To be or not to be metal-free: trends and advances in coupling chemistries
Rick Arneil
D. Arancon
*ab,
Carol
Sze Ki Lin
a,
Carolina
Vargas
c and
Rafael
Luque
*bd
aSchool of Energy and Environment, City University of Hong Kong, Kowloon, Hong Kong
bDepartamento de Quimica Organica, Universidad de Cordoba, Campus Universitario de Rabanales, Edificio Marie Curie (C3), E-14014, Cordoba, Spain. E-mail: q62alsor@uco.es
cDepartamento de Tecnología Química y Ambiental, Universidad Rey Juan Carlos, C/Tulipán s/n, 28933, Móstoles (Madrid), Spain
dDepartment of Chemical and Biomolecular Engineering (CBME), Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong
First published on 30th October 2013
Abstract
Coupling reactions have been part of several extensive studies in order to develop innovative and greener protocols that can generate a wide range of compounds with applications in pharmaceuticals, agrochemicals and biologically active compounds. Metal-free couplings are an important and increasingly trendy field that has attracted a significant deal of interest in recent years, generating a lot of controversy on the issue of whether metal free is really free. Aside from focusing on such a controversial topic itself, this contribution aims to provide a brief introduction on coupling chemistry to point out the transition of this technology from metal-catalyzed to metal-free. This is followed by a range of key selected synthetically useful metal-free processes and a brief commentary on the current debate of whether metal-free reactions are really metal-free and the required experiments for a full understanding of a metal-free coupling process.
Introduction
Metal coupling chemistry has played a very important role in organic synthesis over the past 50+ years, particularly in the development of biologically active compounds and the exploration of many other green routes to synthesis. The early discoveries of reactions including Suzuki–Miyaura, Heck, Stille, Sonogashira and Negishi couplings paved the way for the development of a wide range of chemistries for the synthesis of valuable organic compounds with important applications for pharmaceuticals, synthesis of important intermediates, drug discovery and many others.1,2Classical coupling protocols employed Pd(0) systems as catalysts in a variety of versions (e.g. with co-catalyst salts), providing good to excellent yields of target products.3 The development of alternative catalysts including a Pd-based heterogeneous catalyst,4 Pd nanoparticles,5 and transition metal catalysts6 have attracted many research endeavours in coupling chemistry around the world, all for the common aim of improving the metal loading (that is, reduce the amount of transition metal needed), reaction yield, functional group tolerance, and even reaction rates.7
With the recent rise of green chemistry, scientists are working towards alternative synthetic strategies that could lessen, if not remove the amount of waste generated by traditional synthetic methods. Although traditional coupling methods proved effective in industrial applications, they are also generating harmful metal waste and many other organic waste by-products that are not easy to treat. With this rationale, this contribution aims to provide a brief overview on traditional coupling methods and a comparison of their respective metal-free coupling counterparts (chosen and assessed based on researchers’ efforts to prove them fully metal-free). Particularly, the review will touch two ways to produce a metal-free coupling protocol – one via microwaves and the other using photochemistry.
Microwaves have been shown to have the ability to increase the solubility of many organic compounds in water, thereby opening the chemistry of aqueous coupling reactions.8 Since microwave irradiation is known to be much faster and more efficient than traditional heating processes, the rate of conversion of the target compound becomes much faster. On an application perspective, aside from microwaves, the utilisation of light as a promoter is a green method as it allows the potential utilisation of sunlight. Photocatalytic reactions are, however, known to co-generate a myriad of other side products due to the randomness of radical reactions. In any case, we believe that careful functional group control and optimized irradiation time can possibly create a future clean photocatalytic coupling method that takes advantage of direct sunlight exposure.
In the light of the aforementioned premises, this contribution has aimed to be unique in its kind, providing an accessible tutorial review with perspective on the basics, trends and advances of coupling reactions with a particular emphasis on metal-free couplings. Rather than being very comprehensive, the manuscript will focus on key literature examples to illustrate trends on metal-free couplings and future research avenues to pursue in the future. For detailed overviews on coupling reactions, readers will be kindly referred to recent literature reports in the different sections of this contribution.
Advances in metal-catalysed traditional couplings (Suzuki and Sonogashira)
Sonogashira coupling is one of the most widely used C–C bond-forming reactions today.9 It is an efficient route to synthesize important intermediates with an internal alkyne moiety, for the preparation of fine chemicals or polymers.10,11 The process generally involves the cross-coupling of a vinyl or aryl halide and a terminal alkyne in the presence of a transition metal, and is closely related to the Heck coupling (alkenes are utilised as substrates in the Heck reaction). The original Sonogashira protocol, reported in the 1970s (Scheme 1), also used copper(I) iodide as a co-catalyst to in situ convert the alkyne into a copper acetylide that was subsequently substituted by a palladium complex.12 It has been shown that Sonogashira couplings can also be accomplished with Pd catalysts in the absence of any copper salt13 or by using a non Pd-based transition metal catalyst.14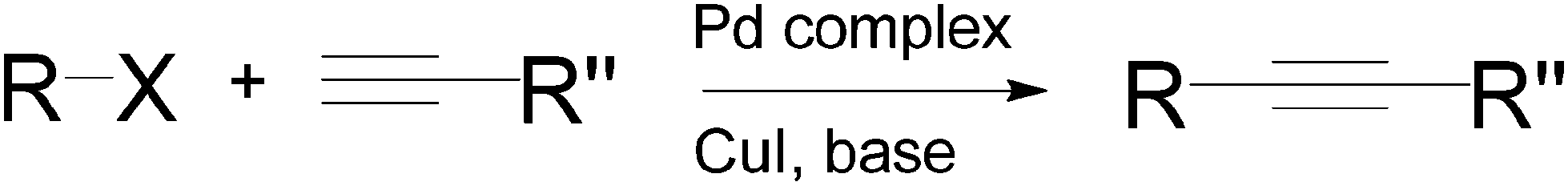 | ||
| Scheme 1 The Sonogashira coupling reaction between halides and alkynes. | ||
The mechanism of a traditional Sonogashira coupling involves the oxidative addition of an alkyl halide to a Pd(0) catalyst (Scheme 2). The transmetallation reaction between the Pd-alkyl halide complex and the Cu-alkyne forms the Pd complex of both the starting alkyl halide and the alkyne. The coupled product is readily formed via reductive elimination. The addition of a basic reagent in this case is an important step as it deprotonates the terminal alkyne to form an alkyne anion that binds easily to the oxidized copper.
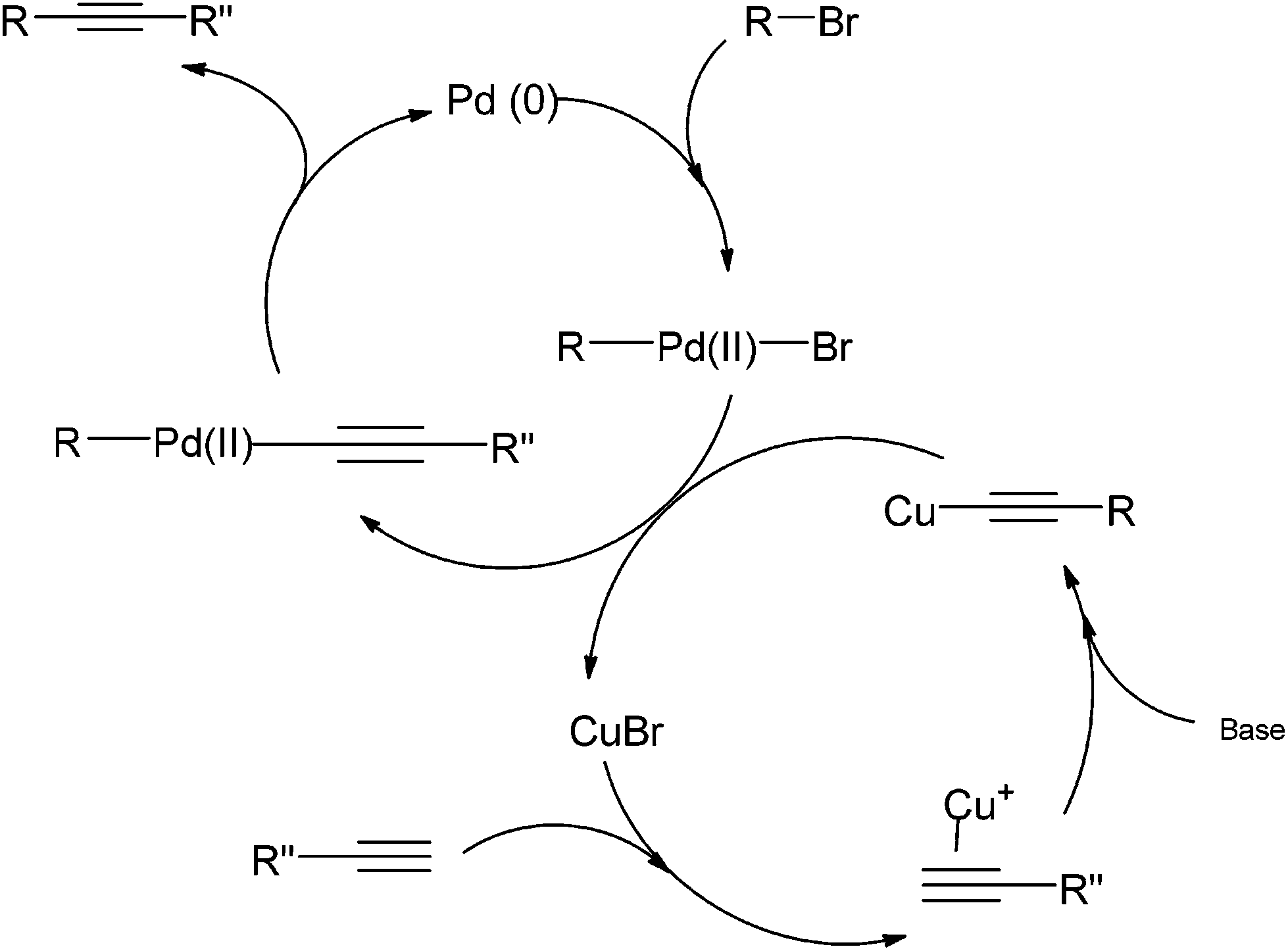 | ||
| Scheme 2 Proposed catalytic cycle of Sonogashira coupling.15 | ||
Many different solvents have been attempted to improve yields in Sonogashira coupling chemistries. Since most coupling partners are only soluble in hazardous organic solvents, substitute greener solvents can offer a series of advantages and could be considered as a major advance towards greener synthetic coupling. The use of water has already been investigated in these reactions.16 An example of a Pd-catalysed coupling reaction in aqueous media deals with the synthesis of alkynyl ketones from an alkyne, alkyl halide, and carbon monoxide.17 This coupling method works in the presence of pyrrolidine and triethylamine as bases (Scheme 3).
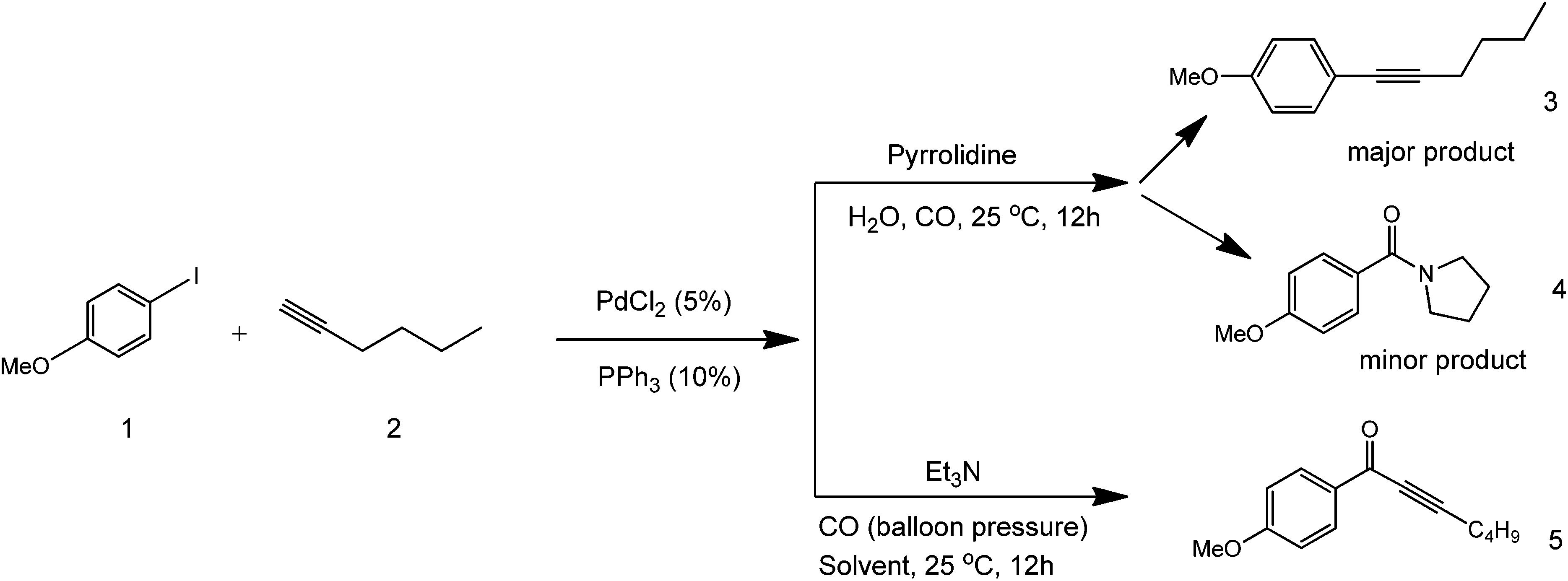 | ||
| Scheme 3 Aqueous phase coupling in the presence of a base (pyrrolidine or triethylamine) to form three different coupling products. | ||
As illustrated in the above scheme, three different products can be formed in this reaction, depending on the utilised base. Both pyrrolidine amide and alkyne coupled products are formed using pyrrolidine as a base since the amide product is also stable (structures 3 and 4 in Scheme 3). With triethylamine, the keto-alkyne product selectively forms, as the amine used is tertiary (structure 5). The mechanism of this process depends on the rate of the iodobenzene species to add oxidatively to the Pd complex. As such, the more electron deficient the benzene becomes, the higher the yield of the carbonyl product (structure 5). Alternatively, the coupled product (structure 3) forms when the alkyne adds to the Pd complex faster than an electron-rich benzene species. This study showed that the solubility of organic compounds in a polar solvent can be increased to make the reaction cleaner under increased pressure conditions. Bases play important roles in the generation of intermediates that dictate the structure of products formed in coupling reactions.
Aside from water as a solvent, the use of ionic liquids as solvents in coupling processes is also currently being explored, offering a simple separation between products (and by-products) and the starting transition metal catalyst for enhanced recyclabilities. The use of a 1-butyl-3-methylimidazolium hexafluorophosphate [BMIm][PF6] (structure 6) was reported for this purpose, showing excellent yields in the coupling of iodobenzene and phenylacetylene using a Pd complex catalyst and a Cu salt as a co-catalyst (Scheme 4).18
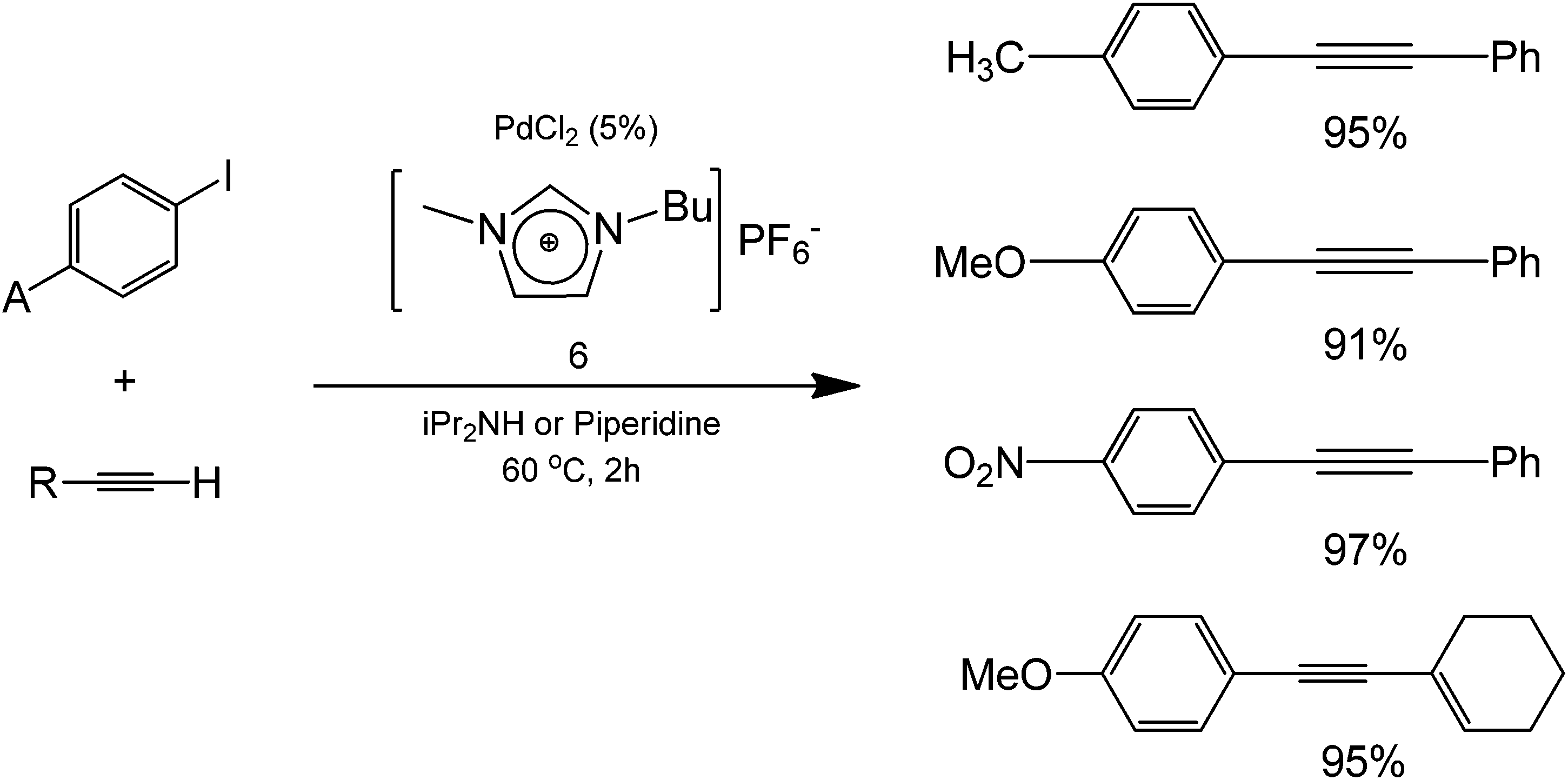 | ||
| Scheme 4 The coupling reaction using an ionic liquid solvent [BMIm][PF6] along with amine bases (di-isopropyl amine and piperidine) shows the tolerance of the protocol to different electron donating and withdrawing groups attached to the starting aryl halide. | ||
The type of Pd complexes were varied in this protocol and the yield for the coupling was the highest (96%) using PhCl2(PPh3)2 as a starting Pd complex. When this complex was used in different organic solvents, reduced yields were obtained (48% with toluene, 55% with THF, and 81% with DMF), highlighting the improvement of the reaction yield conducted under ionic liquid medium. Authors also performed the reaction in the absence of copper as a co-catalyst. Results showed that coupling products were still obtained. These findings seemed to point out the potential ability of the ionic liquid to serve as both a catalyst and an activating agent for the alkyne.
Phosphonium-based19 ionic solvents have also been reported to serve as reaction media in a step-wise Sonogashira-type coupling, carbonylation and hydroamination steps for the one-pot synthesis of 3-methyleneisoindolin-1-ones.19 Phosphonium based ILs are especially attractive solvents due to their thermal and chemical stability and resistance to degradation. The optimized reaction conditions show the use of a PdCl2(PPh3)2 complex and [P+][Br−] ionic liquid solvent, 1 atm CO, at 110 °C for 18 h (Scheme 5). In the presence of Et3N, the reaction is believed to occur via a stepwise mechanism to produce structure 8 as an intermediate instead of going directly to structure 7.
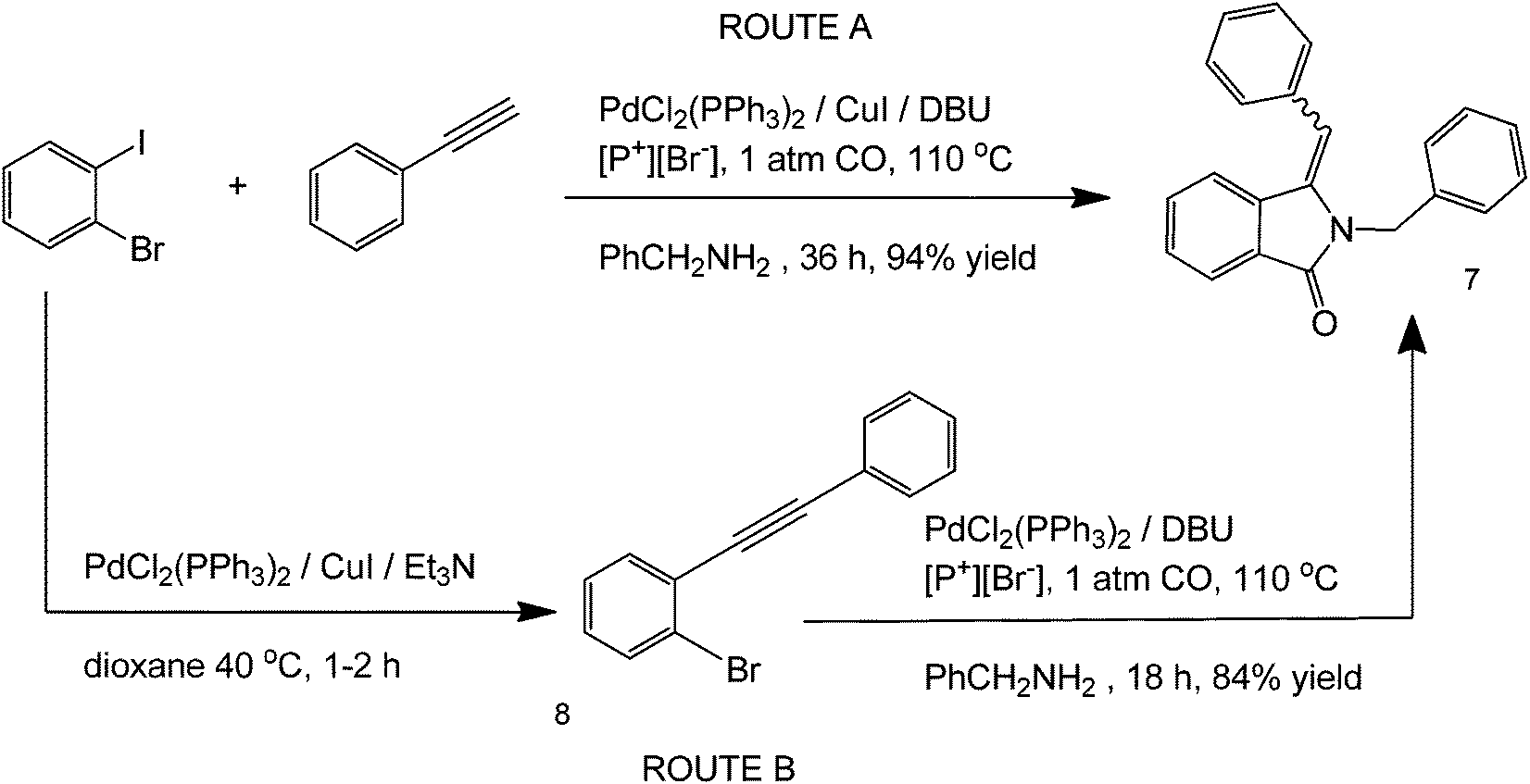 | ||
| Scheme 5 Reaction scheme and the optimized reaction conditions for the one-pot synthesis of 3-methyleneisoindolin-1-ones using a phosphonium-based ionic liquid. | ||
The highest product yields in this reaction were obtained under atmospheric conditions using [P+][Br−] IL. The carbon monoxide here, although important in the carbonylation reaction, was added in excess and did not have an effect on the reaction rate. The use of IL in both of these cases is especially attractive because it makes the reaction green by (a) greatly simplifying product separation, and (b) serving as both a solvent and a promoter ligand for one-pot reactions.
