heavily-doped semiconductors (mainly metal oxides and sulfides) offer a readily-expanded range of LSPR energies and even the possibility of self-sensitization in the case of wide band gap oxides.The motivations for using plasmonic nanostructures for photocatalysis are described in detail elsewhere.39–41 In brief, plasmons have large volumetric extinction coefficients that make plasmonic nanostructures excellent sensitizers from which electrons or energy can be transferred. Excited LSPRs also generate large field enhancements in the local environment of the plasmonic particle, which may enhance electron–hole generation of nearby materials. And, similar to plasmon-enhanced photovoltaics, light scattering of plasmonic nanostructures increases the path of light through a film.
New photocatalytic systems employing plasmonic nanostructures are typically plasmon-enhanced rather autonomous plasmonic photocatalysts. Enhancement has been described in multiple modes: local field enhancement of generation in a semiconductor and generation from the plasmon itself. Several groups have reported photoelectrodes of Au/TiO2 (ref. 42–44) or Au/Fe2O3 (ref. 45) which show a strong spectral dependence of photocurrent or activity coinciding with the LSPR feature of the gold component. Cronin et al. attributed this spectral dependence to enhanced electron–hole generation at the Au/TiO2 interface, which accounts for the large enhancements (>50×) in visible light photocatalytic activity. Other groups have claimed direct plasmon sensitization, with as much as 95% of electrons derived from the LSPR feature of a sensitizer.42
Priebe et al. have explored this debate in detail using a Au/P25 TiO2 system studied by in situ photo-EPR.46 Similar to previous reports, the authors verify that in contrast to pure TiO2, gold–titania nanostructures are capable of photocatalytic hydrogen production with only visible light. In situ electron paramagnetic resonance is used to elucidate the dynamics of electrons excited in the gold LSPR. TiO2 loaded with gold under illumination shows a strong paramagnetic signal, which they suggest is caused by electrons derived from the LSPR and trapped at surface sites on the titania. This signal diminishes in the presence of O2 flow, indicative of the formation of O2−. Unlike signals from paramagnetic Ti(III) species, the trapped electron signal which occurs only for the Au/TiO2 structure is still apparent at 290 K under visible illumination. Using the in situ EPR probe strongly suggests that the enhancements of visible-light photocatalytic activity in tailored plasmon/titania nanostructures are a direct consequence of electron generation and transfer from the plasmonic metal particle.
Most work has focused on plasmon-enhanced photocatalysis on semiconductor surfaces, but photocatalytic reactions have been reported directly on the metal surface of supported plasmonic nanoparticles. In this sense, although some works are not related to hydrogen production, they show great promise for the field because of other recent reports on splitting of H2 over gold particles that demonstrated how these plasmon-based catalysts represent a new and distinctive class of photocatalysts. Rather than rely on the conduction and valence band of semiconductors, these plasmonic metallic nanostructures can photocatalyze reactions by injection of resonantly-excited electrons into adsorbed species. Excellent synthetic control of gold, silver, and other plasmonic nanostructures can be exploited to tune the energy of the plasmon to match the adsorbed species. Linic and co-workers introduced silver nanocubes impregnated on alumina as a photocatalyst for ethylene epoxidation that acted both using thermal and radiative energy.37,47 The authors showed that the silver nanocubes under illumination by a visible light source at 450 K can perform catalytic oxidations at lower temperatures than conventional thermal-only systems and with higher selectivity. From wavelength-selective illumination, photocatytic rate on the silver nanocubes demonstrates a wavelength-dependence nearly superimposable with the plasmon absorption feature. The reactivity of the silver nanocubes is attributed to electron transfer from electrons excited in the nanocube LSPR donating to an O2 π* orbital, forming a transient O2− species (Fig. 5). More recent work demonstrates superlinear catalytic response to flux and higher quantum efficiency at higher fluence and temperature, in contrast to semiconductors.47 Such plasmon-active metal nanostructures offer much promise as a new class of photocatalysts.