Bases have also been used as solvents in coupling reactions.20–22 The use of bases is particularly attractive due to the ability of the base to serve as both a reaction medium and an activating agent of the alkyne. The amount of this base is one of the most important parameters to optimize in a Sonogashira coupling reaction, as bases are generally involved in the rate-determining step. Amines include a family of commonly employed bases in coupling chemistries as their basicities can be easily fine-tuned by varying the different groups around the nitrogen. Recently, the role of amines in copper-free couplings have been discussed. The activation of the alkyne coupling partner could happen in two possible ways:23 Path A (Scheme 6) takes place when the alkyne is a much better ligand than the amine (as is the case for alkynes attached to electron-donating groups), and the Pd-complex selectively coordinates with the alkyne to form the highly reactive complex [Pd(Ar)(X)(L)(RC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH)]. A subsequent deprotonation step of the alkyne by the amine yields an acetylide intermediate that is more susceptible for insertion.
CH)]. A subsequent deprotonation step of the alkyne by the amine yields an acetylide intermediate that is more susceptible for insertion.
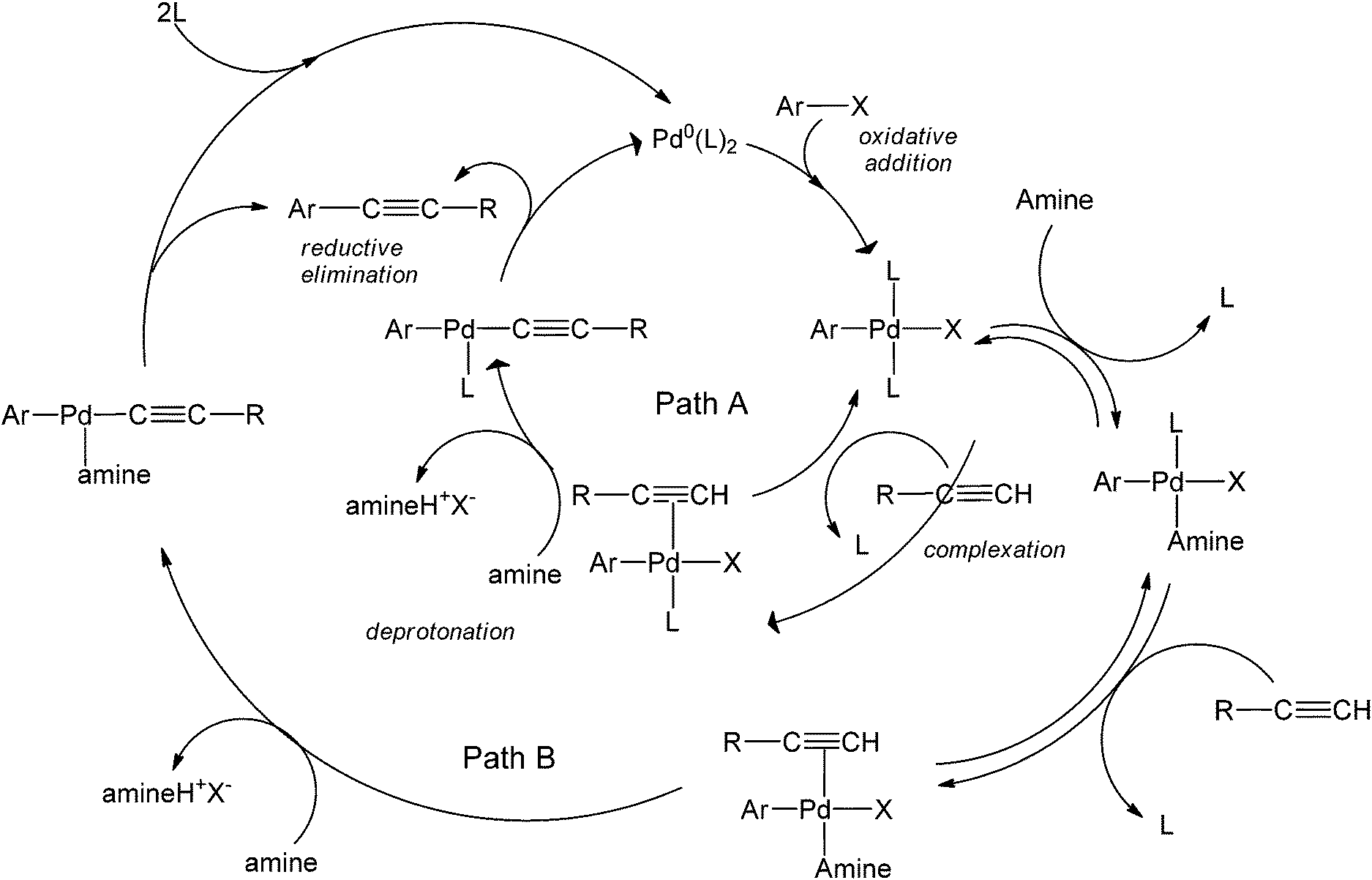 | ||
| Scheme 6 The catalytic cycle showing the two possible roles amines play in Cu-free C–C bond formation.23 | ||
The alternative mechanism shows an amine that forms a more stable complex with Pd as compared to that with the alkyne, so that the alkyne coordinates with the Pd-amine complex to then form the coupled product. Path A occurs with PPh3 as a ligand (since it is not a good leaving group with respect to AsPh3). Comparably, the formation of the Pd-amine complex happened much faster with AsPh3, thereby following Path B. Because of the formation of this stable Pd-amine complex, AsPh3 might not serve as a good ligand in such coupling processes. Aside from its obvious toxicity, the ligand reduces the efficiency of the reaction by generating a very stable intermediate with Pd.
For further reading on Sonogashira coupling developments, readers are kindly referred to the excellent and highly comprehensive reviews reported in recent years by Chinchilla et al.14 and Brown et al.24 Similarly, for developments in Heck-type couplings, readers are suggested to consult the recent overview of McCartney and Guiry,25 mainly on the current role of microwave irradiation in speeding up the usually slow Heck coupling.
The Suzuki–Miyaura coupling reaction can also be considered as one of the most versatile and commonly used C–C forming reactions.26–28 It involves the reaction between an organoboron compound (generally a boronic acid) and an aryl (or alkenyl, or alkynyl) halide in the presence of a metal catalyst and a base (Scheme 7), forming a wide range of biaryl-type molecules that find applications as pharmaceuticals, agrochemicals and dyes.
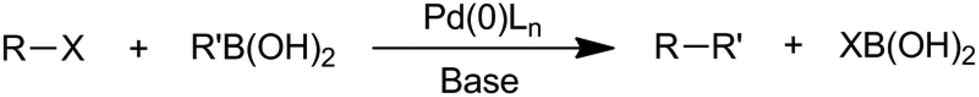 | ||
| Scheme 7 The Suzuki–Miyaura cross-coupling reaction between halides and boronic acids. | ||
The mechanism (Scheme 8) involves the oxidative addition of a phenyl halide to a metal (typically palladium) as the rate-determining step. This step is immediately followed by the transmetallation of a substituted boronic acid to produce a complex containing only the alkene and the phenyl group. Reductive elimination (cis configuration) then produces a coupled product. This reaction is considered one of the most versatile C–C forming reactions in organic synthesis today, being able to couple both sp2 and sp carbons with great tolerance, for steric hindrance and an electronic environment.30,31
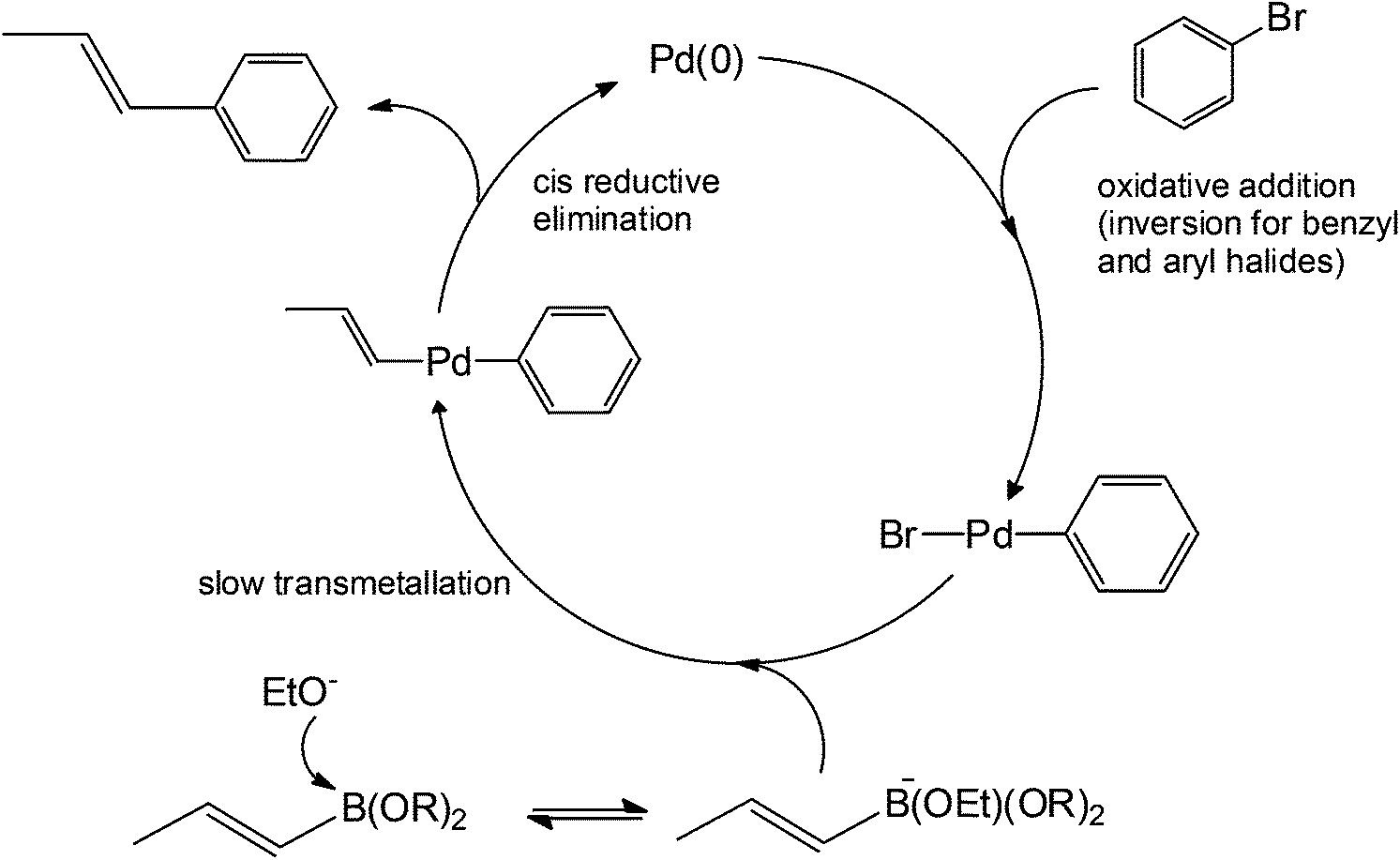 | ||
| Scheme 8 Proposed catalytic cycle of Suzuki coupling.29 | ||
The ability to couple two different groups in one single step provided organic chemists a convenient way to synthesize large molecules with multi-functional properties. Although both Sonogashira and Suzuki-type coupling reactions have changed the way we devise the synthesis of various bioactive compounds, they are not without drawbacks. These reactions use transition metals as catalysts that are usually not recyclable. Depending on the type of reaction, they can possibly generate metal–organic complex by-products that might have several unknown toxicities.
In order to develop greener synthetic protocols for Suzuki coupling, several strategies have been proposed. Apart from the well known heterogeneous catalysts and Pd-derived systems, ultralow metal catalyst loadings with aryl and vinyl boronic acids as coupling partners were proposed in recent years.32 Some of these methods worked by adding only 1 mg L−1 of Pd complex to the reaction mixture in the presence of either ethanol or tetrabutyl ammonium bromide (TBAB) under microwave heating at 150 °C for 7 minutes (Scheme 9). Importantly, the study showed that microwave heating can significantly improve coupling reaction times at much lower concentrations. Because of this, as will be demonstrated by the next sections, microwave heating has been employed as a primary tool in the development of ultralow-metal loading or even metal-free coupling methods.
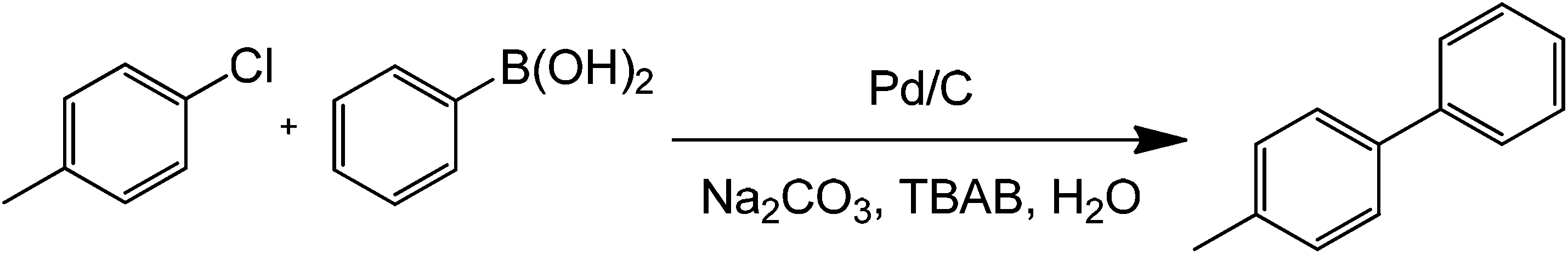 | ||
| Scheme 9 Traditional Suzuki–Miyaura coupling of p-chlorotoluene and benzylboronic acid in the presence of a base, a phase-transfer agent, water, and ultra-low palladium concentration. | ||
This type of coupling was also reported to be accomplished using an iron catalyst, in an apparent move to avoid the usually expensive Pd catalyst. The mechanism with iron catalysis is believed to occur similarly to a living radical polymerization, where a single electron is transferred from the iron to the C–X bond, which readily reacts with a pre-coordinated arene (to an iron).33 However, this study did not fully investigate the presence of relevant noble metals (e.g. Pd) in sub ppm/ppb quantities in the protocol. A similar coupling strategy is also demonstrated using an Fe–Ni alloy34 (Scheme 10). For further reading on developments of Suzuki couplings, readers are kindly referred to a recent overview in the field by Fihri et al.35
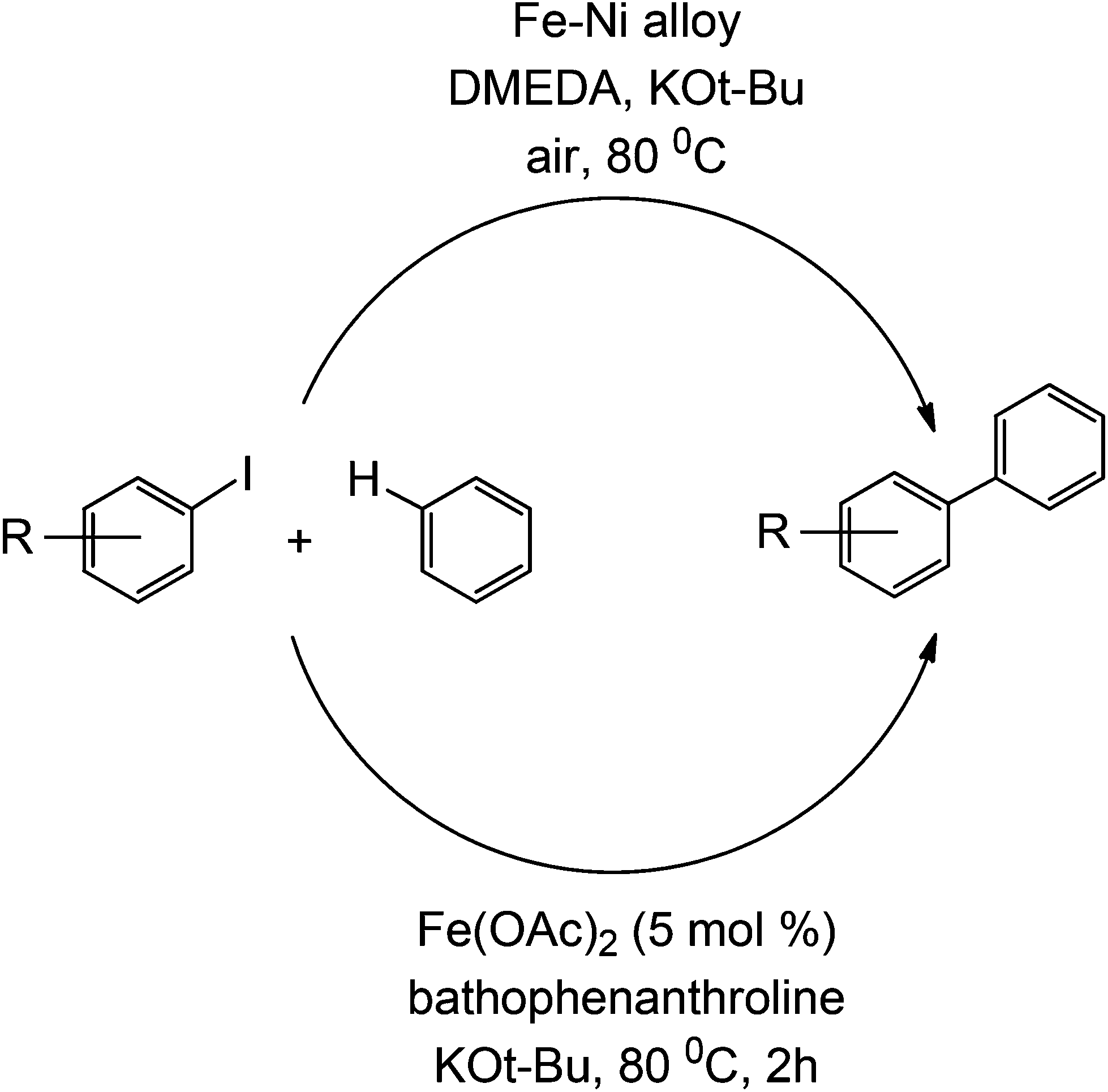 | ||
| Scheme 10 Two coupling reactions involving iron catalysts (Suzuki-type coupling). | ||
The advances in transition-metal catalysed coupling reactions show an important trend of finding greener approaches to carry out the coupling reactions. Coupling methods have evolved through the years from mainly Pd-catalyzed to iron and nickel to being “in principle” metal free.36 Included in the many improvements of these methods are the use of different bases and ionic liquids as both solvents and catalysts that make the reaction operationally simple. Both ionic liquids and bases have been shown to have a possibly dual role in a coupling reaction by acting as a reaction solvent and as an effective C–H activating agent. As previously mentioned, the presence of a strong polar base and a phase-transfer agent can drive a Suzuki coupling even by using very minor amounts of Pd. By using bases of different strengths, as will be shown in the next sections, coupling reactions can be performed under metal-free conditions. The next sections in this review will deal with this important ability of bases to activate strong C–H bonds. Furthermore, the use of alternative protocols including microwave-assistance and photocatalysis can provide an additional greener platform for the successful development of greener coupling chemistries.
Transition metal-free couplings
Metal-free couplings have been the dream of scientists from the early stages of coupling reactions, a fact that can easily be translated to many types of organic transformations. The development of metal-free protocols in coupling processes can indeed be considered a significant step forward towards advanced greener synthetic methodologies in organic synthesis. A series of metal-free coupling reactions were reported in the past years from the early report of the Leadbeater group on a transition-metal free Suzuki coupling.37 These seminal findings were later reassessed by the same group to find out that the reported Suzuki coupling was actually catalysed by traces of Pd as low as 50 ppb that were present in the base (sodium carbonate) utilised in their reactions.37 In the presence of a suitable phase transfer agent, base, and microwave heating, the metal-free coupling of aryl halides with alkynes was also shown to be possible.38 The reaction conditions were optimised by studying the coupling of 4-bromoacetophenone with phenylacetylene (Scheme 11).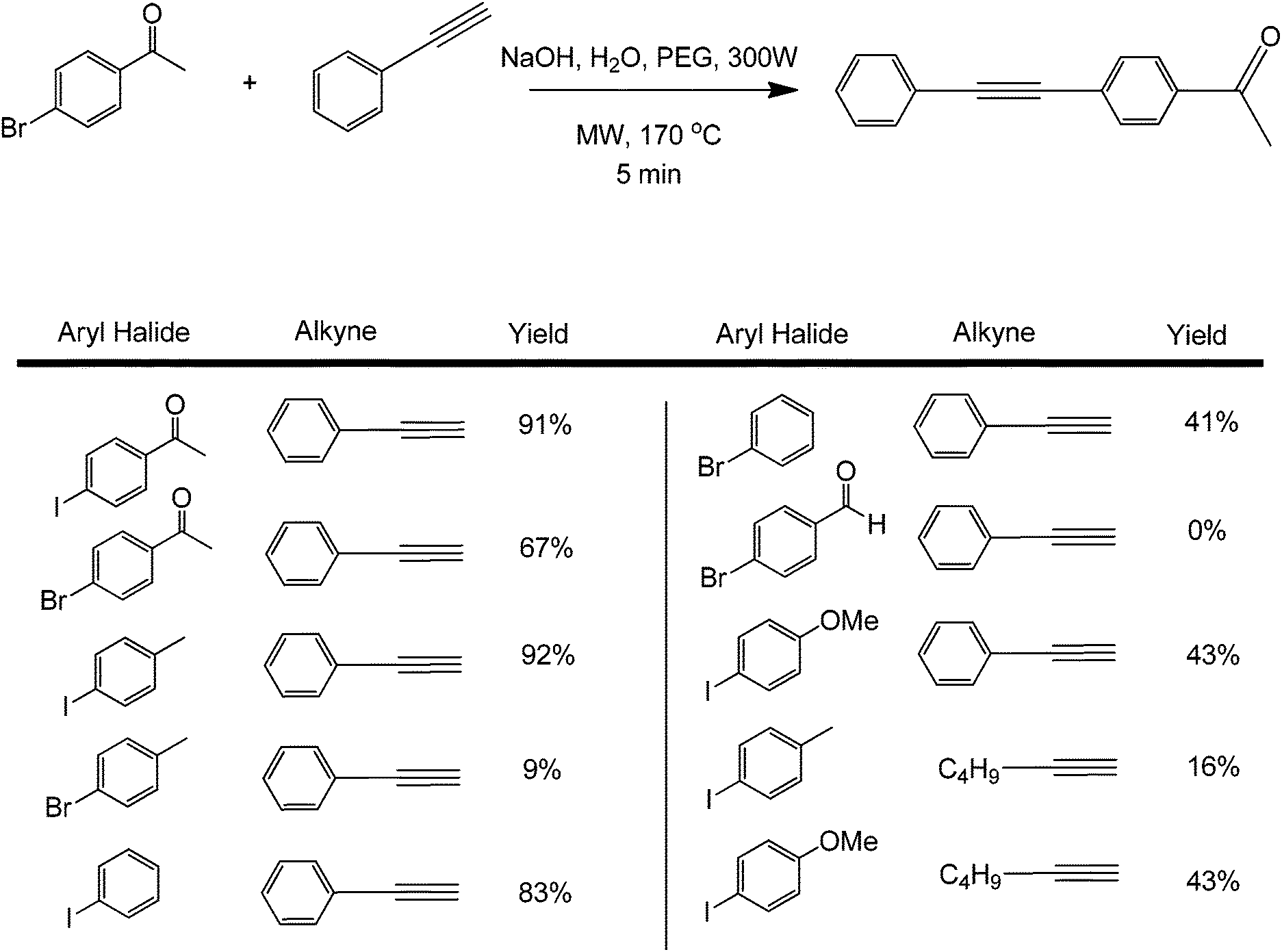 | ||
| Scheme 11 Transition metal-free coupling of 4-bromoacetophenone and phenylacetylene and its scope. | ||
An 85% conversion could be achieved by increasing the microwave power to 300 W and the reaction temperature to 170 °C. The quantity of PEG used was very important in the reaction. Small variations from the optimum PEG amount led to lower conversions, which is due to the lower solubility of the organic precursors in water. A range of coupling substrates were tested under optimum reaction conditions (Scheme 11). As expected, aryl iodides gave better product yields as compared to aryl bromides. The mechanism of the process is believed to involve a possible acid–base reaction of the alkyne with the base, which is subsequently followed by a nucleophilic aromatic substitution at the carbon bonded with the halide. With this mechanism, a better leaving group also means faster formation rate of the product.
When results obtained with phenylacetylene and 1-hexyne were compared, lower yields were achieved with the latter. The yield for the metal-free Sonogashira cross-coupling between 4-iodotoluene and phenylacetylene was over 90%, while the corresponding yield with the 1-hexyne reaction was under 50%. These results are in agreement with the literature, stating that phenylacetylenes are easier to couple than aliphatic alkynes in metal-free protocols;32,39 mainly because of the former's ability to stabilize the formed intermediates for nucleophilic aromatic substitution. No reaction (or very low yield) was found when chlorobenzene derivatives were tested for this coupling. Low yields were also found when p-methoxyhalobenzene derivatives were used as aryl halides. These results show that microwave heating can easily speed up the generally slow Sonogashira coupling by promoting deprotonation of the terminal proton of the alkyne. Under a similar reaction protocol, 2-bromonaphthalenes were used as aryl halide substrates with phenylacetylene. The aryl halide–acetylene ratio was optimized at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in the presence of a weak base in Na2CO3 and microwave heating for 15 minutes (Scheme 12).40
:1 in the presence of a weak base in Na2CO3 and microwave heating for 15 minutes (Scheme 12).40
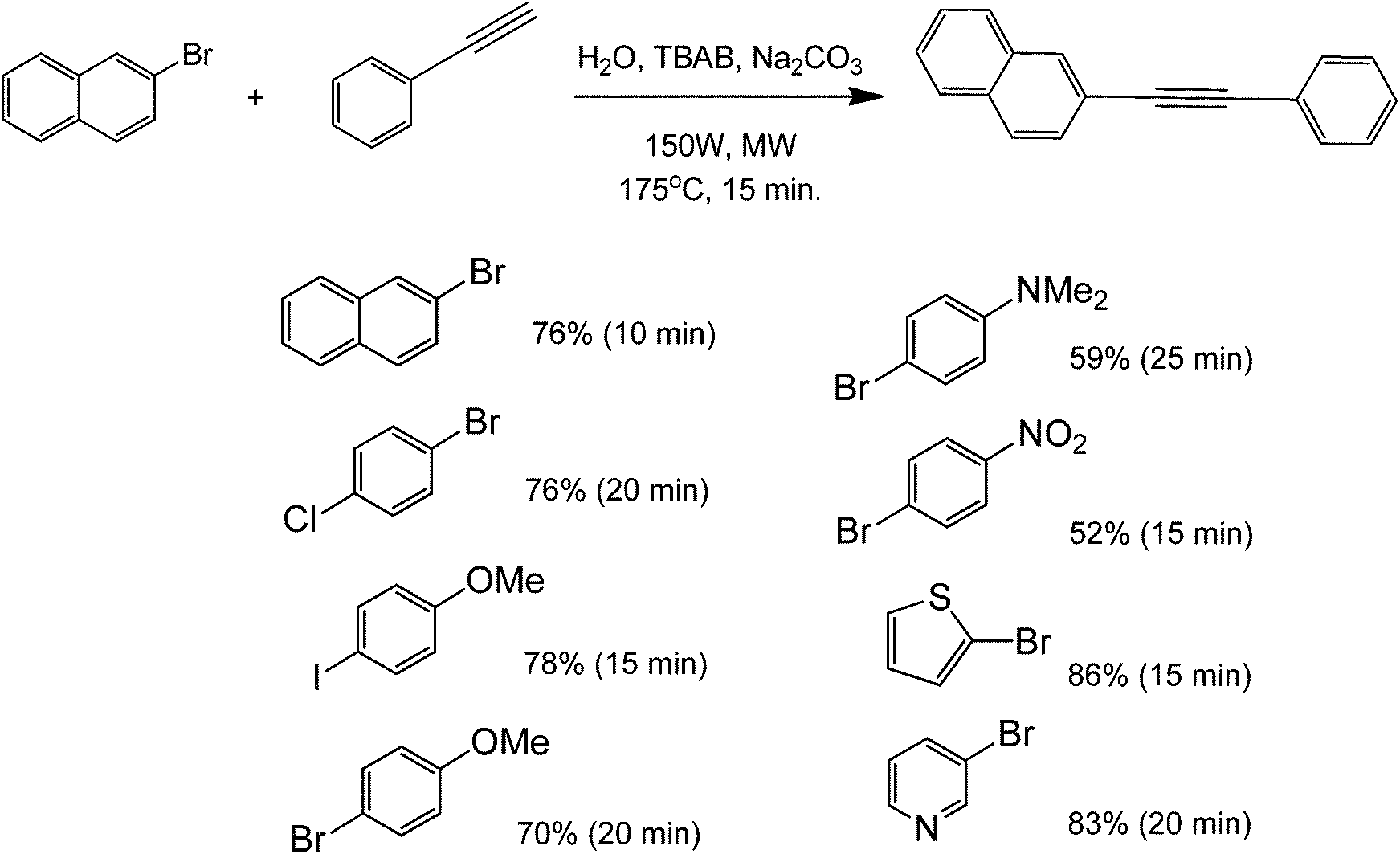 | ||
| Scheme 12 Microwave-promoted Sonogashira-type coupling of 2-bromonaphthalene and phenylacetylene in water and its reaction scope. The percentages reflect the reaction yield with the particular aryl halide while the times indicate the reaction time under microwave. | ||
In general, this strategy may be applied to the coupling of deactivated aryl halides. Heterocyclic thiophene and pyridine halides are also suitable substrates for this reaction with phenylacetylene. The partial reaction scope shows that the yield of the coupled product decreased with increasing electron-withdrawing ability of the para-substituent. Although the substrate scope (for aryl halides) is different for both microwave-mediated metal free coupling protocols previously mentioned, it is clear that substituents of aryl halides do play a major role in the promotion of better yields. Careful selection and optimization of the type of substituents can lead to the creation of a good yielding metal-free coupling method.
As in the Hamlet-kind of duality (to be or not to be metal-free), these examples set the basis for careful investigations on whether traces of any metal impurities could catalyse reported metal-free or even non-traditional metal coupling processes, which in some cases do not even guarantee a definite metal-free transformation.37 In any case, the next sections have been aimed to highlight key examples and advances on transition metal-free protocols of a range of coupling chemistries which have the potential to become the basis of future synthetic organic coupling protocols.
Reactivity of halide substrates with base-activated alkynes
In any case, a series of metal-free coupling protocols has also been more recently reported under a series of different chemistries. Bases also play important mediating roles in coupling processes. In many metal-free coupling protocols, bases have been shown to significantly affect a reaction yield by serving as a primary C–H activating agent. An example that illustrates this point is the use of 1,4-diazobicyclo(2.2.2)octane (DABCO) as both the base and the catalyst41 (Scheme 13).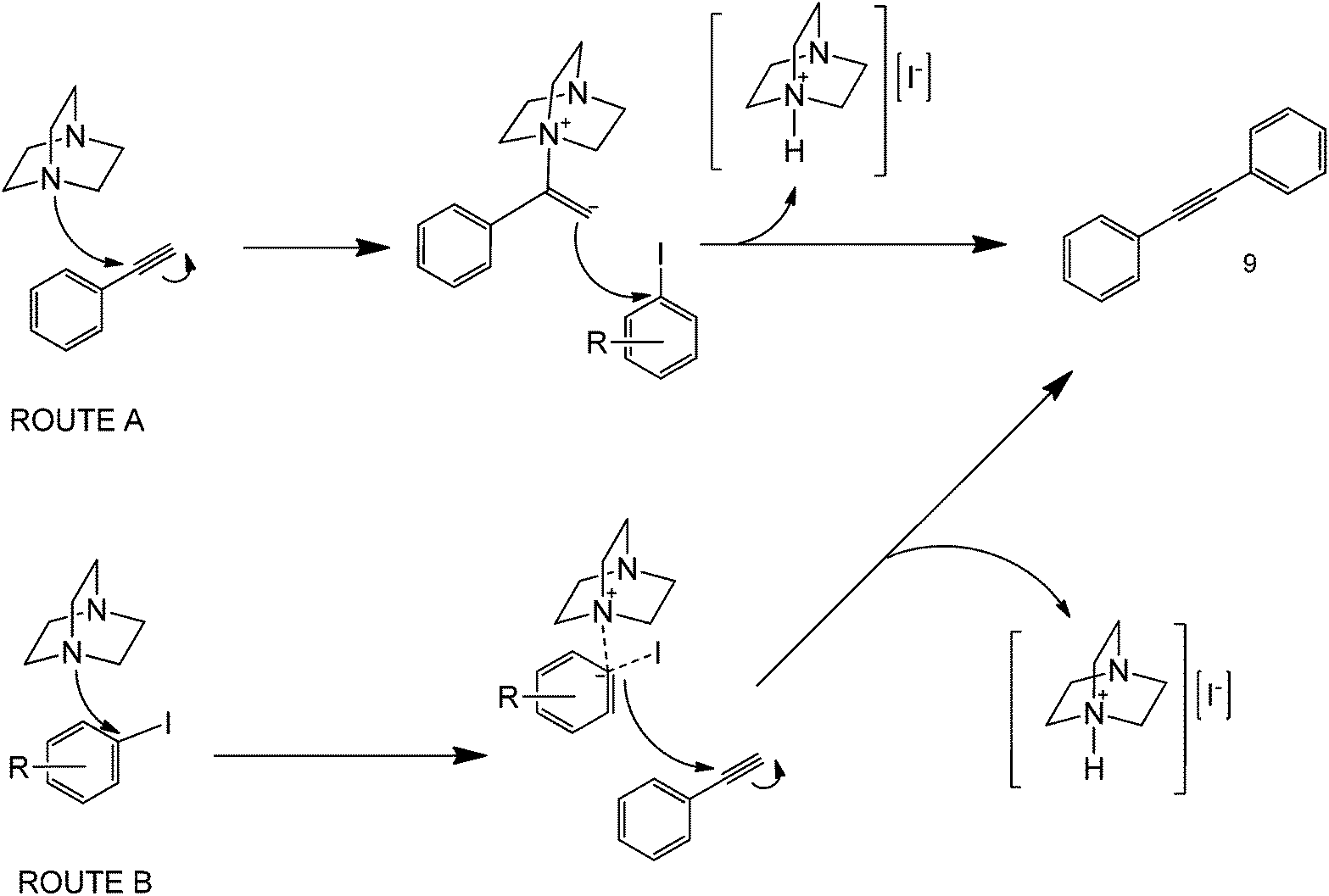 | ||
| Scheme 13 Proposed mechanism for the metal-free coupling of phenylacetylene and iodobenzene in the presence of a base (DABCO).41 | ||
The reaction had a very narrow scope of substrates (only iodoarenes and phenylacetylene worked in the protocol) and rather long reaction conditions (48+ h reaction, only improved under microwave irradiation in some cases) since the base used is highly sterically hindered. The proposed mechanism was suggested to involve a potential activation of the terminal carbon of the triple bond in phenylacetylene to promote a nucleophilic attack of this carbon to that iodide-bonded from iodobenzene to yield a range of cross-coupled products (Scheme 13, Route A). For this case, the steric environment of the base is a basis for the formation of the products, since the attack of the nitrogen group to the carbon of the alkyne will form a tetrahedral intermediate. The direct attack of a DABCO nitrogen to the ipso carbon of iodobenzene as the first step is rather unfeasible due to geometry considerations (Scheme 13, Route B). Microwave heating could clearly promote the coupling reaction in various ways, either by increasing the reactants’ solubility in water, or increasing the collision frequency of the basic activating agent and the alkyne. To address this, the strength of amines could be altered using different alkyl groups, but this very same concept can also increase the steric hindrance of the molecule, that easily limits the deprotonation in the first step.
As such, the general recommendation is to come up with an optimized base to alkyne ratio to balance the two limiting factors. The formation of coupling products also depends on the leaving group ability of the halide used; although this is generally true, the real dependence lies on the compatibility of the base catalyst to the reaction conditions brought about by the presence of both an aryl halide and an alkyne in the reaction mixture. In this particular example, the authors double checked the presence of Pd or other metal impurities in the substrates that were not detected above 0.5 ppm. However, to ensure that this protocol is indeed metal-free, it is necessary to analyse the Pd concentration with a more sensitive analytical technique.
As with classical coupling reactions, alkyl halides are primary choices of coupling partners because of the polarity of the C–X bond, with halogens being very electronegative atoms. Because of this, C–X is very susceptible towards insertion and nucleophilic attack. Interestingly, the ability of halogens to act as reactive substrates in the metal-free synthesis of symmetrical 1,3-diyne compounds in the absence of base and oxidant was recently demonstrated (Scheme 14).42 The method showed excellent functional group compatibility and high yields using dimethylformamide (DMF) as the only solvent. The homocoupling of 1-iodoalkynes was found to be more effective for the synthesis of symmetrical 1,3-diynes, as compared to 1-bromoalkynes. The mechanism of the proposed reaction involved the substitution of the C–Br bond with an iodine. This promoted the formation of a high energy radical that spontaneously coupled to form a symmetric product. This reaction is of particular interest because of its simplicity and potential applicability. Although not stated in the original work, the formation of the alkyne radical might have been due to the interaction of the solvent with the highly polar C–X bond. However, it is important to note that the starting reagent, KI, might have been contaminated with another metal even if it was directly from a fresh reagent bottle. Also, no evidence was presented by the paper itself that this protocol is completely metal-free.
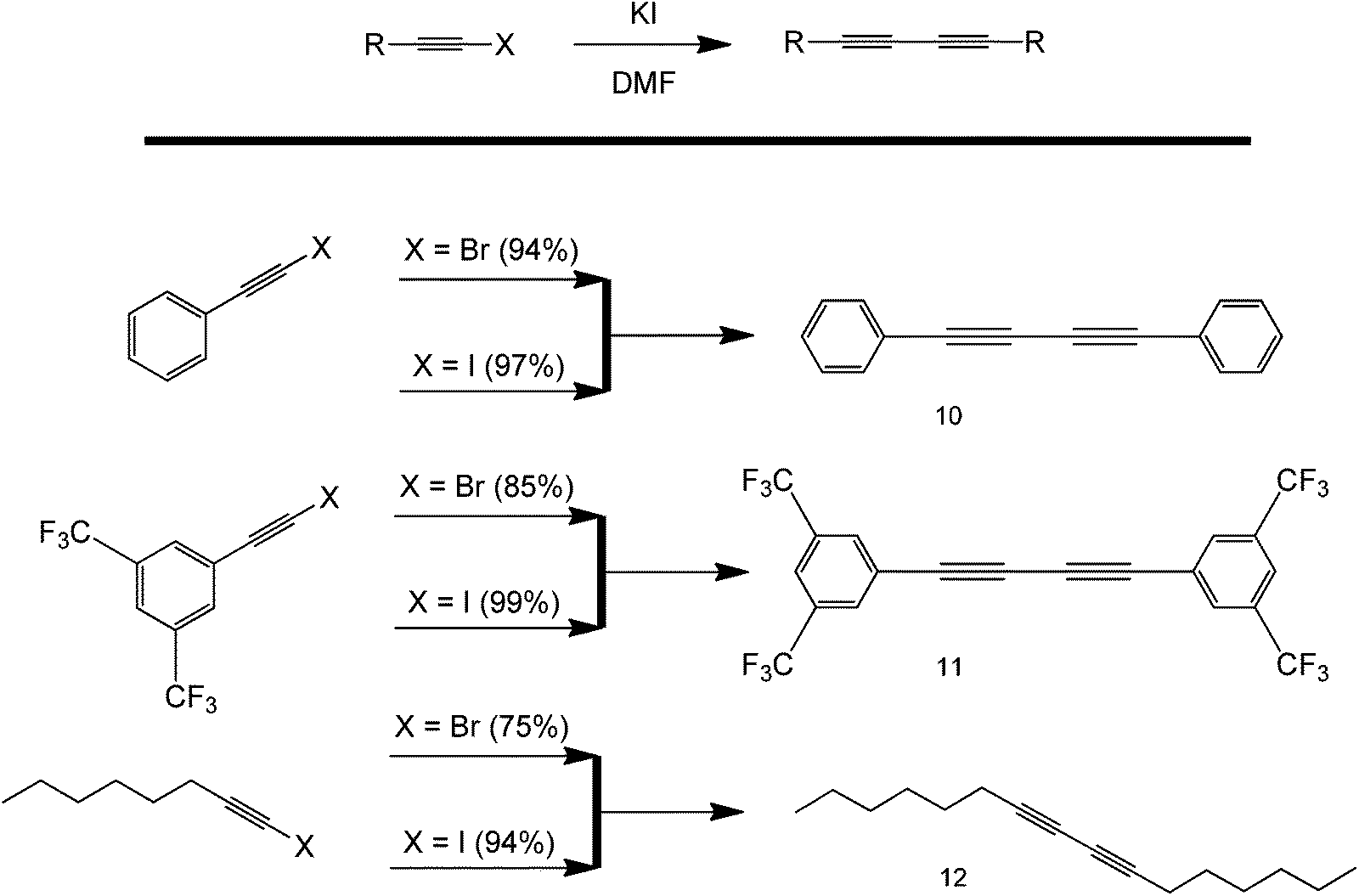 | ||
| Scheme 14 Reaction scope of a metal-free coupling protocol using only KI and solvent DMF. | ||
A similar coupling reaction was also carried out in the presence of a very strong base such as tBuOK (Scheme 15), which acts as an activator for the alkyne.43 Originally, this Glaeser-type coupling was performed using copper, as C–H can easily add in an oxidative fashion to this metal and the symmetric product is formed in the presence of oxygen. However, studies reported by Itami and co-workers pointed out that the same activation can be carried-out using a simple proton abstraction by a base, to make the entire process greener.44 Their work focused on the use of tBuOK in the coupling of various deactivated heterocycles and haloarenes.45 A very strong base (tBuOK) has also been shown to mediate the coupling of unactivated arenes using nitrogenous organocatalysts, such as 1,10-phenanthroline46,47 and N,N′-dimethylethane-1,2-diamine.48
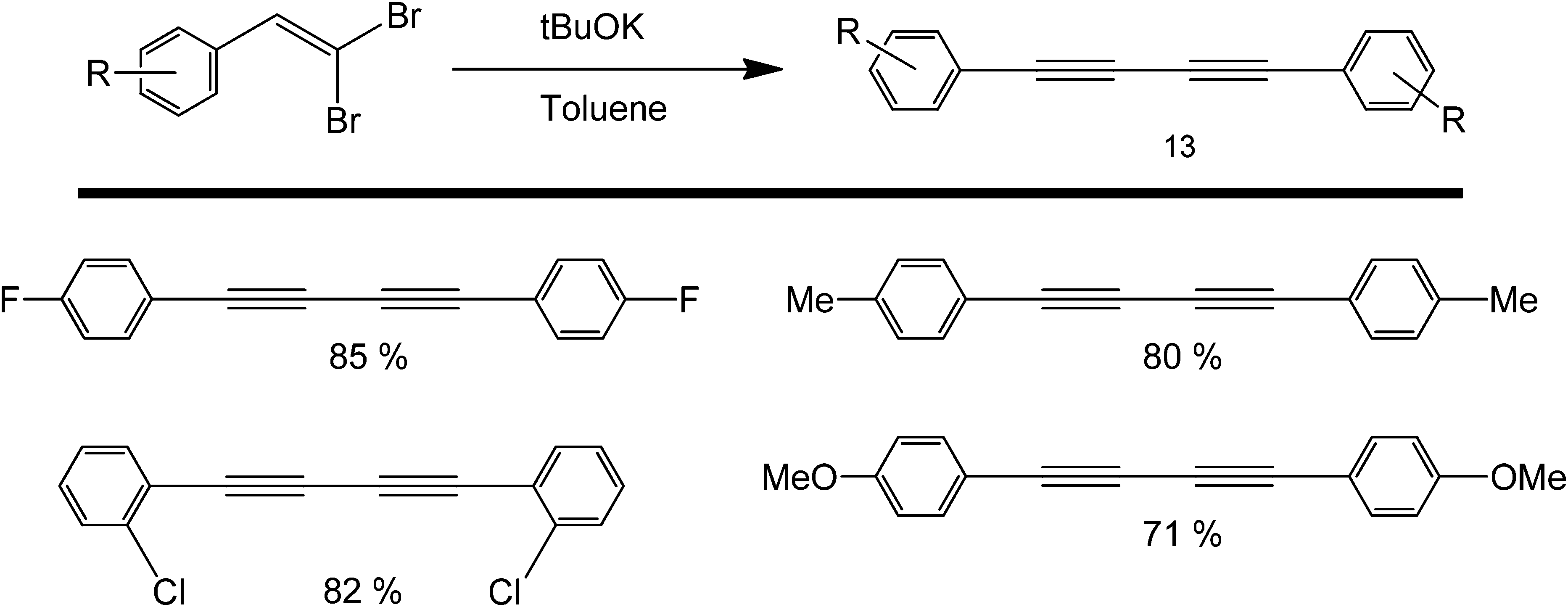 | ||
| Scheme 15 Transition metal-free homocoupling of 1-haloalkynes using a sterically hindered base in tBuOK. | ||
One drawback of the protocol is that tBuOK is a very sterically hindered, strong base, thereby limiting its possible use as an activating agent of sterically demanding alkynes. These studies were in good agreement with the previous assertion that electron withdrawing groups can increase the yield of a metal-free coupling reaction.