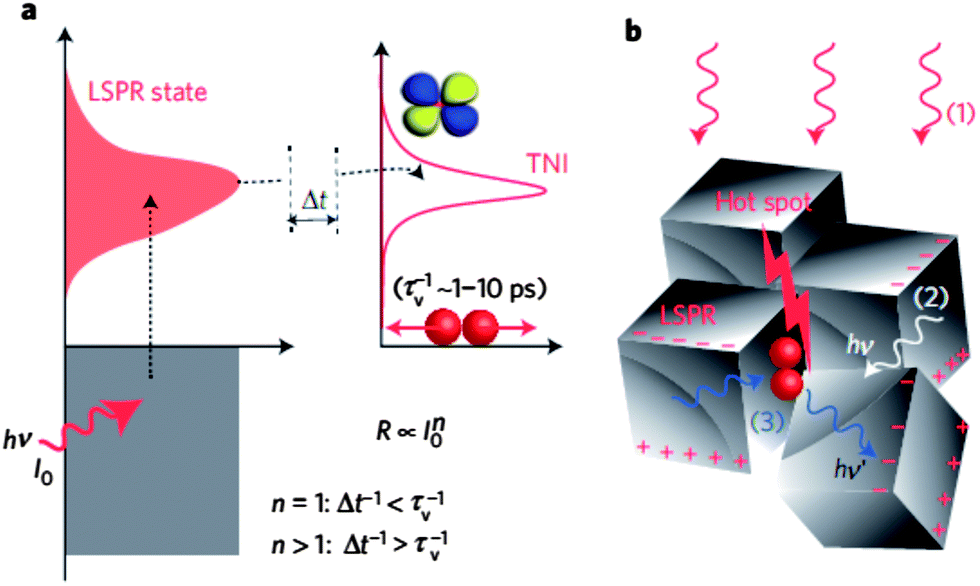 | ||
| Fig. 5 (a) Cartoon demonstrating the transfer of electrons from the LSPR of the plasmonic silver nanoparticles to the empty transient negative ion (TNI) anti-bonding orbital of oxygen. The transfer rate depends on the number of photoexcited carriers (n). (b) Cartoon depicting the localization of the LSPR at hot spots on the silver nanocube surface. Such spots are proposed as the active sites for oxygen splitting. Reprinted by permission from Macmillan Publishers Ltd: Nat. Mater. (ref. 47), copyright 2012. | ||
5. Nanocomposites
As the synthetic control of lithographic and colloidal techniques for making nanostructures improves, increasingly complex architectures combine the unique advantages of several materials for enhanced photocatalytic effect. The motivating example for work on complex nanostructures for photocatalytic water splitting is the leaf, which uses the so-called Z-scheme of two visible band-gap absorbers with electron transfer. Using a photovoltaic cell and appropriate catalysts for oxidation and reduction, an autonomous “artificial leaf” has been realized with 2.5% efficiency in solar water splitting.48Many complex nanostructures employ a Z-scheme by using two semiconductors with offset bands forming a type-II heterojunction. Type-II heterojunctions allow for efficient splitting of the photogenerated electron and hole. The enhanced photocatalytic performance of type-II nanostructures was recently reviewed.49 Among the more recent, creative interpretations of the type-II junction is the twin-induced type-II homojunction demonstrated by Liu et al.50 The authors demonstrate a controllable synthesis of Cd0.5Zn0.5S nanorods of alternating zinc blende and wurtzite crystal structures due to a high number of twin planes. The composition of the wires is tuned to adjust the overpotential for proton reduction and maintain some visible light absorption. The authors show that the presence of stacking faults generates a conduction band offset between the wurtzite and zinc blende phases along the same nanowire, allowing charge separation explaining photocatalytic generation of hydrogen at remarkable quantum yields as high as 62%.
Increasingly elaborate “nanosystems” have sought to use vapor-grown semiconductor nanowires as the basis for light-absorption and charge separation in photocatalysis. Analogous to the field of photovoltaics, these systems have substantial potential advantages over planar junctions, which have relatively low surface area and require much longer carrier diffusion, often necessitating more expensive fabrication. By thinning the semiconductor active layer, shorter carrier diffusion lengths and therefore lower-purity semiconductors can be effectively used for photogeneration of electron–hole pairs. The pillar array of a nanowire forest has the additional benefit of very effectively scattering light, increasing the path length of light in the active layer and partially compensating for the relatively poor absorption of indirect semiconductors like silicon. Separate groups have used silicon-based nanowire architectures to create high surface area photocatalysts, either by the formation of radial p–n junctions51 or Si/TiO2 junctions.52 After formation of the nanowire forest, the semiconductor layer can be decorated by hydrogen- and/or oxygen-evolving materials. Modular design of complex photocatalytic systems, although it increases the difficulty of fabrication, allows the ideal material to be used for each step in the photocatalytic reaction pathway—similar to the complexity of biological water splitting.52 Despite such rational design, total water splitting “nanosystems” still offer relatively modest water-splitting efficiency vis-à-vis traditional photovoltaic cells coupled to water-splitting catalysts.
Stepping away from traditional semiconductors, Mubeen et al. recently reported a complexly-tailored nanostructure which typifies the possibilities of modular design.53 The authors combined relatively new advances in fabrication and knowledge of plasmonic photoanodes42,54 to make gold rod absorbers decorated with TiO2/Pt nanoparticles on the tip and an electrodeposited cobalt oxygen-evolving complex on the sides. Absorption, reduction, and oxidation are each treated separately using an optimized material choice. By varying the excitation wavelength, the authors demonstrate that absorption due to the LSPR of the gold nanorod is responsible for photocatalytic water splitting.
The nanotube architecture is also convenient for the transport of charges to/from electrodes (in the case of photoelectrochemical reactions) or to improve charge separation in case of photocatalysts with multiple components. As a particular case, carbon nanotubes (CNTs) possess exceptional conductivity and are therefore of great interest in this field.55 Additionally, high surface area and the possibility of acting as photosensitizers are also appealing elements for using CNTs in photocatalysis. Many recent works report sol–gel syntheses of CNTs–TiO2 composites in which CNTs are usually randomly dispersed inside a TiO2 matrix. Better control of the CNT–TiO2 interface might create an extended Schottky barrier that retards electron–hole recombination dynamics by electron injection from the semiconductor to the CNTs. Following this idea, uniform coating of multi-wall CNTs (MWCNTs) with titania, ceria or zirconia was reported by Cargnello et al.56 Oxidation of the outer MWCNTs layers produced carboxyl groups able to bind metal alkoxide precursors in solution. The use of Pd or Pt nanoparticles exposing carboxyl groups on the monolayer edge in a similar way allowed to obtain final nanostructures where MWCNTs were embedded inside oxide layers containing metal particles within the oxide. Given the versatility in the compositions, the structures demonstrated to be active for a variety of heterogeneous reactions. In particular, the role of MWCNTs in enhancing the photocatalytic properties of the structures was demonstrated by the fact that the system comprising the MWCNTs evolved H2 during methanol photoreforming at a rate almost double than samples without the nanotubes (Fig. 6).