Palladium-catalyzed symmetrical couplings have been reported for structures 10, 11, 12 and 13. Structure 9 in particular has been synthesized using a Pd(0) catalyst in the presence of an amidine base, benzene solvent, and ethynyltrimethylsilane.49 The role of the base in this reaction follows the same principle to that proposed for DABCO in the presented metal-free coupling – that is as a proton shuttle. On the other hand, homocoupling products similar to structures 11, 12, and 13 have been synthesized using a base and Pd(OAc)2.50 Both Pd-catalyzed and metal-free couplings mentioned thus far used organic bases in the reaction which could lead to some important questions on the ability and role of these bases to drive the coupling reaction forwards in the presence and absence of a metal.
Oxidative C–C and C–heteroatom couplings
Oxidative cross-coupling reactions take place when the carbon couples with a more electronegative atom, such as oxygen and nitrogen in the presence of an oxidizing agent or when C–H bonds are converted to C–C. The choice of the oxidant should be carefully made to establish control of the reaction and not affect the other functional groups present in the starting substrate. One common oxidant used for cross-coupling is dicyanobenzoquinone (DDQ) which has been reported as an efficient oxidizing agent for the oxidative coupling of benzylic and allylic sp2 C–H bonds and with active methylenic sp3 C–H bonds under metal-free conditions.51 Although, there has been extensive research on sp3 carbon activation, metal-free counterparts have remained underexplored due to their obvious difficulty – that is the stable nature of the bond.52The reaction was shown to proceed smoothly for various 1,3-dicarbonyl compounds with a range of benzylic and allylic substrates in good to excellent yields. Since the method is mainly microwave mediated, no metal is required for the reaction and it can proceed without any additional reagent. Also, simple benzylic and allylic substrates can be used directly without pre-functionalisation or pre-activation. Scheme 16 shows an example of this reaction under relatively clean conditions using only the oxidant and a solvent.53 This novel direct cross-dehydrogenative-coupling (CDC) between allylic compounds and oximes provides a highly efficient, fast and convenient approach to synthesise oxime ethers.
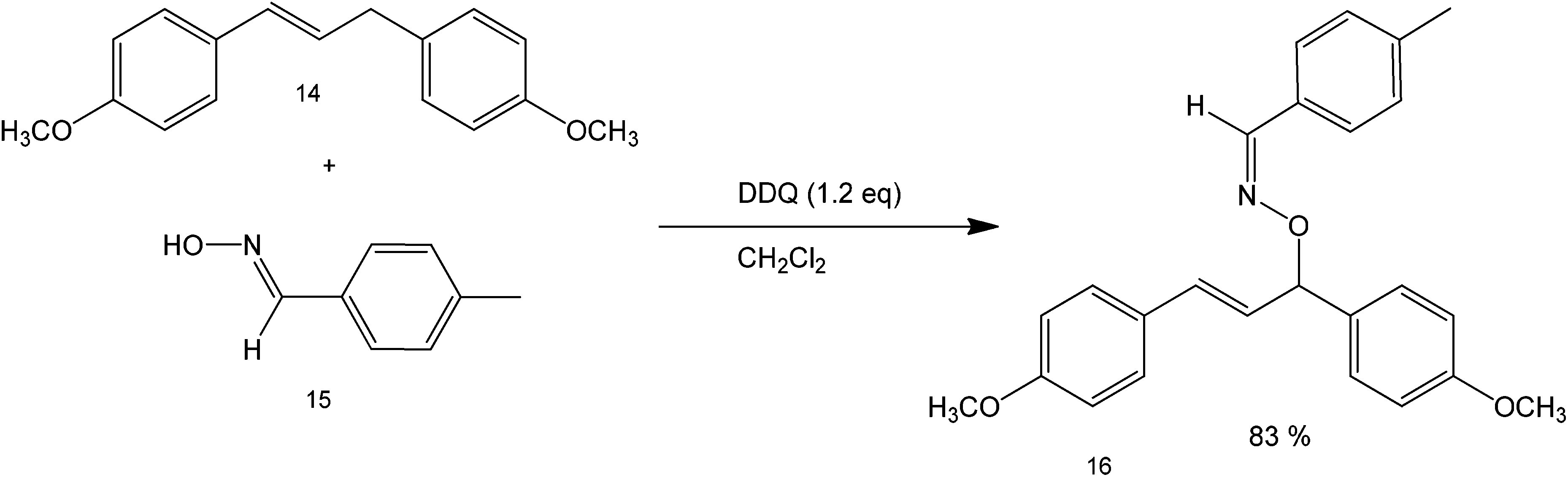 | ||
| Scheme 16 Coupling reaction between (E)-4-methylbenzaldehyde oxime (structure 15) and (E)-4,4′-(prop-1-ene-1,3-diyl)bis(methoxybenzene) (structure 14) in the presence of 1.2 eq. of DDQ and methylene chloride as solvent. | ||
The mechanism of the reaction starts with a single electron transfer initiated by the DDQ reagent to produce a biphenyl radical which undergoes another electron transfer to form a carbocation (Scheme 17), that is the interaction between structure 17 and 18. DDQ comparably forms a stable hexa-substituted aromatic benzene that undergoes another acid–base reaction with the oxime to form an anionic species that is reactive towards the biphenyl carbocation.
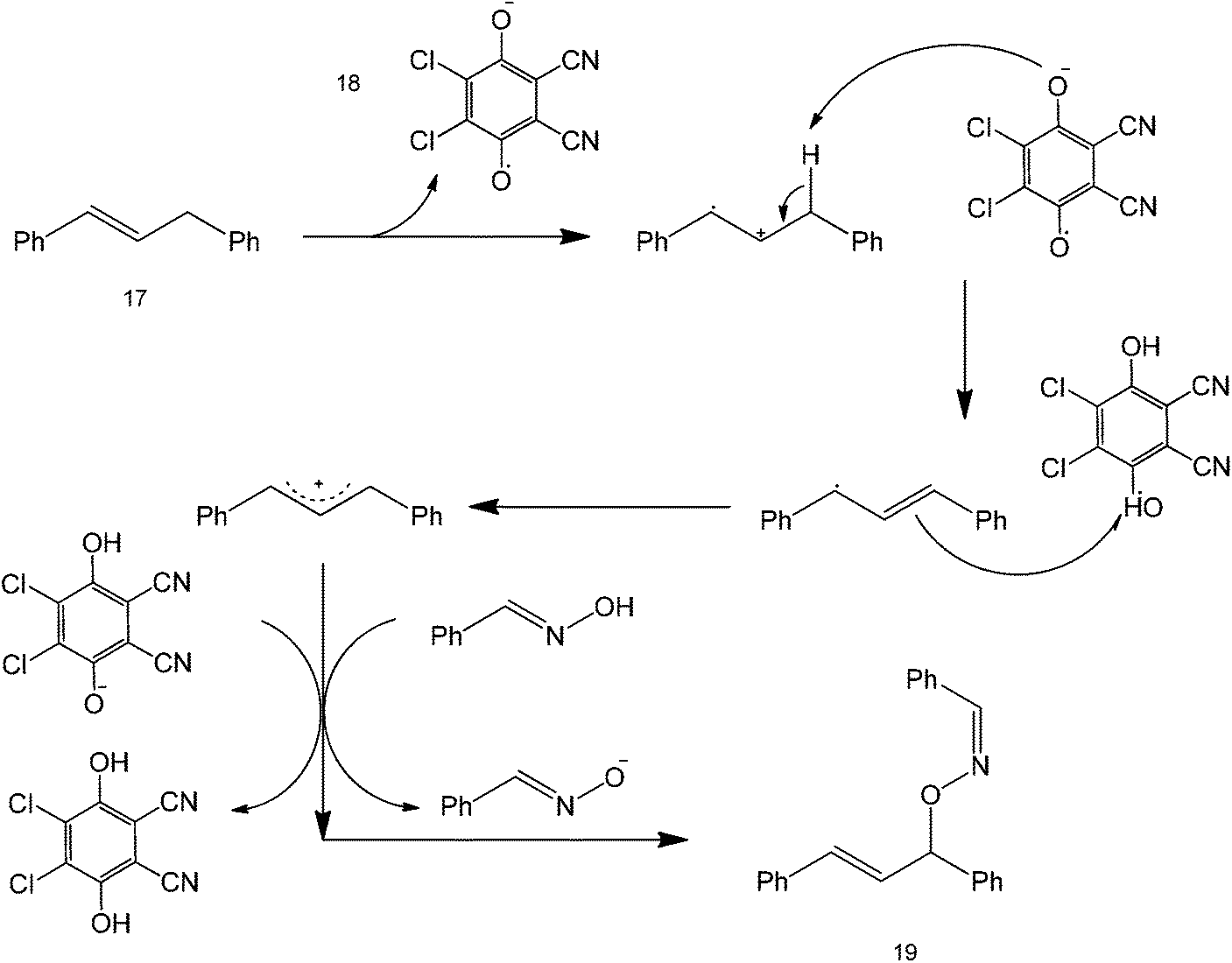 | ||
| Scheme 17 Metal-free synthesis of oxime ethers using DDQ via a radical mechanism. | ||
Although not shown here, the mechanism implies that reaction yields can be increased by functionalizing the starting oxime structure with electron-withdrawing groups, as they could stabilize the oxygen anion formed after DDQ reduction. Coupling of the oxime anion and the biphenyl carbocation forms structure 19.
A similar strategy can also be used to synthesize diarylallyl ethers or thioethers.54 Diarylallyls can be coupled with an alcohol or with a thioester to produce an ether product (Scheme 18). The coupling yield increased with increasing electron-withdrawing ability of the alcohol (thiol). A study on the scope of the reaction was also presented by varying the substituents around the diarylallyl (structure 20). The increase in yield could be attributed to the increasing electron withdrawing ability of R1 and R2 (as they have the ability to stabilize an otherwise unstable radical intermediate). This protocol has been shown to work in diketone couplings (coupling happens at the most reactive alpha carbon) and diarylallyls.55
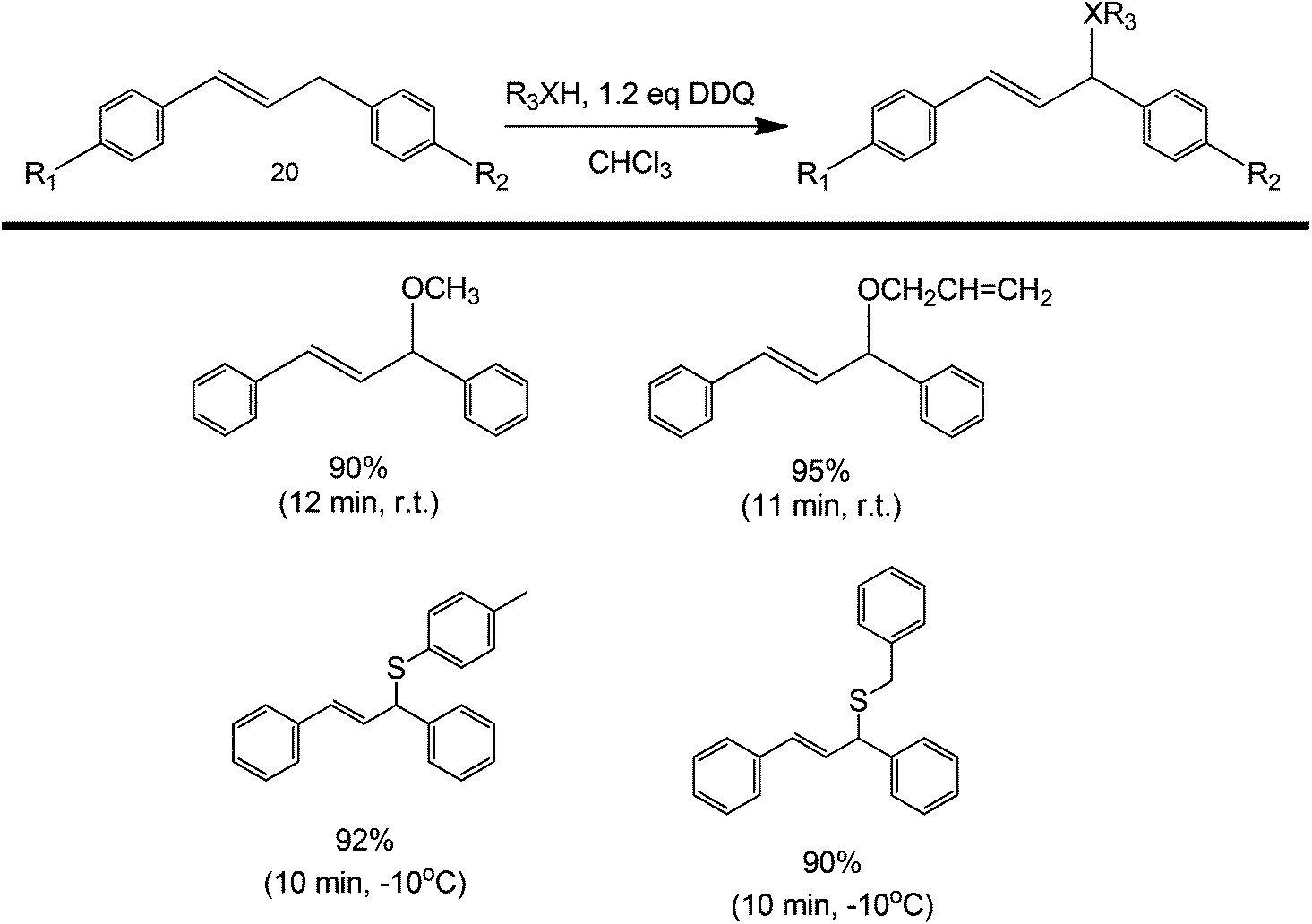 | ||
| Scheme 18 Metal-free diarylallyl ether/thioether formation via oxidative C–H activation. | ||
A different approach for oxidative metal-free couplings aside from using DDQ is through a tosylhydrazone pathway.56 This recently developed method for the synthesis of ethers involves the formation of a reactive hydrazone intermediate between an amine and a ketone. This metal-free C–O bond-forming reaction proceeds through a base-promoted decomposition of tosylhydrazones in the presence of phenols or alcohols, and results in the insertion reaction of the carbene into the O–H bond (Scheme 19). Notably, the starting materials are readily available and stable. Furthermore, the operational simplicity of the process and the high functional group tolerance are remarkable.
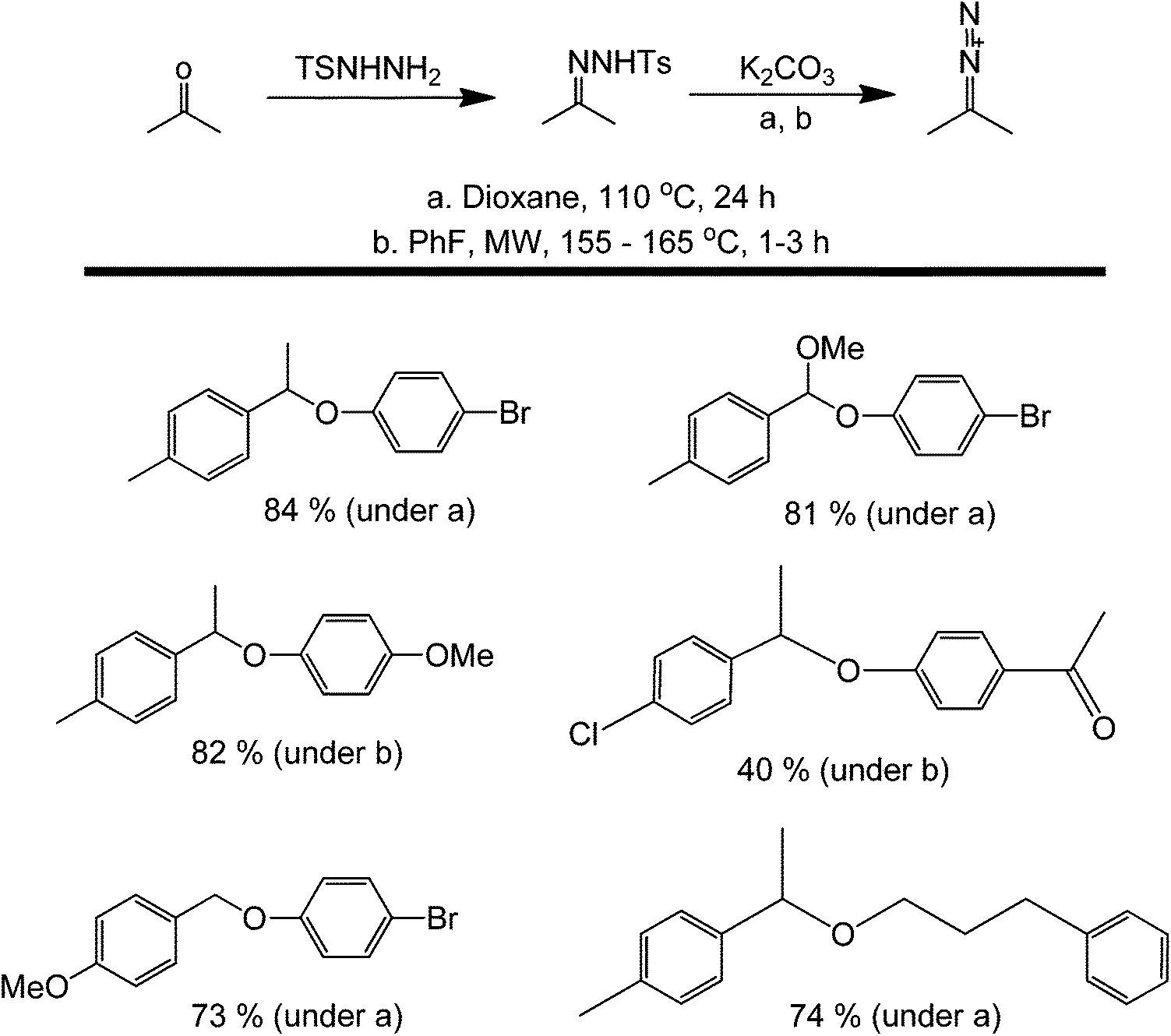 | ||
| Scheme 19 Metal-free reductive etherification of carbonyl compounds. | ||
Organocatalytic properties of tosylhydrazones for C–C and C–O bond forming reactions has long been demonstrated. However, these compounds have recently attracted a great deal of attention because they can also perform a metal-free coupling reaction with a range of starting substrates.57 A metal-free method using boronic acids and N-tosylhydrazones has been reported by Barluenga et al. for C–C bond formation58 (Scheme 20). From a synthetic point of view, this transformation represents an extremely simple way to carry out the reductive arylation or alkylation of carbonyl compounds, a task that otherwise requires several synthetic steps. Moreover, it was not necessary to isolate the tosylhydrazone formed as an intermediate and the reaction could be conducted in one pot directly from carbonyl compounds (Scheme 21 for mechanism). The scope of the reaction could also be extended to the use of alkenyl boronic acids or unsaturated tosylhydrazones. In these cases, mixtures of regioisomeric alkenes are obtained. The reported protocol is a novel, unusually simple and a general procedure to manipulate carbonyl compounds creating an intermolecular C–C linkage. For these reasons, we believe that this reaction meets the requirements to become a widely used key transformation in future organic synthetic protocols.