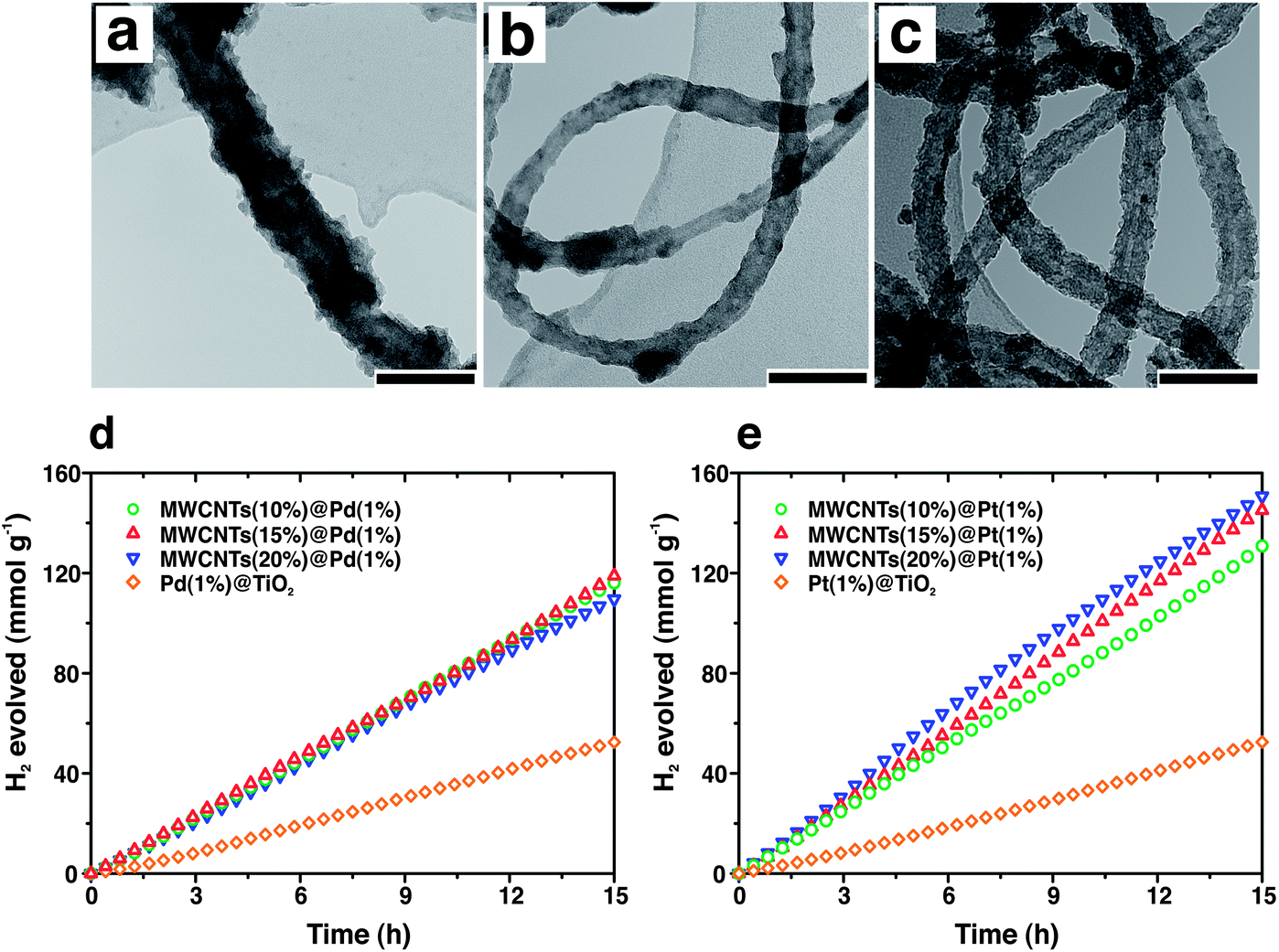 | ||
| Fig. 6 (a–c) TEM images of MWCNTs@Pd/TiO2 at increasing concentration of MWCNTs from 5 (a) to 10 (b) to 15 wt% (c). Photocatalytic activity for H2 production from methanol photoreforming on MWCNTs@Pd/TiO2 (d) and MWCNTs@Pt/TiO2 (e) nanocomposites. Adapted with permission from ref. 56 Copyright 2012 American Chemical Society. | ||
Carbon nitride (C3N4), which forms graphitic two-dimensional sheets from tri-s-triazine units, has been recently explored as another potential organic photocatalyst for hydrogen production.57,58 Graphitic carbon nitride can be produced on large scale with abundant, sustainable elements and unlike most carbonaceous polymers, is relatively stable. Wang et al. demonstrated that the band structure of an ideal graphitic carbon nitride sheet is suitable for performing both oxidative and reductive half-reactions of water splitting.59 They further demonstrated that with 3 wt% Pt, C3N4 powder is a stable hydrogen evolution catalyst. Since this report, exfoliation techniques have been used to show the increased activity of the high-surface area sheets, compared to the bulk-C3N4.60 Its membrane qualities have been exploited as the wall of a vesicle wall scaffold and light absorber in hydrogen evolution.61 Combined with cobalt oxide nanoparticles, carbon nitride can also generate oxygen from visible light with as high as 1% efficiency.62 Similar to the plasmonic autonomous water splitting device, both half reactions have been demonstrated using graphitic carbon nitride sheets and await combination.
6. Conclusions
Novel approaches to tailor the structure of photocatalysts to manipulate photogenerated charges are emerging as methods to improve the activity of the systems. Lithographic techniques, assembly of nanocrystals, precise size and shape control, synthesis of well-controlled heterostructures, and the use of plasmonic building blocks are being applied with great ingenuity to improve current photocatalysts, including enhancement of absorption, charge separation, and surface reactivity. In this feature article we highlighted the most exciting systems that have been applied to the sustainable production of hydrogen. Manipulation of nanostructures will achieve an even better degree of control that will help face important challenges in the broader energy scenario.Acknowledgements
M.C. was supported by the National Science Foundation through the Nano/Bio Interface Center at the University of Pennsylvania Grant Number DMR08-32802. B.T.D. was supported by the Department of Energy Office of Basic Sciences, Division of Materials Science and Engineering, under award no. DE-SC0002158.Notes and references
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26 CrossRef CAS PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS.
- M. Cargnello, A. Gasparotto, V. Gombac, T. Montini, D. Barreca and P. Fornasiero, Eur. J. Inorg. Chem., 2011, 2011, 4309 CrossRef CAS.
- D. I. Kondarides, V. M. Daskalaki, A. Patsoura and X. E. Verykios, Catal. Lett., 2008, 122, 26 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746 CrossRef CAS PubMed.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856 CrossRef CAS PubMed.
- U. Diebold, Surf. Sci. Rep., 2003, 48, 53 CrossRef CAS.
- J. Tao, T. Luttrell and M. Batzill, Nat. Chem., 2011, 3, 296 CrossRef CAS PubMed.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638 CrossRef CAS PubMed.
- X. Q. Gong and A. Selloni, J. Phys. Chem. B, 2005, 109, 19560 CrossRef CAS PubMed.
- X. Han, Q. Kuang, M. Jin, Z. Xie and L. Zheng, J. Am. Chem. Soc., 2009, 131, 3152 CrossRef CAS PubMed.
- S. Liu, J. Yu and M. Jaroniec, Chem. Mater., 2011, 23, 4085 CrossRef CAS.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, J. Pan, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2009, 131, 12868 CrossRef CAS PubMed.
- J. Pan, G. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133 CrossRef CAS PubMed.
- T. R. Gordon, M. Cargnello, T. Paik, F. Mangolini, R. T. Weber, P. Fornasiero and C. B. Murray, J. Am. Chem. Soc., 2012, 134, 6751 CrossRef CAS PubMed.
- A. Dong, X. Ye, J. Chen, Y. Kang, T. Gordon, J. M. Kikkawa and C. B. Murray, J. Am. Chem. Soc., 2010, 133, 998 CrossRef PubMed.
- V. Gombac, L. Sordelli, T. Montini, J. J. Delgado, A. Adamski, G. Adami, M. Cargnello, S. Bernal and P. Fornasiero, J. Phys. Chem. A, 2009, 114, 3916 CrossRef PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806 CrossRef CAS PubMed.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096 CrossRef CAS PubMed.
- R. Rossetti, S. Nakahara and L. E. Brus, J. Chem. Phys., 1983, 79, 1086 CrossRef CAS.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Science, 2012, 338, 1321 CrossRef CAS PubMed.
- S. E. Habas, P. Yang and T. Mokari, J. Am. Chem. Soc., 2008, 130, 3294 CrossRef CAS PubMed.
- T. Mokari, E. Rothenberg, I. Popov, R. Costi and U. Banin, Science, 2004, 304, 1787 CrossRef CAS PubMed.
- Y. Shemesh, J. E. Macdonald, G. Menagen and U. Banin, Angew. Chem., Int. Ed., 2011, 50, 1185 CrossRef CAS PubMed.