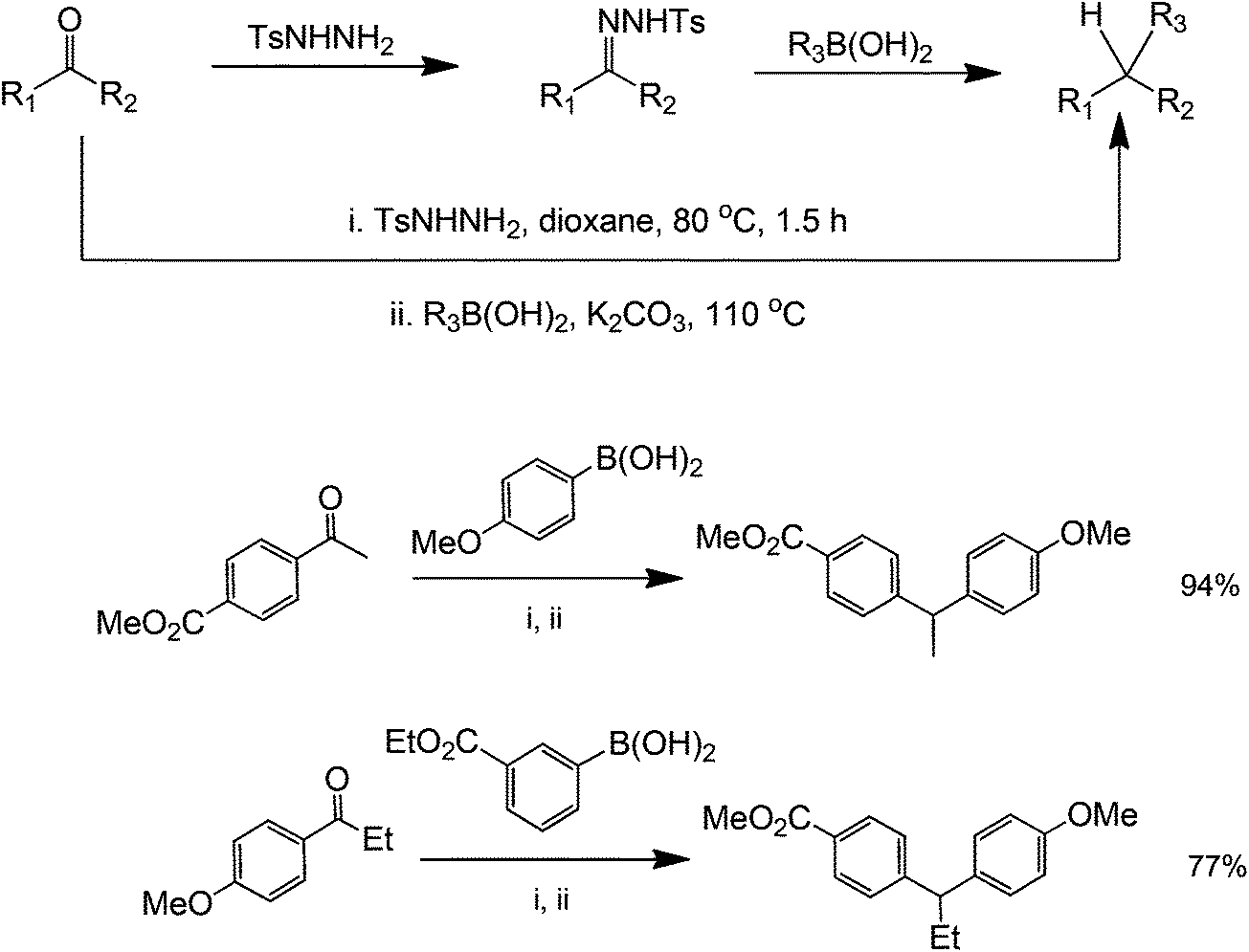 | ||
| Scheme 20 Boronic acid-mediated coupling of ketones and aryl groups. | ||
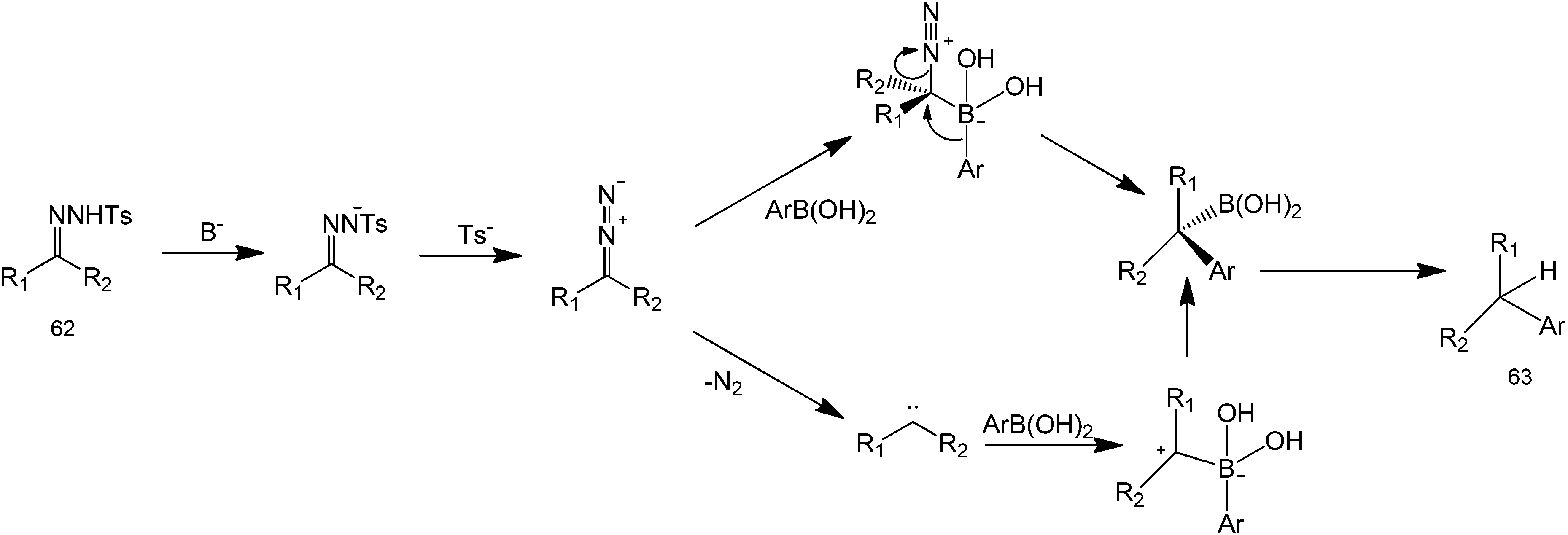 | ||
| Scheme 21 Proposed mechanism of the coupling of ketones and aryl groups using tosylhydrazone and boronic acid. | ||
Another promising C–C forming reaction involves the direct coupling of an aldehyde with a heterocyclic compound (Scheme 22). This transformation is particularly interesting owing to the potential to directly attach a carbonyl group to the C2 of an aromatic nitrogen compound. In the presence of a mild oxidant (e.g. (bis(trifluoroacetoxy)iodo)benzene (PhI(OCOCF3)2) and sodium azide), this reaction is able to tolerate a wide range of alkyl and aromatic aldehydes as well as monocyclic and polycyclic N-heterocycles. The reaction is believed to occur via a reaction cascade which starts with the formation of trifluoroacetic acid and the subsequent generation of the azide and trifluoroacetoxyiodobenzene radicals. Trifluoroacetic acid activates the N-heterocycle via protonation while the azide radical will form an aldehyde radical. The activated aldehyde and N-heterocycle will couple and subsequently react with the generated trifluoroacetoxybenzene radical to form another molecule of trifluoroacetic acid, iodobenzene, and the coupled product (Scheme 23).59
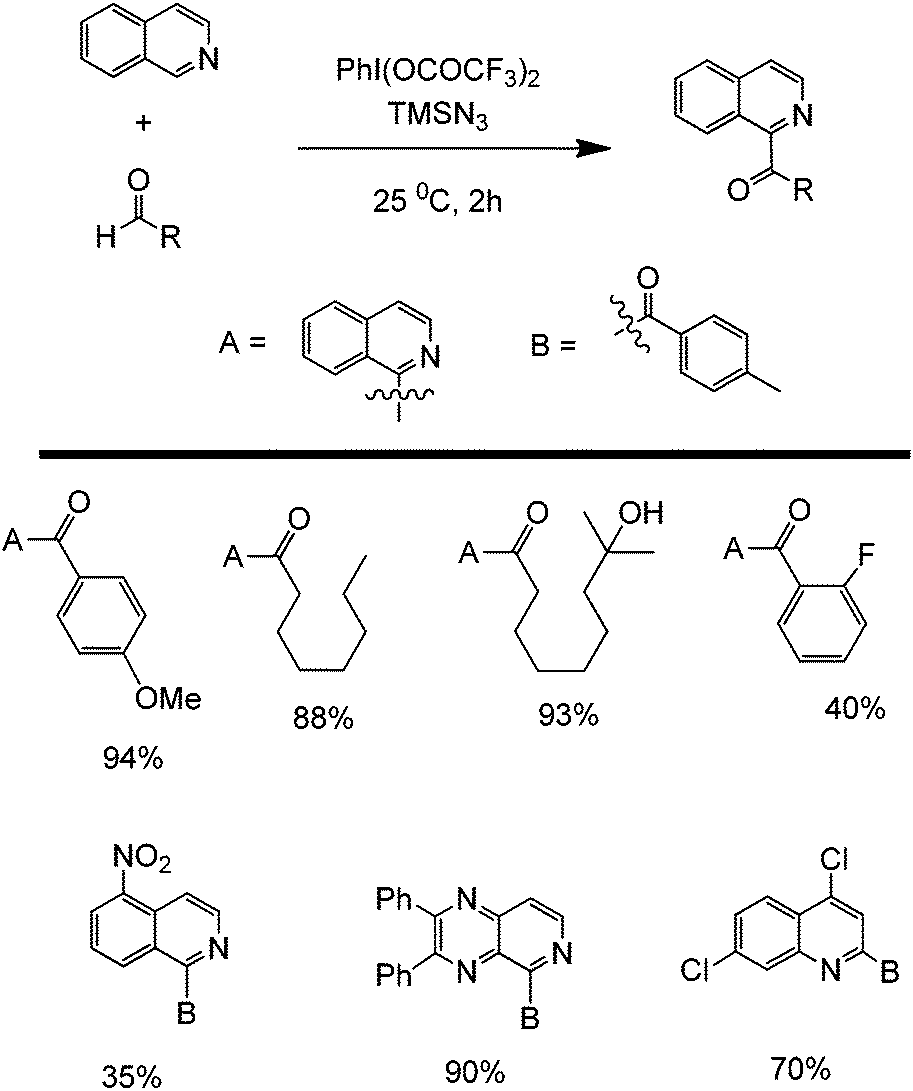 | ||
| Scheme 22 Partial scope of the cross-dehydrogenative coupling of N-heterocycles and aldehydes. | ||
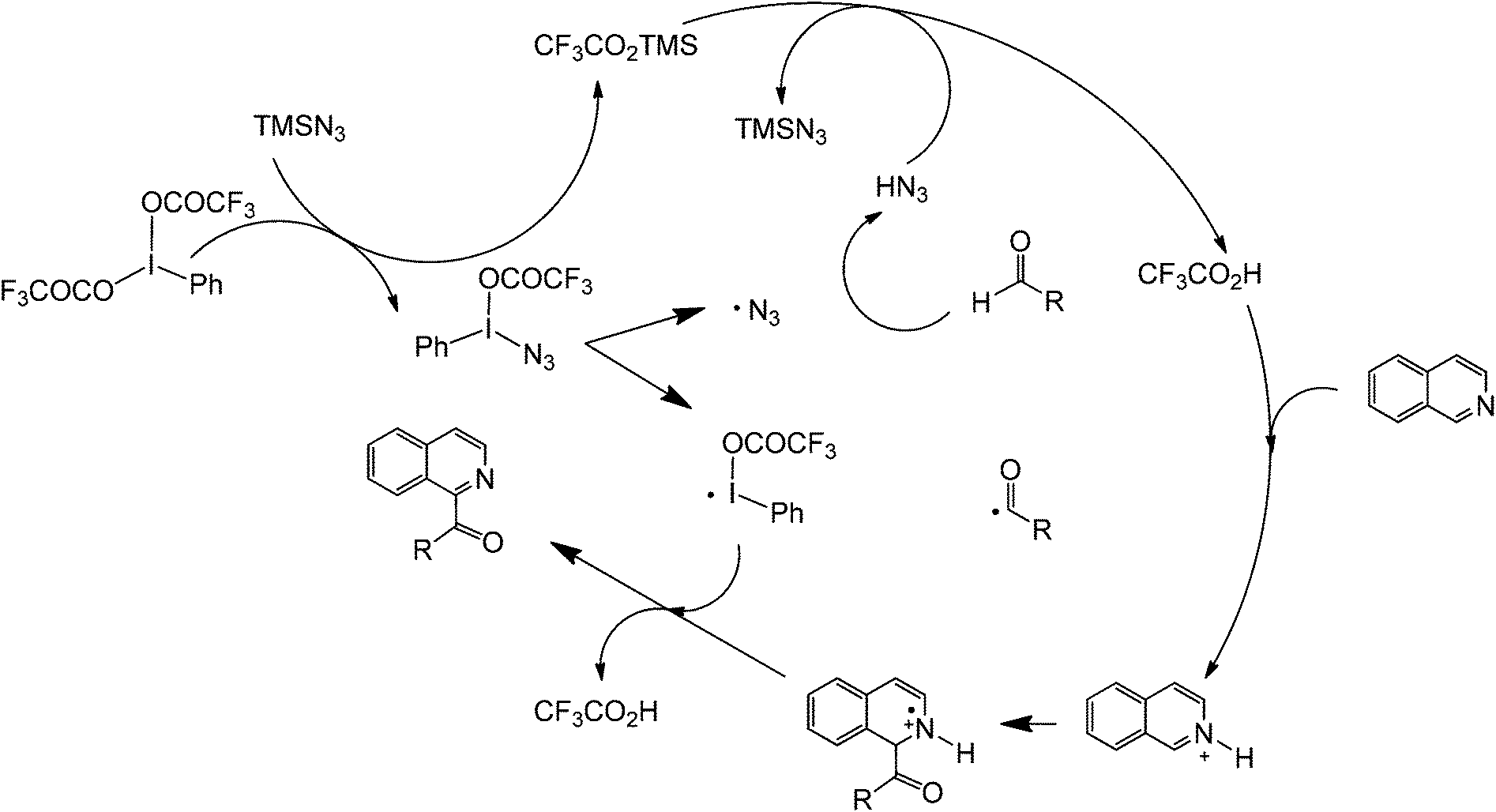 | ||
| Scheme 23 Proposed mechanism of the cross-dehydrogenative metal-free coupling of N-heterocycles and aldehydes. | ||
Aside from being used as a leaving group in tBuOK-mediated reactions, iodine's hypervalency has also been shown to be advantageous in metal-free coupling chemistries. Hypervalent iodine reactions can be used to carry out oxidative cross coupling reactions of heterocyclic compounds (Scheme 24).60,61 This protocol is a simple and efficient approach to the clean synthesis of polymeric 3-substituted thiophenes and pyrroles. The iodine reagent used in the study has a tetrahedral core that has the capability to bond with more carbons. The yield of the reaction was found to increase depending on the substituent, that is, the more electron-donating the substituent, the higher the yield – a departure from base catalyzed metal-free couplings where product yields generally increase with improved electron-withdrawing abilities of substituents. One important advantage of iodine as a catalyst is its recyclability by means of methanol precipitation, as well as the possibility to direct the synthesis to specific sites and specific orientations (regioselectivity and stereoselectivity).
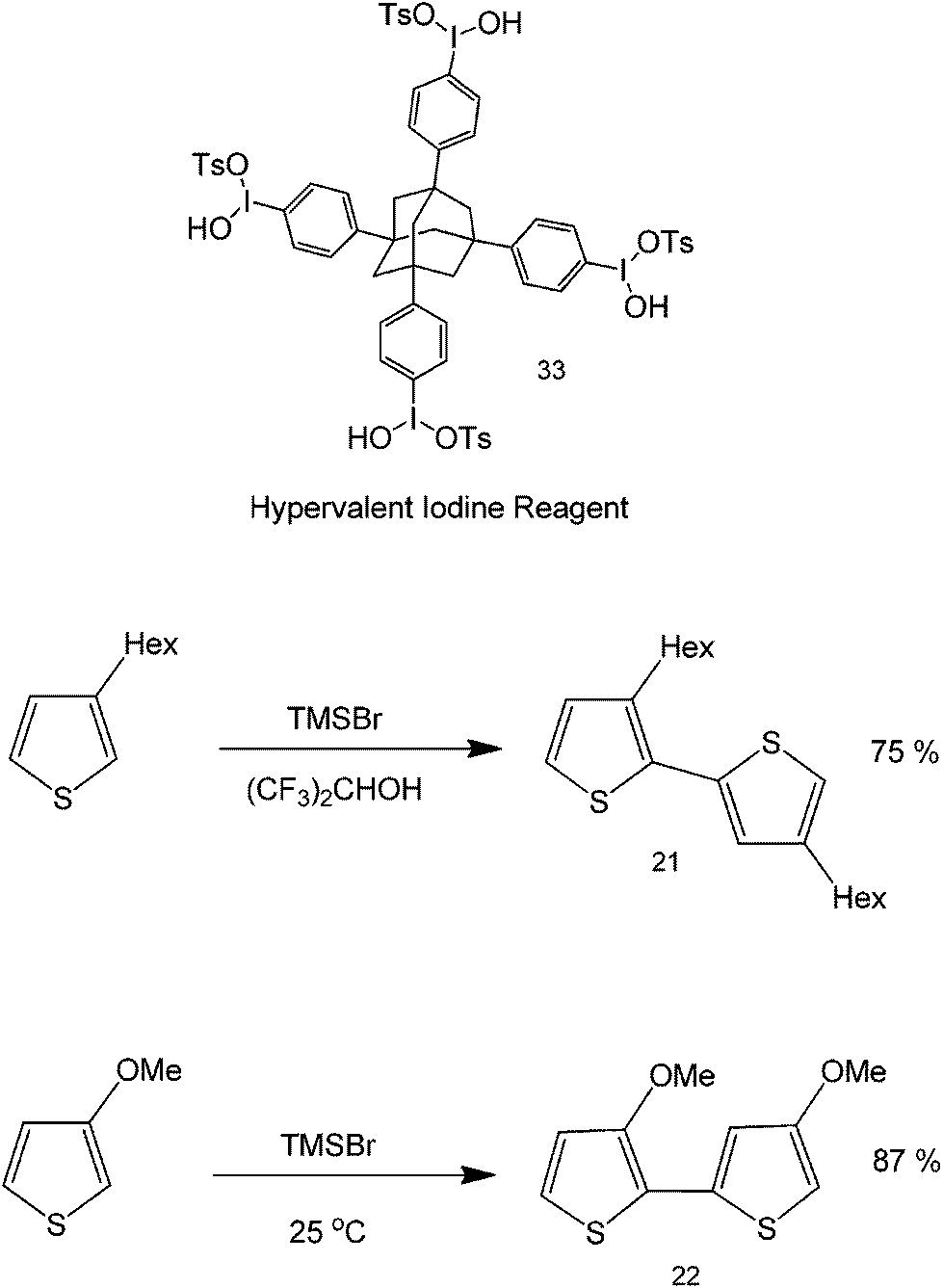 | ||
| Scheme 24 Coupling reactions with an iodine reagent as a catalyst. The synthesis of a H–T methoxy substituted polythiophene was performed with hypervalent iodine and an organic solvent. | ||
Commonly, this coupling can be accomplished via functionalization of thiophene at C2 with a halide and then 2-halothiophene is used to produce a Grignard reagent, which in the presence of Ni(dppe)Cl2 will form a head-to-tail polythiophene polymer.62
As seen in Scheme 24, thiophene polymerization can be differently accomplished depending on the functionality within the C3 of the heterocycle. In the presence of a weak electron donating bulky functional group, the coupling takes place via head-to-tail (H–T) conformation but with alternating orientations of the alkyl groups (structure 21). Using a stronger electron donating group (such as a methoxy group), the H–T coupling can be directed in such a way that methoxy groups can stay to the same side of the polymer (structure 22). The synthesis of a highly ordered polythiophene is very important since these functionalities play important roles in natural products and organic materials because of their interesting properties. Polystyrene, like polythiophene, has also been prepared using a slightly similar process using silane radical atom abstraction.63 The metal-free coupling of heterocyclic compounds, including their transition-metal catalyzed counterparts, have paved the way to the direct synthesis of highly ordered heterocycles that find relevance in major areas of medicine and materials science.64
Most coupling reactions are so far accomplished using a direct one-pot approach. Interestingly, Pruger et al. reported the indirect synthesis of a Sonogashira-type coupling product using a reactive sulfone intermediate.65 These couplings are based on the nucleophilic aromatic substitution of beta-carbonyl sulfones (structure 24) with a partially deactivated fluorobenzene (structure 23) to either produce aromatic alkynes or alpha-aryl carbonyl compounds (Scheme 25). The study was explored under both basic and acidic conditions to ascertain the reaction mechanism. The sulfone intermediate was found to readily cyclize with the nearby nitro group to produce a stable ketone (structure 25) which could drive the reaction forward (upon base activation) by forming a very stable SO2, the coupling product (structure 26), and an amide side product.
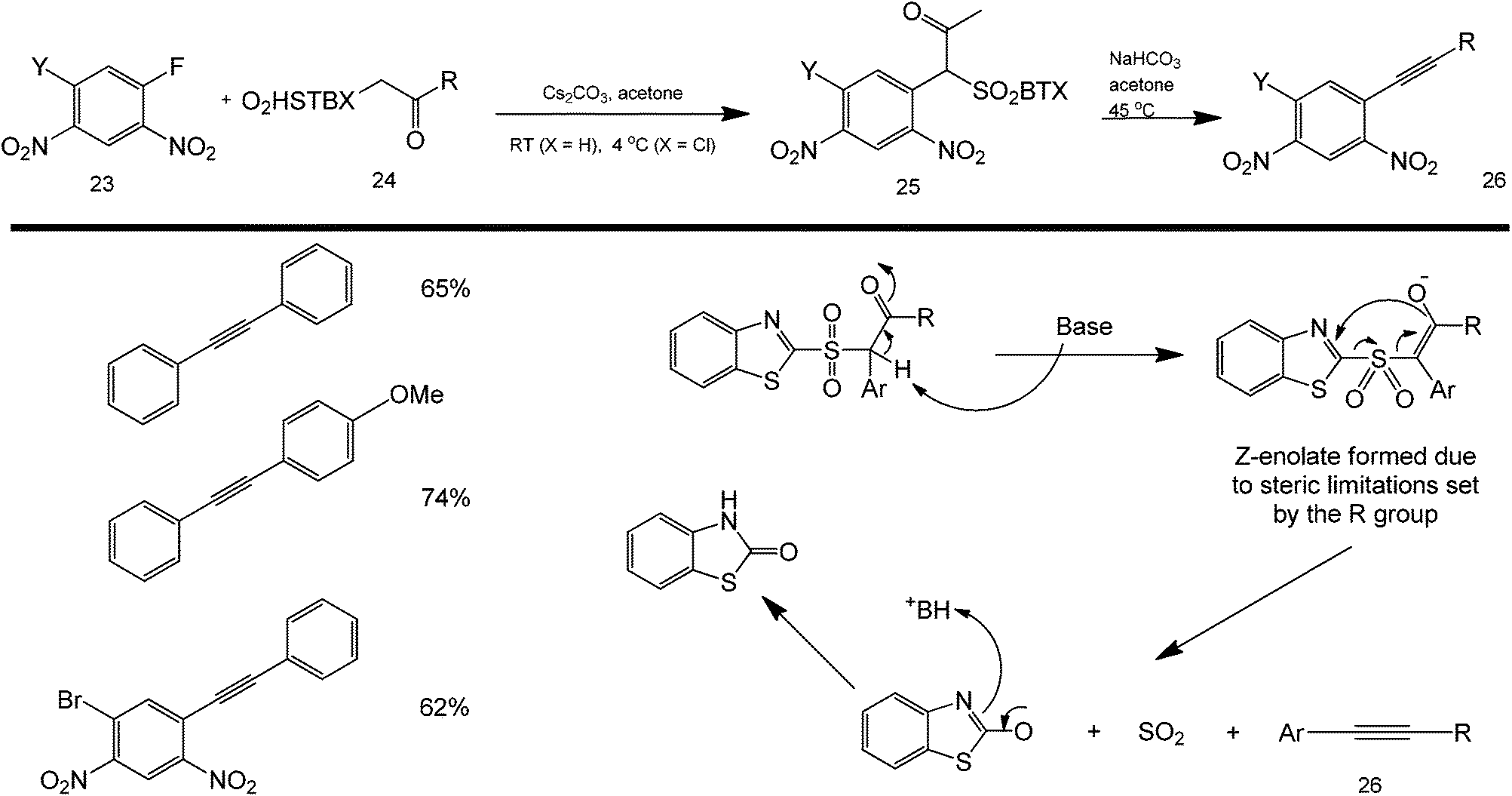 | ||
| Scheme 25 Scope of the reaction between fluorobenzene and sulfonyl ketones in a two-step coupling of aromatic and alkyne groups. | ||
The scope of these reactions is demonstrated by the synthesis of Sonogashira and alpha-carbonyl arylated products from a range of electron-deficient aryl fluorides with a variety of functional groups and aryl-, heteroaryl-, alkyl- and alkoxy-substituted sulfone nucleophiles – clearly demonstrating the remarkable synthetic utility of this reaction.
Another coupling of interest mainly in drug synthesis is the direct C–N coupling. This synthetic challenge can be performed using a route presented by Poirier et al.66 involving a metal-free amination of 2- and 3-haloindoles to form novel N-linked 2-or 3-(azo-1-yl) indoles via a metal-free and solvent-free acid-catalysed process (Scheme 26). On first inspection, the reaction can be inferred to proceed via a simple nucleophilic aromatic substitution. However, based on preliminary studies, the product was not isolated with increasing basicity of the nucleophile. The initiation of the reaction is therefore believed to proceed via formation of an acidic halide product that donates a proton to form a more reactive HN+ intermediate. The C–Cl carbon then becomes more susceptible towards nucleophilic attack of the heterocyclic amine. A similar coupling reaction can also be performed using nitrogen heterocycles and alkanes providing direct carbon activation of an otherwise unreactive alkane (Scheme 27).67
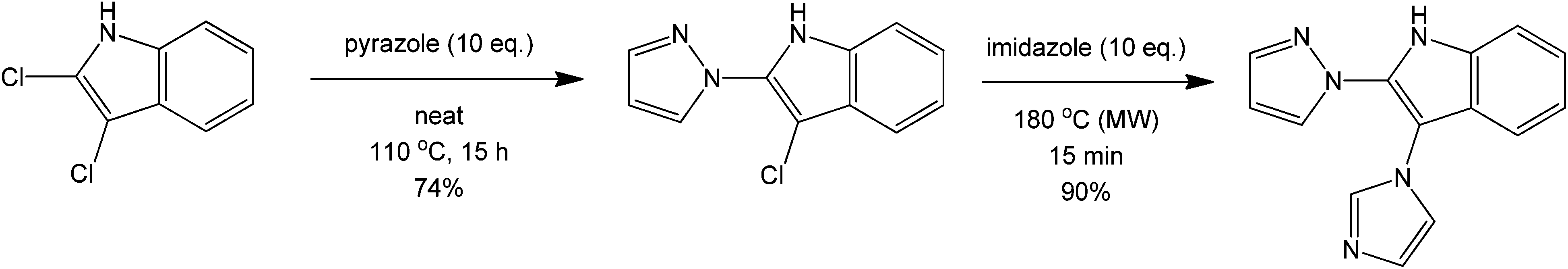 | ||
| Scheme 26 Reaction scheme for the direct synthesis of aminated indoles using a haloindole precursor. | ||
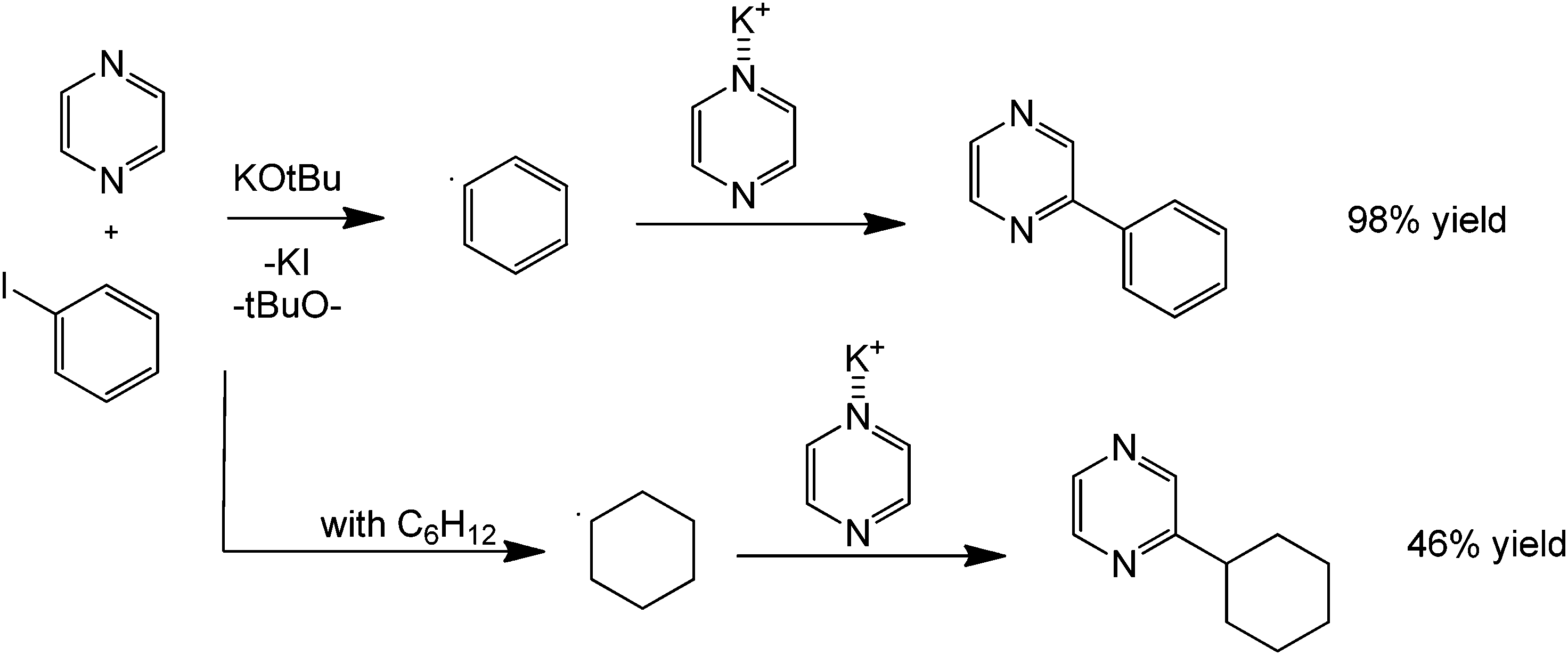 | ||
| Scheme 27 Reaction scheme for the direct alkylation of a heterocyclic amine with an alkane (cyclohexane) in the presence of a base and potassium iodide. | ||
Direct functionalization of amines with an aryl compound is a challenging synthesis. The nitrogenous substrate itself, as previously mentioned, could act as a basic activating agent in many metal-free homocouplings. However, it has been shown that even the direct coupling of carbon and nitrogen can be carried-out in a clean and efficient manner. A notable example is the work of Bolliger and Frech68 about the synthesis of various N-arylated amines derived from aryl halides. Dioxane and KN(Si(CH3)3)2 were found to be the ideal solvent and base for this transformation. The conversion rates and yields observed are excellent as shown by the relatively high yields. The selective synthesis of 6-halopyridin-2-amines and asymmetrical pyridine-2,6-diamines (derived from consecutive reactions of 2,6-dibromopyridine and 2,6-dichloropyridine, respectively with different amines) is possible in almost quantitative yields within very short reaction times.
This method also allows the synthesis of pyridine-2,6-diamines in one-pot. However, catalysts are in many cases not required to efficiently and selectively couple aryl halides with amines, making a transition metal-free version of the Buchwald–Hartwig reaction extremely attractive for the synthesis of N-arylated amines with substrates configuration substituents on the aryl halide, which either promote regioselectively and/or do not require regioselective aminations (Scheme 28).
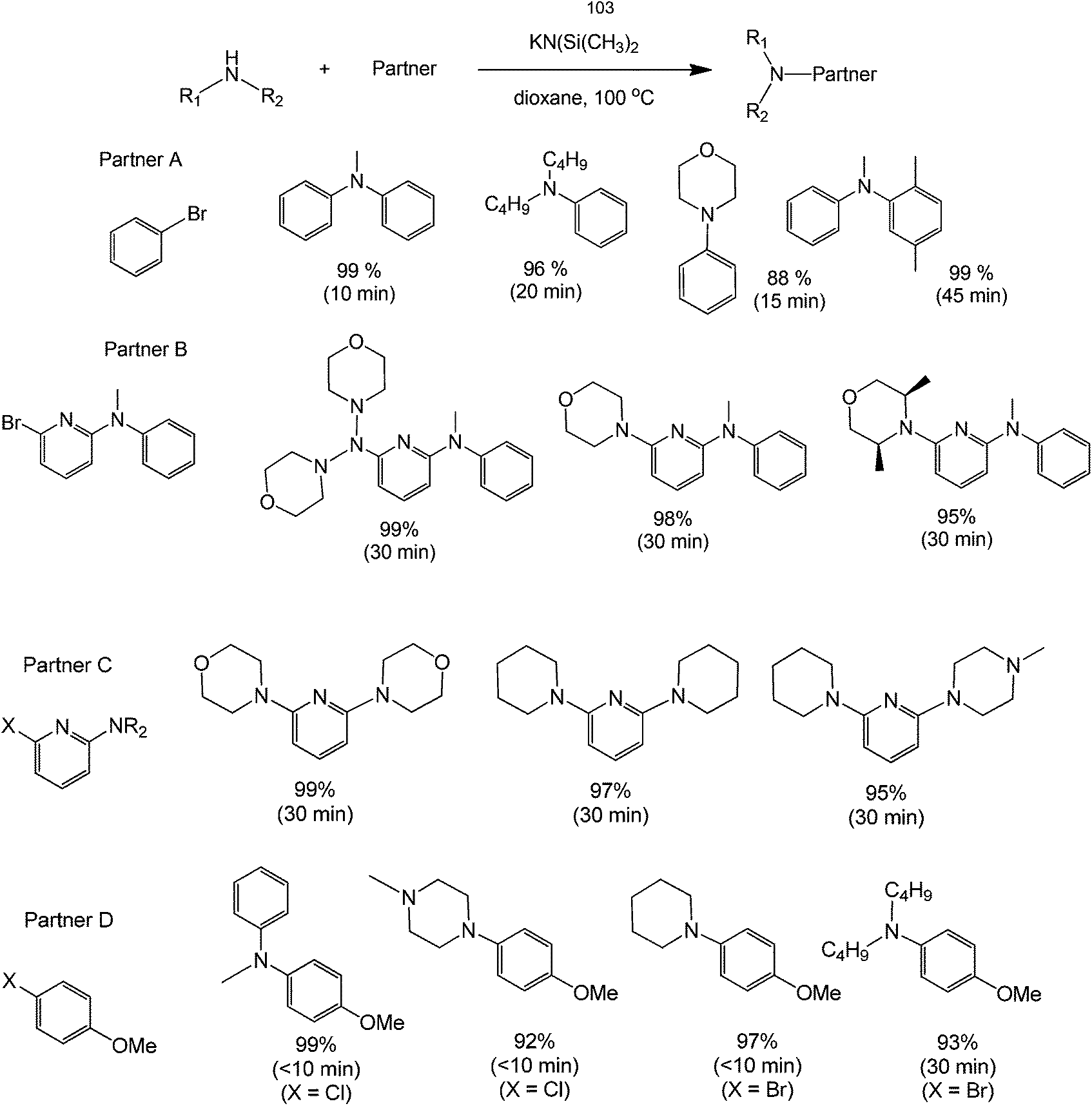 | ||
| Scheme 28 Reaction scope of the direct coupling of an amine with an alkyl group in the presence of potassium (dimethylsilylene)amide and dioxane. | ||
Organometallic precursors as promoters
Although strictly speaking not completely metal-free, organometallic precursors are good starting materials for coupling reactions.69 As these have been previously discussed, Grignard reagents, along with transition metals, make an efficient coupling protocol.70 As previously shown, the sole presence of halogens in a molecule can provide specificity to a reaction. Coupling two different moieties with a halogen therefore becomes very difficult if the halogen needs to be left untouched. These synthetic issues have been shown to be overcome by using a butyl lithium precursor. The mechanism of the reaction is believed to occur via a very unstable benzyne intermediate after the butyl lithium reagent abstracts the bromine. This proposed benzyne intermediate rapidly couples with an unreacted bromobenzene to form a dibromobiaryl (Scheme 29).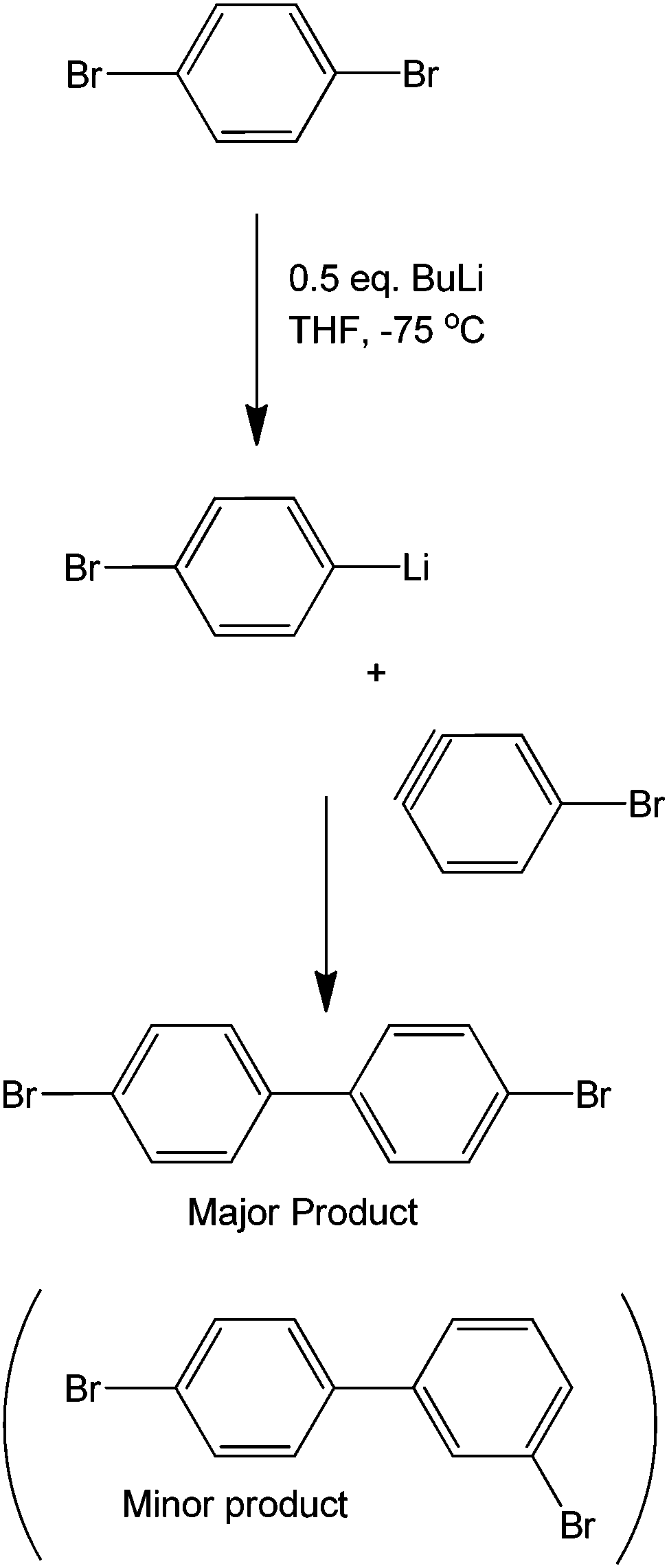 | ||
| Scheme 29 Proposed mechanism for the formation of dibromobiaryls. | ||
As seen in the scheme, the formation of the benzyne intermediate easily explains the observation for the two types of products formed. The metallation process is exemplified by the synthesis of phosphine ligands (Scheme 30) that can be used for further coupling, therefore opening the door for the synthesis of multi-aromatic compounds with various functionalities.
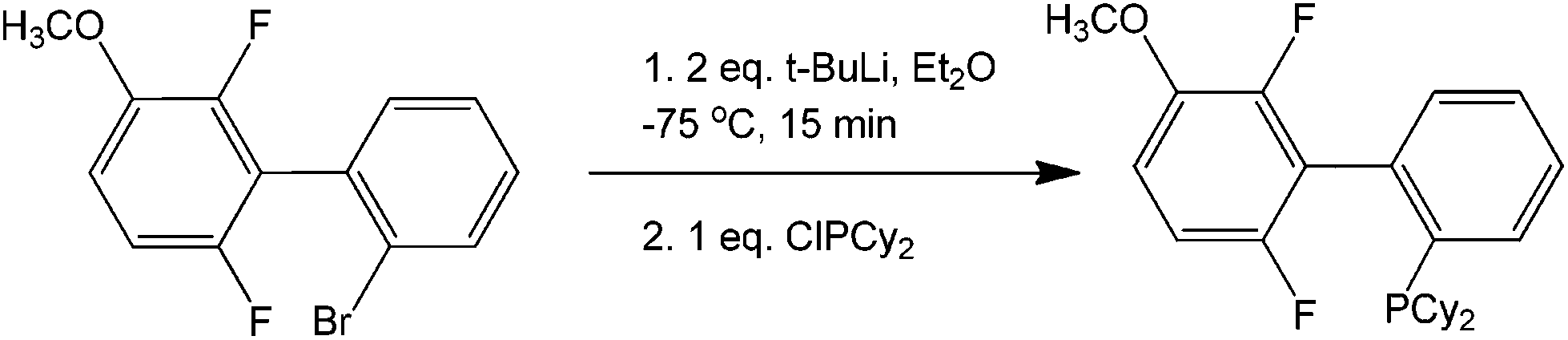 | ||
| Scheme 30 Reaction protocol for the synthesis of ligand intermediates using the transmetallation pathway of lithium. | ||
Grignard reagents are mainly used as nucleophilic substrates in many organic reactions.71–73 Using diphenoquinone 1 (structure 27) as an electron acceptor, the oxidative dimerisation of alkenylmagnesium reagents can proceed with complete retention of stereochemistry. As seen in Scheme 31, the tolerance of the reaction towards the presence of electron withdrawing and donating substituents is good. This concept allows the coupling of various aryl agents using main-group metal derivatives only. Thus, mono- and diorganomagnesium compounds, which are complexed with LiCl, undergo efficient homocoupling in the presence of phenoquinone DPQ, which acts as a two-electron acceptor.
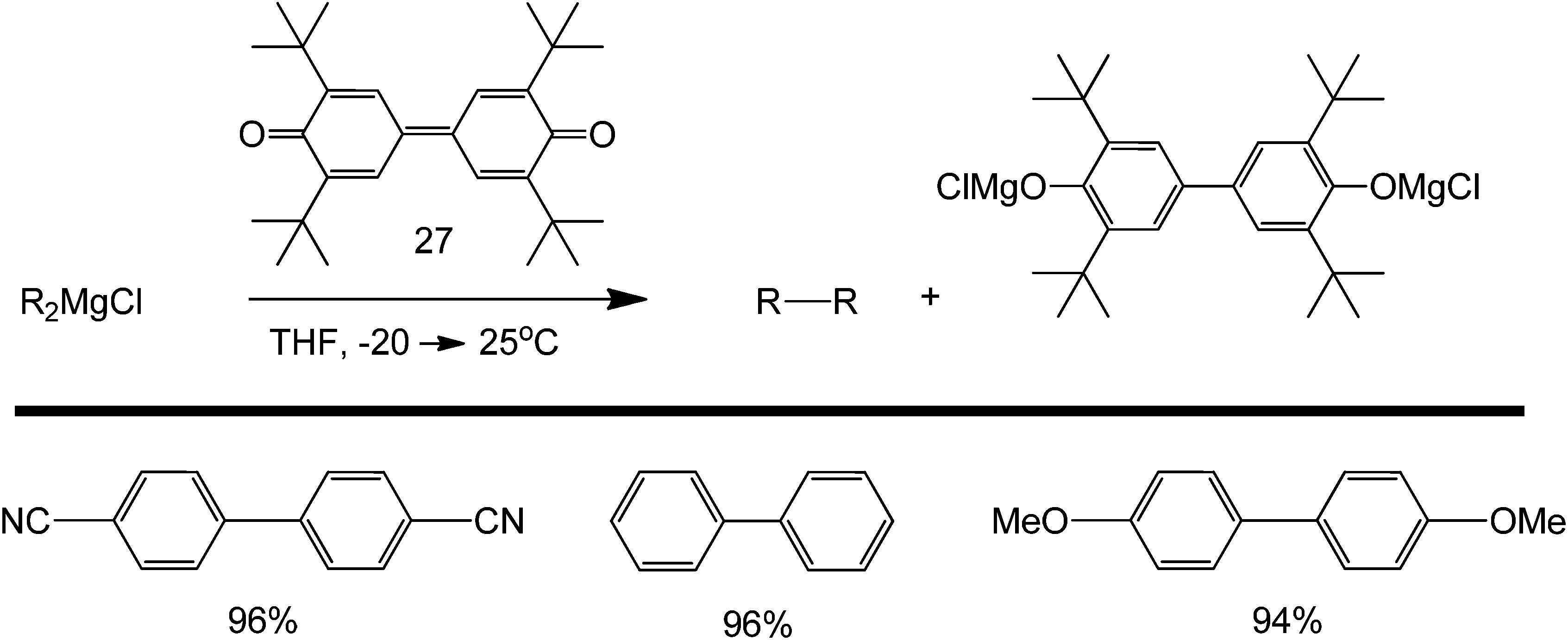 | ||
| Scheme 31 Homocoupling of Grignard reagent in the presence of 3,3′,5,5′-tetra-tert-butyldiphenoquinone (1) as a two-electron acceptor. | ||
A broad range of functionalised biaryls, diynes and dienes are also obtained. Increasing the electron donating ability of the para-substituent of the phenyl group increases the reaction yield (Scheme 31). Interestingly, great specificity towards the C–Br bond is shown since the benzene ring contains a δ-alkene (Scheme 32). In the final product, the reactive alkene outside the aromatic ring and the Br group attached to benzene were left untouched. These Br groups can be used for further coupling reactions. With this protocol, it is possible to perform polymerization reactions with highly ordered structures. The reaction also facilitates the homocoupling of multi-functional group substrates with very good specificities, opening the possibility to synthesize highly symmetric multi-functional coupled products (Scheme 33) with excellent retention of double bond configuration. The proposed methodology was also found to be amenable to both aromatic and aliphatic substrates.