- L. Amirav and A. P. Alivisatos, J. Am. Chem. Soc., 2013, 135, 13049 CrossRef CAS PubMed.
- L. Amirav and A. P. Alivisatos, J. Phys. Chem. Lett., 2010, 1, 1051 CrossRef CAS.
- H. Zhu, N. Song, H. Lv, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2012, 134, 11701 CrossRef CAS PubMed.
- D. Tanaka, Y. Oaki and H. Imai, Chem. Commun., 2010, 46, 5286 RSC.
- F. Andrew Frame, E. C. Carroll, D. S. Larsen, M. Sarahan, N. D. Browning and F. E. Osterloh, Chem. Commun., 2008, 2206 RSC.
- F. A. Frame and F. E. Osterloh, J. Phys. Chem. C, 2010, 114, 10628 CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706 CrossRef CAS.
- M. A. Holmes, T. K. Townsend and F. E. Osterloh, Chem. Commun., 2012, 48, 371 RSC.
- M. W. Knight, H. Sobhani, P. Nordlander and N. J. Halas, Science, 2011, 332, 702 CrossRef CAS PubMed.
- M. Maillard, P. Huang and L. Brus, Nano Lett., 2003, 3, 1611 CrossRef CAS.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240 CrossRef CAS PubMed.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3, 467 CAS.
- A. O. Govorov, H. Zhang and Y. K. Gun'ko, J. Phys. Chem. C, 2013, 117, 16616 CAS.
- S. C. Warren and E. Thimsen, Energy Environ. Sci., 2012, 5, 5133 CAS.
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612 CrossRef CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911 CrossRef CAS PubMed.
- J. Lee, S. Mubeen, X. Ji, G. D. Stucky and M. Moskovits, Nano Lett., 2012, 12, 5014 CrossRef CAS PubMed.
- Z. Liu, W. Hou, P. Pavaskar, M. Aykol and S. B. Cronin, Nano Lett., 2011, 11, 1111 CrossRef CAS PubMed.
- E. Kowalska, R. Abe and B. Ohtani, Chem. Commun., 2009, 241 RSC.
- I. Thomann, B. A. Pinaud, Z. Chen, B. M. Clemens, T. F. Jaramillo and M. L. Brongersma, Nano Lett., 2011, 11, 3440 CrossRef CAS PubMed.
- J. B. Priebe, M. Karnahl, H. Junge, M. Beller, D. Hollmann and A. Brückner, Angew. Chem., Int. Ed., 2013, 52, 11420 CrossRef CAS PubMed.
- P. Christopher, H. Xin, A. Marimuthu and S. Linic, Nat. Mater., 2012, 11, 1044 CAS.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645 CrossRef CAS PubMed.
- Y. Wang, Q. Wang, X. Zhan, F. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326 RSC.
- M. Liu, D. Jing, Z. Zhou and L. Guo, Nat. Commun., 2013, 4, 2278 Search PubMed.
- E. L. Warren, J. R. McKone, H. A. Atwater, H. B. Gray and N. S. Lewis, Energy Environ. Sci., 2012, 5, 9653 CAS.
- C. Liu, J. Tang, H. M. Chen, B. Liu and P. Yang, Nano Lett., 2013, 13, 2989 CrossRef CAS PubMed.
- S. Mubeen, J. Lee, N. Singh, S. Kramer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247 CrossRef CAS PubMed.
- S. Mubeen, G. Hernandez-Sosa, D. Moses, J. Lee and M. Moskovits, Nano Lett., 2011, 11, 5548 CrossRef CAS PubMed.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233 CrossRef CAS.
- M. Cargnello, M. Grzelczak, B. Rodríguez-González, Z. Syrgiannis, K. Bakhmutsky, V. La Parola, L. Liz-Marzán, R. J. Gorte, M. Prato and P. Fornasiero, J. Am. Chem. Soc., 2012, 134, 11760 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717 CAS.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452 CrossRef CAS PubMed.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 3, 1139 CrossRef.
- J. Zhang, M. Grzelczak, Y. Hou, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Sci., 2012, 3, 443 RSC.
| This journal is © The Royal Society of Chemistry 2014 |