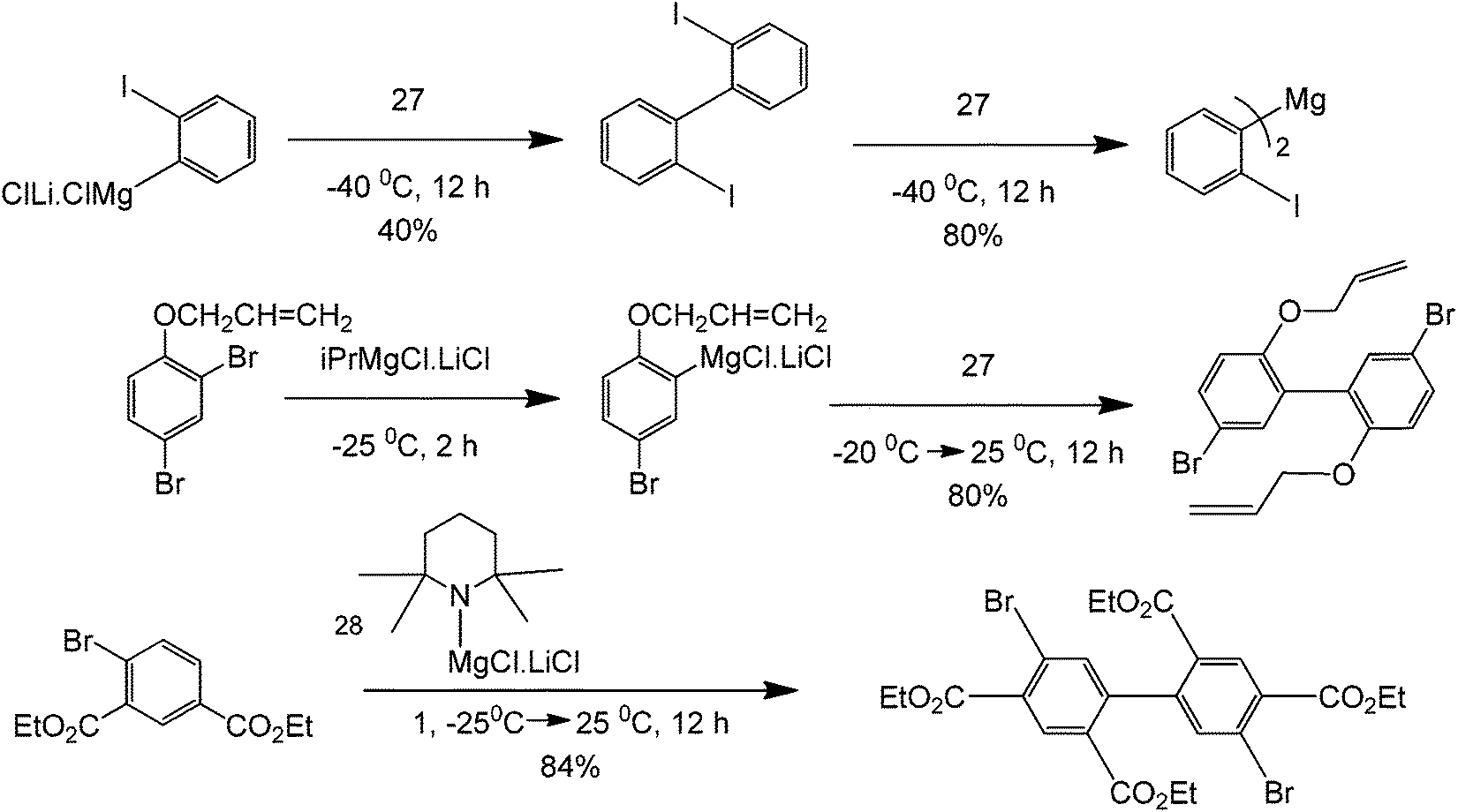 | ||
| Scheme 32 Homocoupling scope of Grignard reagent with a catalyst (structure 27). | ||
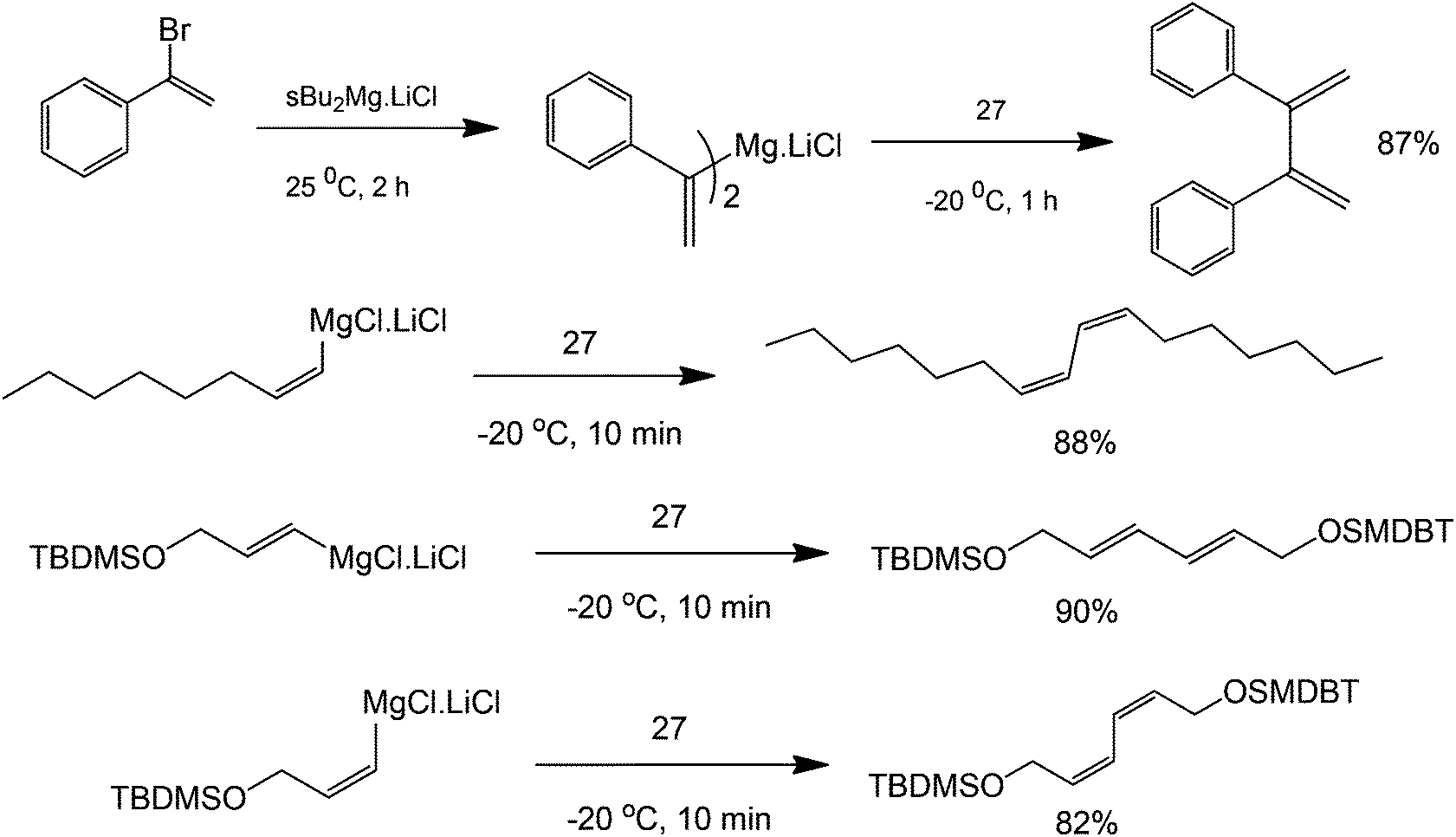 | ||
| Scheme 33 Reaction scope of the coupling of aliphatic and aromatic alkenes with 3,3′,5,5′-tetra-tert-butyldiphenoquinone (structure 27). | ||
Another common oxidant used in organic chemistry mild oxidation reactions is 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO); this oxidant is mild enough to selectively direct the oxidation of primary alcohols into aldehydes (not carboxylic acids).74–76 This oxidant has also been used in the cross coupling of aryl magnesium compounds (Scheme 34).77 The side product isolated after the coupling has been identified as a TEMPOMgX salt which can be easily recovered into TEMPO via purging with oxygen gas. The detailed mechanism of TEMPOMgX reactions has not been reported to date. However, the reaction is believed to proceed through the usual Grignard reagent-mediated coupling in a similar way to that reported by other oxidants (Scheme 35).
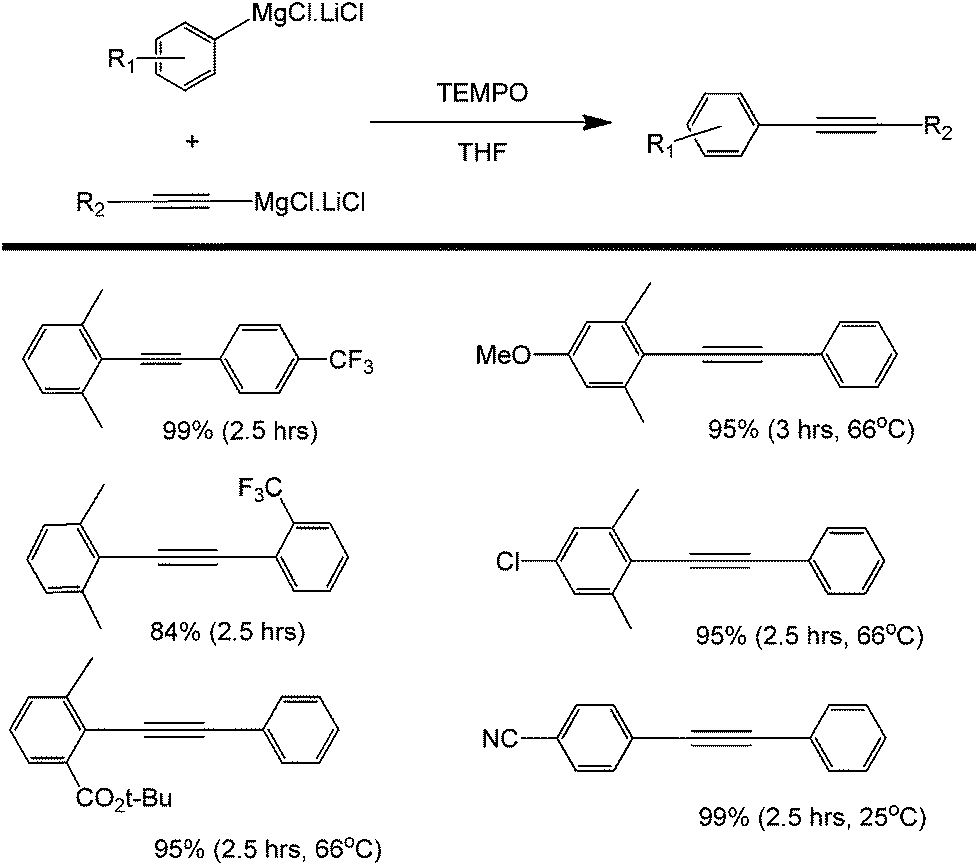 | ||
| Scheme 34 Cross-coupling of ortho-substituted aryl Grignard reagents with alkynyl Grignard reagents using tetramethylpiperidine-N-oxyl radical (TEMPO) as an organic oxidant. | ||
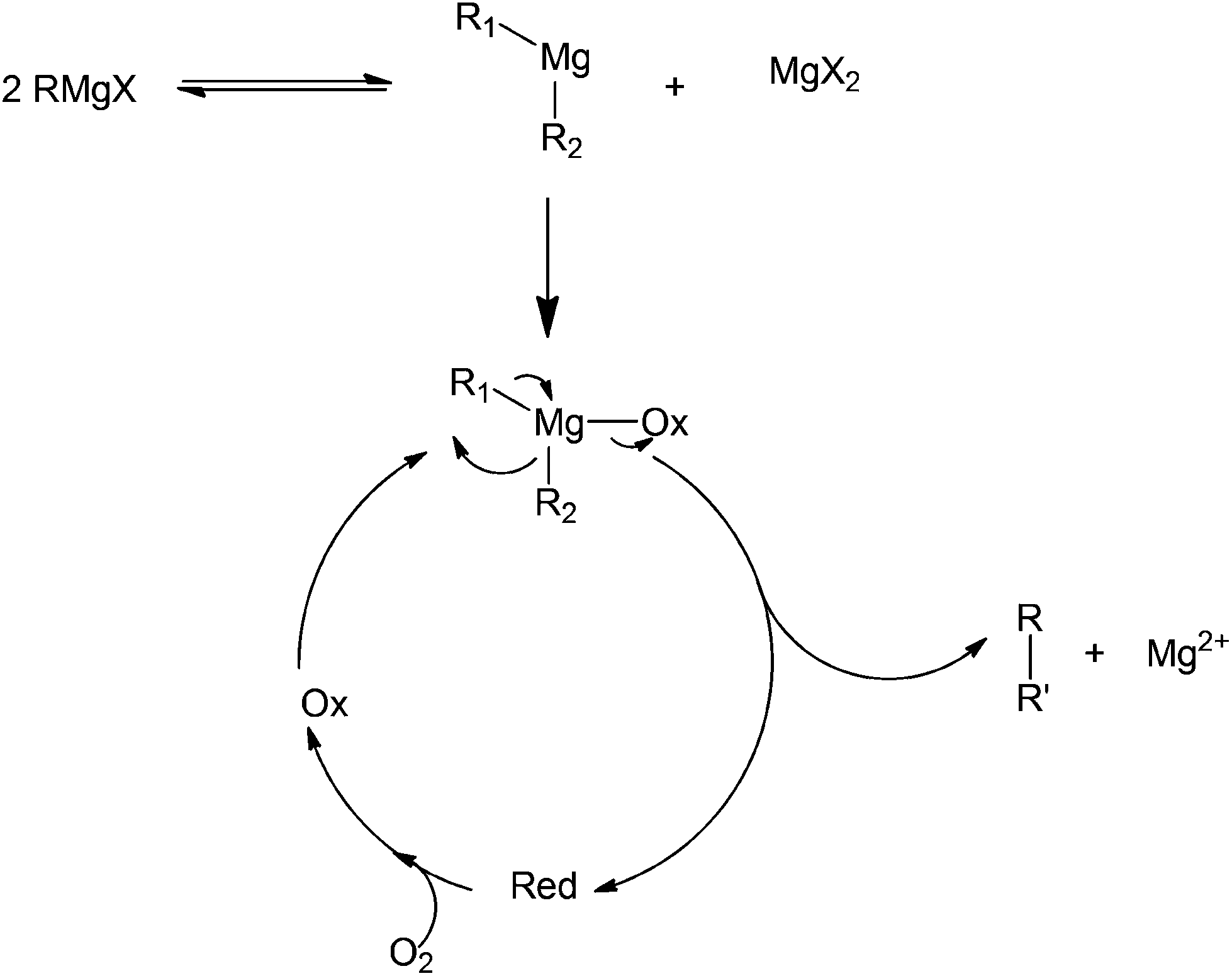 | ||
| Scheme 35 Proposed coupling mechanism involved in the use of a Grignard reagent as a precursor.78 | ||
The yields of coupling products reported in the studies presented were high, but it is important to note that both electron donating and withdrawing substituents at the para position of the starting alkyne were tolerated (Scheme 34). However, if the para substituent is placed in the ortho position (as the case for –CF3), the yield was found to decrease significantly. This observation points to the larger effect of steric hindrance to the formation of the intermediate and eventually the product.
C–C forming photocatalytic reactions: metal vs. metal-free catalysed processed
The reactions so far discussed involve the generation of a cation/radical by heterolytic cleavage of an Ar–X bond initiated by a strong base or by other means. Another set of coupling reactions involving radical generation are mainly photocatalytically mediated by a light-absorbing dye or an organocatalyst.An alkyne and benzene have been coupled in the presence of light, a ketone, and a proton source79 (Scheme 36). The reaction proceeds via a phenyl cation intermediate formed via photoheterolysis, allowing the use of a variety of precursors, such as readily available aryl chlorides as well as aryl esters. Interestingly, aryl fluorides, which are virtually unreactive under metal catalysis in the Sonogashira reaction, could also be used in this reaction, although low product yields are generally obtained. Despite some limitations, this method has distinct advantages with respect to thermal alternatives. Firstly, the reaction is carried out at room temperature, with no need for a sensitive and/or expensive metallic catalyst and is not necessarily sensitive to water and atmospheric oxygen. The thermal-catalysed Sonogashira reaction has been exploited to a limited degree up to now, but the use of a reactive silyl derivative may be advantageous for applications with complex derivatives to establish synthetic control. Also, the mildness of the light-mediated reaction will allow the use of heat sensitive substrates.
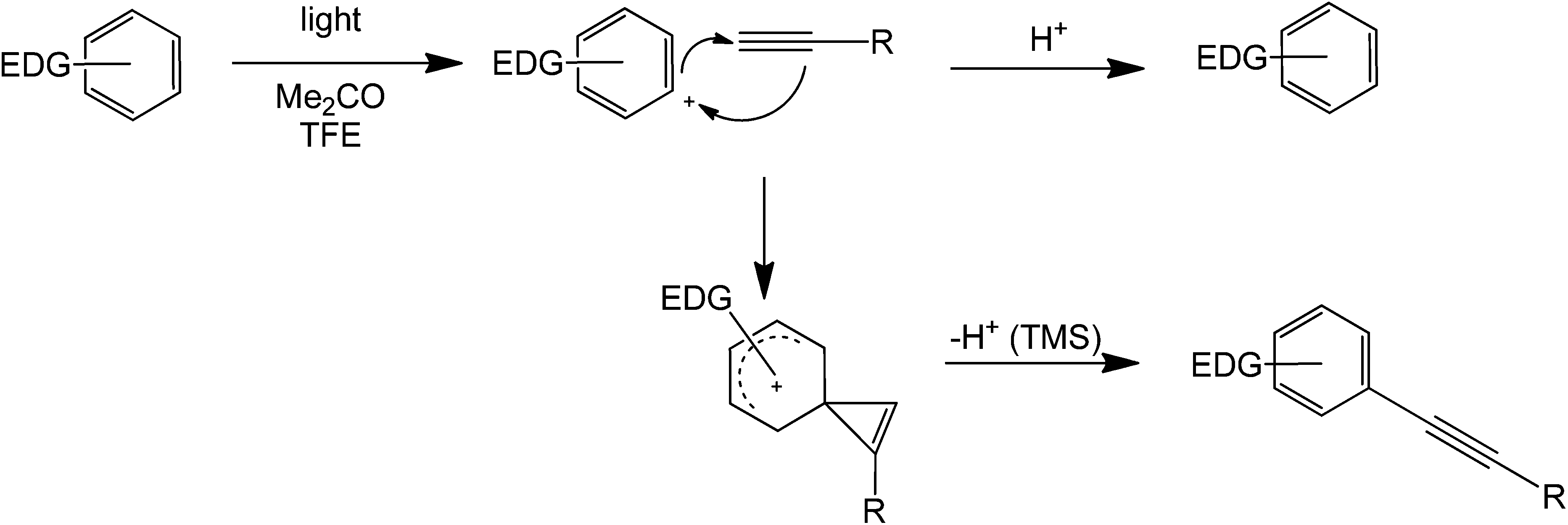 | ||
| Scheme 36 Light catalyzed direct coupling of an alkyne with a benzene compound containing an electron donating substituent. | ||
A photocatalytic approach can also be performed for the phenylation (or allylic functionalization) of alkenes and arenes80 (Scheme 37). The method is general and compares favorably with metal catalysis in terms of mildness of reaction when all the previous findings with phenyl halides and benzene diazonium salts are considered. These reactions are also insensitive to oxygen, moisture, bases and other strong reagents. In addition, a relatively low concentration of the nucleophile is sufficient for an efficient protocol. Because the reaction is essentially initiated by light, expensive and often unrecyclable catalysts are not required.
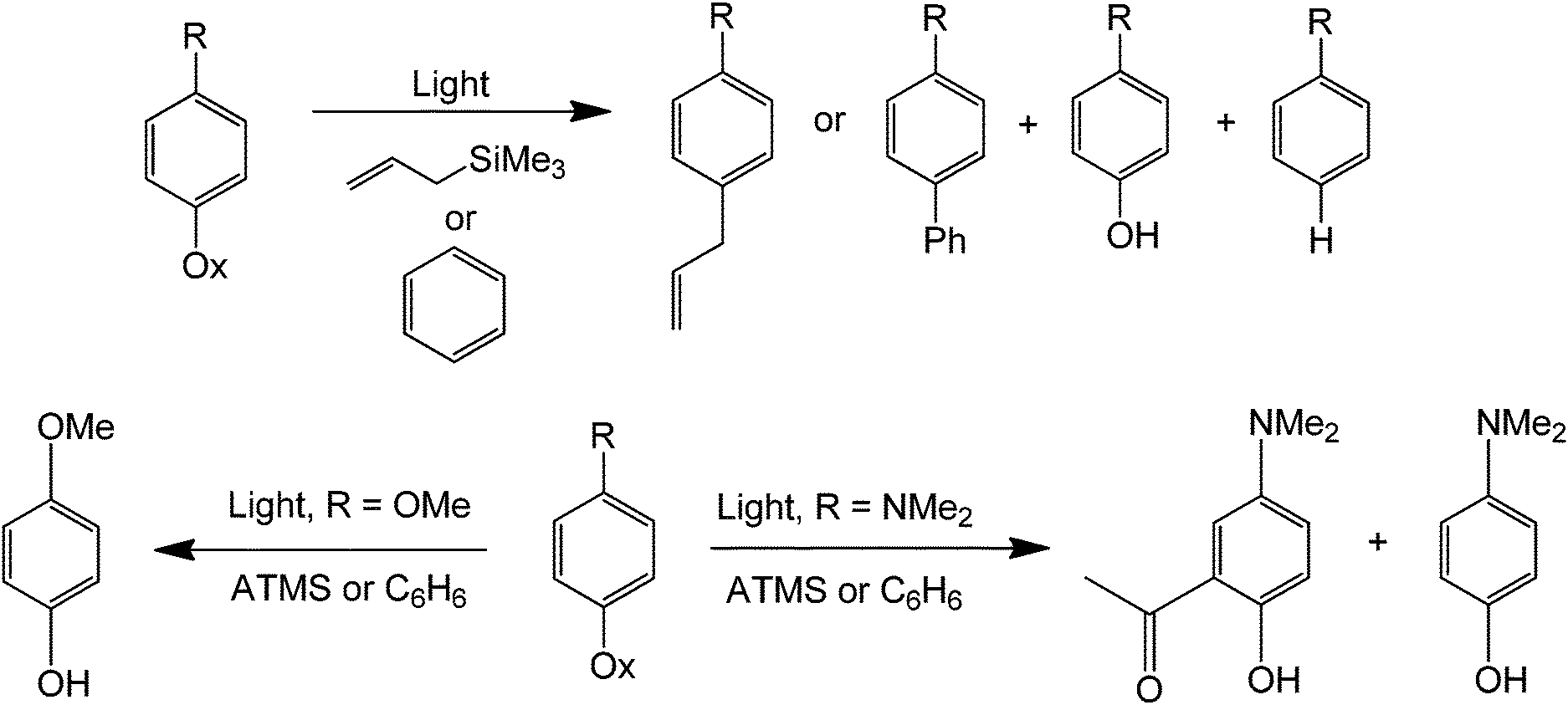 | ||
| Scheme 37 Metal-free light-catalyzed phenylation (or C–C bond formation) using a trimethylsilane or benzene as alkyl sources. | ||
For light-assisted C–C formation reactions, Eosin Y organic dye as a photoredox catalyst can be used to couple heteroarenes and aryl diazonium salts (Scheme 38).81 The Eosin Y acts as an electron donor/acceptor in this reaction since it is highly conjugated. Preliminary investigations using a TEMPO reagent show that this reaction is mainly a radical single electron-transfer. This reaction will allow for the direct coupling of a heterocycle (at the C2 position) with a substituted benzene.
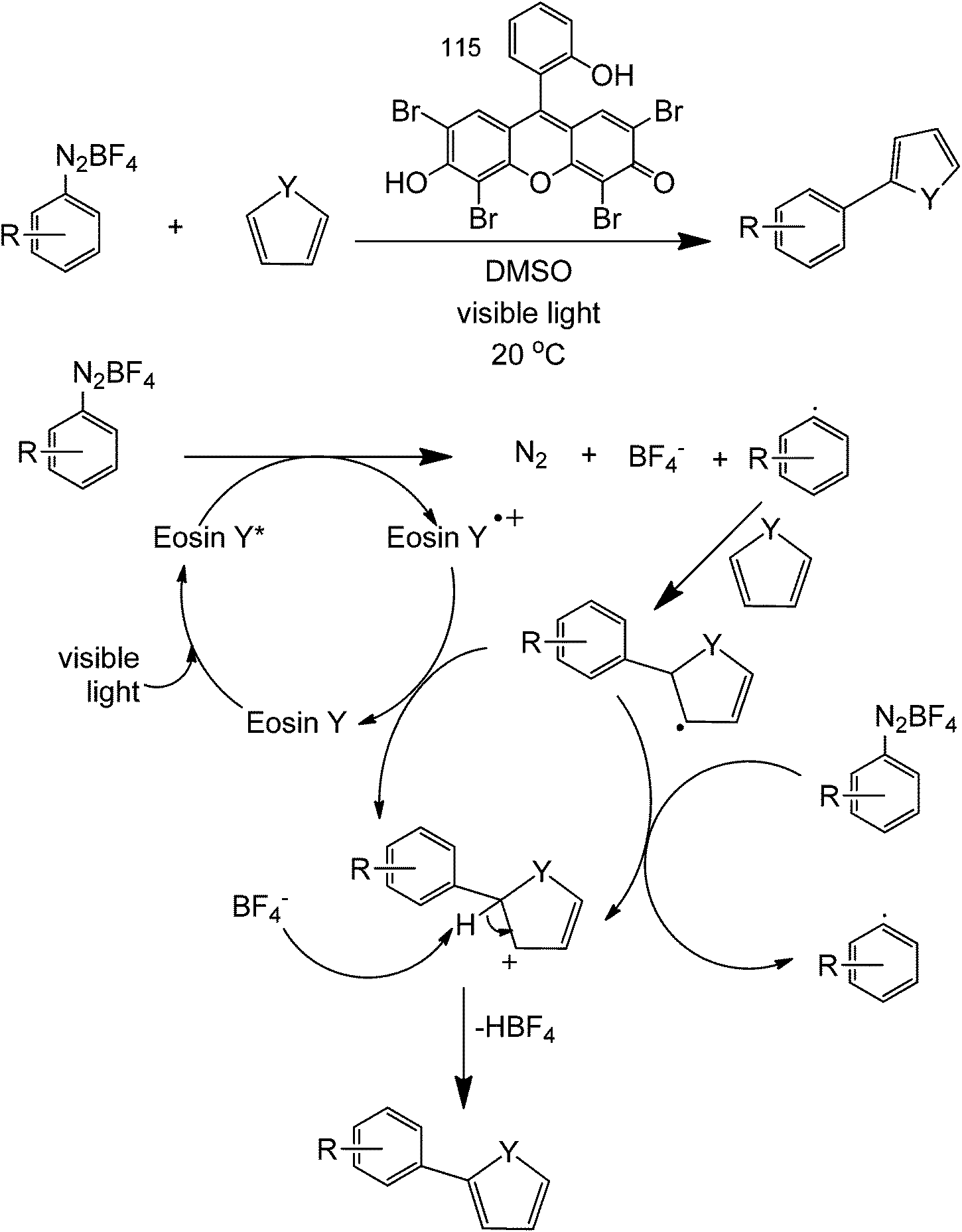 | ||
| Scheme 38 Heteroarene coupling with an aryl diazonium salt in the presence of Eosin Y photoredox catalyst. | ||
Recently, McMillan et al.82 reported an enantioselective α-alkylation of aldehydes which in this case uses a Ru(bpy)3Cl2 photoredox catalyst and an imidazolidinone organocatalyst. The mechanism (Scheme 39) of the reaction starts with the condensation of the aldehyde substrate with a chiral imidazolidinone (trans-methyl-tert-butyl 2,5 disubstituted imidazolidinone) to form an enamine. The alkene at this point stereospecifically reacts with an alkyl radical which was generated by the oxidized form of the Ru complex. Formation of the alkyl radical was done via reduction of the Ru2+ complex to Ru+.
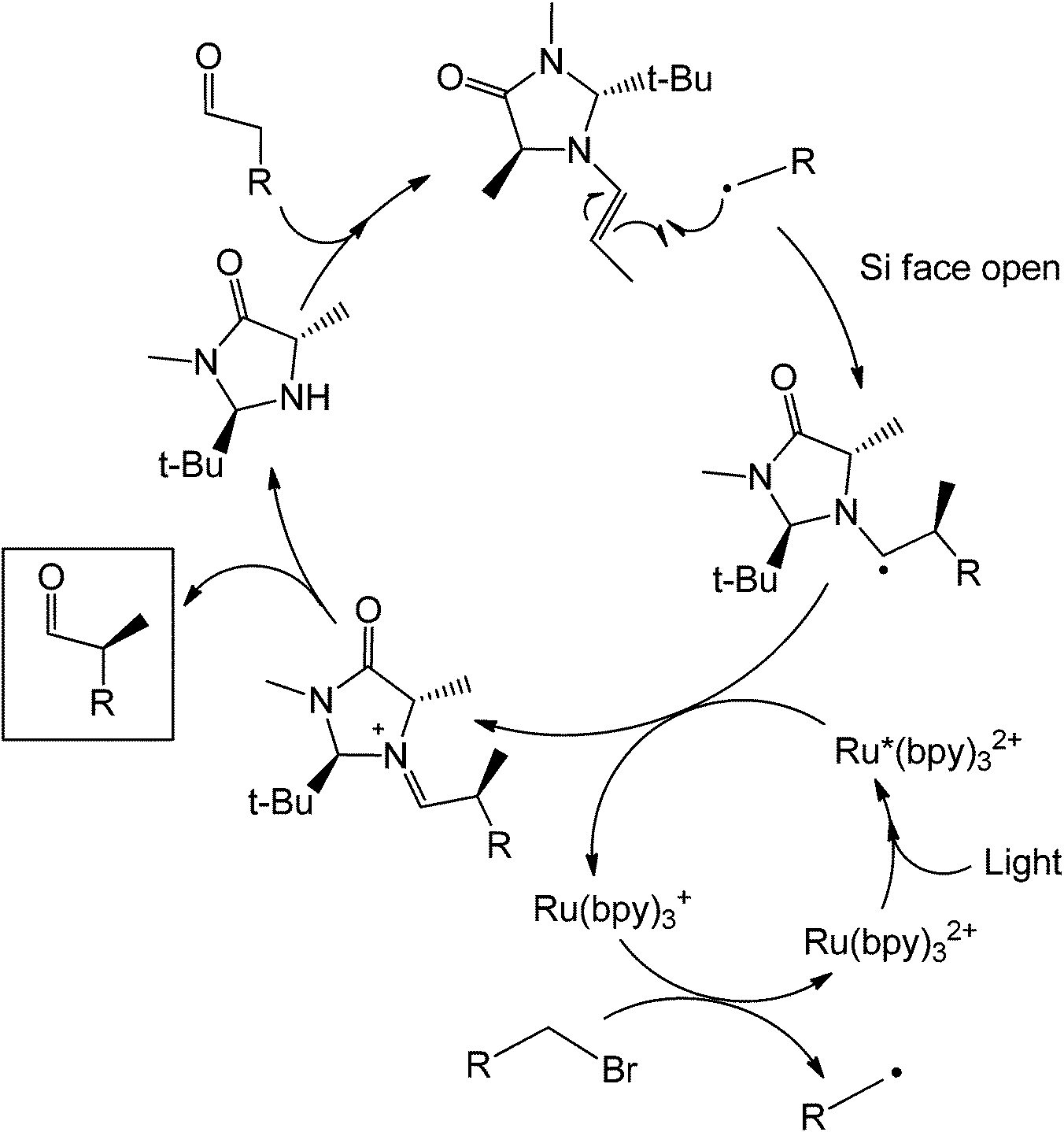 | ||
| Scheme 39 Mechanism of the photocatalytic coupling of the enantioselective alpha substitution of an aldehyde. | ||
The enantioselectivity of the process was achieved due to the presence of the methyl and tert-butyl groups in the imidazolidinone organocatalyst. After condensation with the aldehyde, the olefin will project its position such that it avoids the bulky tert-butyl group. This reaction has also been shown to work with trifluoromethylation on the aldehyde alpha-carbon.83 The mechanism of this reaction is similar with that using the organic dye only that the alpha-substitution here is stereoselective. It is also possible to obtain a different product chirality by varying the steric bulk of the imidazole. Although this example is not totally metal free, the photoredox complex only acts as an electron sink for the entire coupling to happen and maybe can be easily substituted by other conjugated systems which are able to transfer electrons easily. Under a slightly similar condition, a fully metal-free alpha-amination of aromatic ketones has been performed already in the presence of an organocatalyst and light (Scheme 40).84
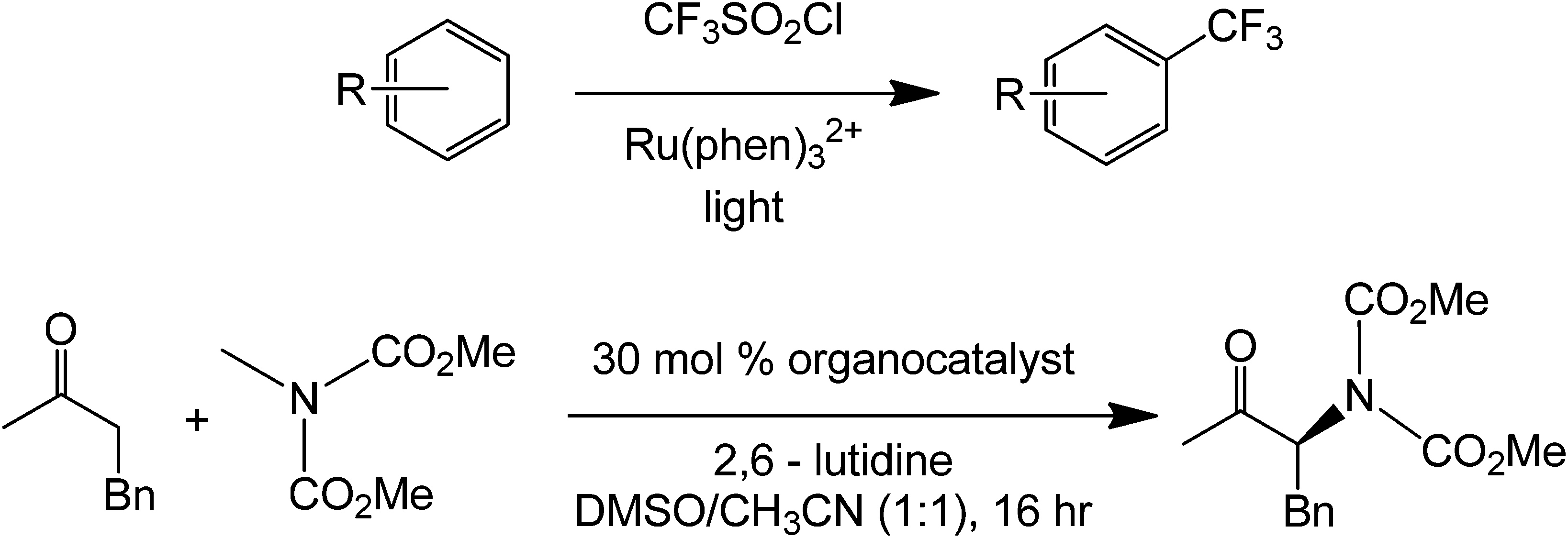 | ||
| Scheme 40 The top part of the figure shows the direct trifluoromethylation of a substituted benzene using photoredox catalysis. The bottom reaction shows the direct alpha-amination of 3-phenylethanaldehyde using an amine organocatalyst. | ||
Metal-free vs. transition metal catalysed processes
Research in transition metal-free couplings is currently a subject of an ongoing debate as to how metal-free the reported metal-free coupling reactions are. As the commentary of Leadbeater in 201037 pointed out, several key steps can be taken to ensure the integrity of a metal-free reaction. These steps include purchasing new materials, qualitative methods such as purification and quantitative detection of transition metals using ICP and ICP/MS have been mentioned in many of these works. However, to maintain and ensure that a coupling reaction is totally metal-free is a very difficult, if not an impossible task due to an almost inevitable contamination with metal traces. Even as low as 50 ppb of Pd could induce the coupling of aryl halides and alkynes.37–39 No study different from that previously mentioned has ever presented and/or validated yet a coupling reaction with ppt levels of Pd or any other transition metal (e.g. the use of Fe-promoted couplings has been proven particularly tricky for a number of reasons).85–87 This section has aimed to compare transition metal couplings from metal-free couplings presented so far.With almost negligible amounts of transition metal present in the protocol, the base has been shown to play a major role in the coupling, as highlighted in various studies. An example of this is the Sonogashira-type coupling of aryl halides and aryl alkynes using DABCO. This particular base has been used in both Pd(OAc)2-catalyzed49 coupling and metal-free coupling.41 In the presence of a Pd complex, the reaction proceeds via the usual Sonogashira mechanism, while the mechanism has been suggested to proceed via alkyne activation and nucleophilic aromatic substitution without a metal complex (Scheme 41). The reaction proceeds smoothly with a wide range of aryl halides and alkynes under transition metal catalysed conditions, while a very restricted scope (only aryl iodides) under particular conditions (high temperatures, microwave irradiation, large quantities of base, etc.) are required to provide even lower conversions under metal-free conditions.41,42 The yields of both transition metal-catalyzed and metal-free coupling reactions are almost comparable. Another observation is the use of quinones in both transition metal catalyzed and metal free couplings;47,50 the main role of quinones in both types of reactions as it is acting as an electron sink.
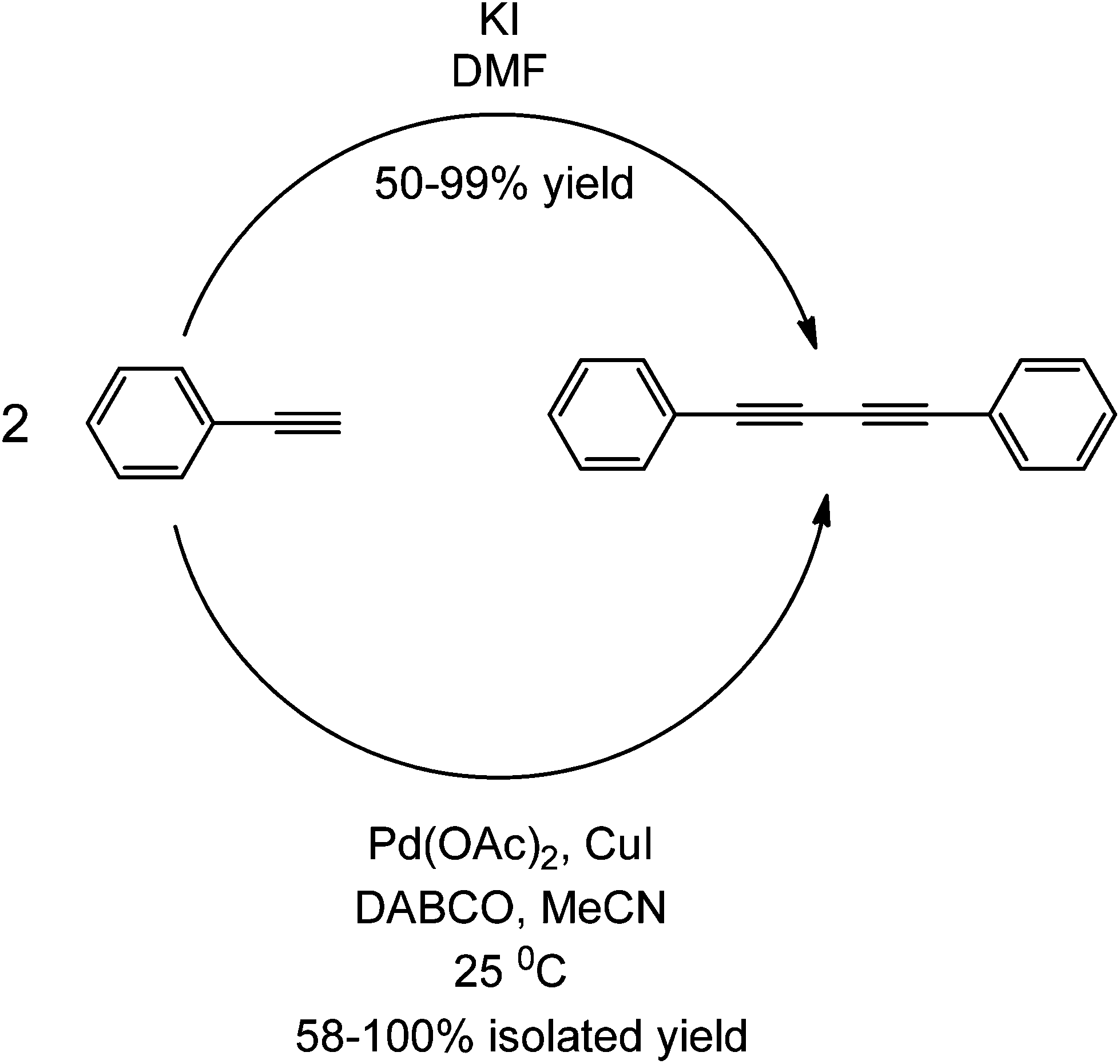 | ||
| Scheme 41 Comparison between a metal-free and a transition metal-catalyzed coupling forming the same symmetric coupled product. | ||
Comparing an organocatalytic approach between a traditional Grignard ketone addition for coupling, we observe that the organocatalytic approach is a much simpler method due to the efficiency of the imine formation in the first step (Scheme 42). We believe it is important to validate if the reaction is indeed metal-free, especially on the second part involving the organoboron. The formation of the second intermediate (the cyclic boron complex) can also proceed in the presence of a transition-metal complex. In terms of yield, the organocatalytic approach appears to have a more superior performance than the traditional Grignard coupling. This is so because reactions with Grignard reagents have the tendency to go uncontrolled, forming homocoupled products aside from the addition products.
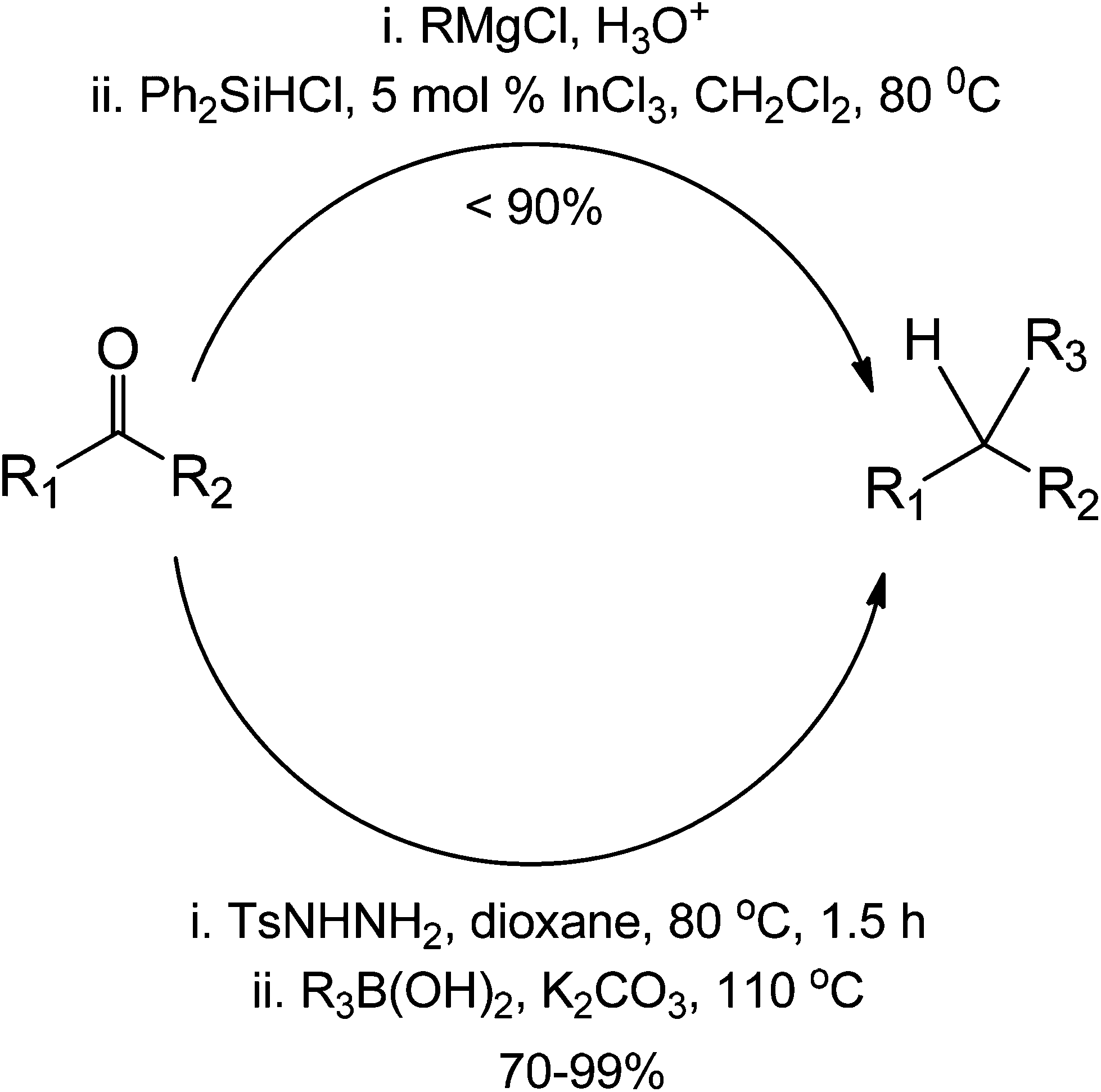 | ||
| Scheme 42 Comparison between an organocatalytic coupling reaction and a common metal-mediated reduction for the formation of a reduced ketone. | ||
Lastly, polythiophenes are traditionally synthesized using Ni(ddpe)Cl2. The route for the synthesis involves the formation of a Grignard reagent to generate the nucleophile for the coupling. The metal-free coupling on the other hand, involves the sole use of nucleophilic substitution. Notably, a metal-free reaction can be, in principle, performed in the absence of a halide functionalization at C2 of the thiophene. This therefore makes the metal-free counterpart operationally simple.62 Yields of products in this particular case (75%) were also similar to those obtained under transition metal catalysis conditions (Scheme 43).
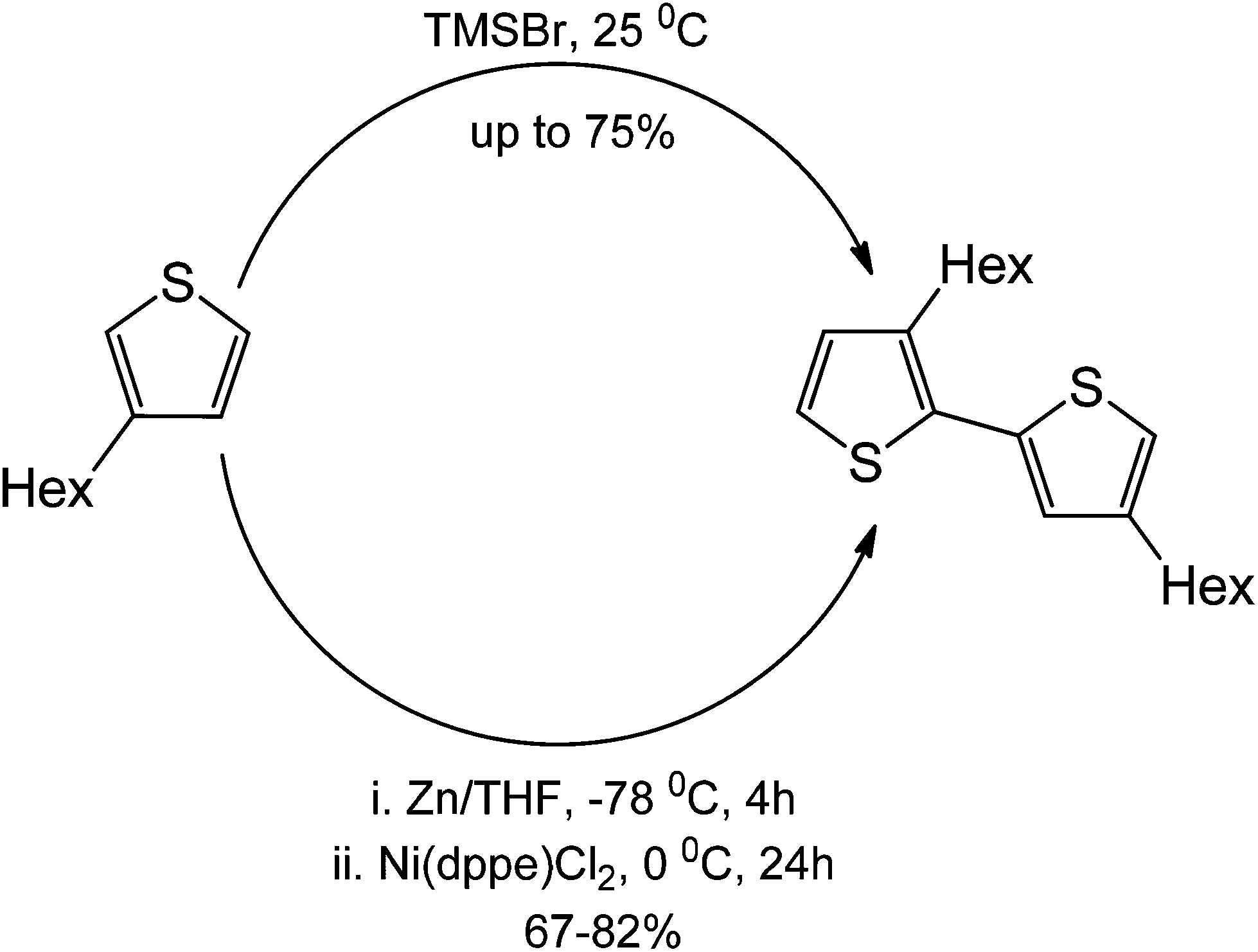 | ||
| Scheme 43 Comparison between a metal-free thiophene H–T polymerization against a traditional metal-free method. | ||
In any case, from these and other examples, it is clearly difficult to prove the metal-freeness of a metal-free reaction since we cannot also rule out the catalytic activity contribution of certain metal traces. In all of the three comparisons made, it is obvious that a metal contamination could have easily catalyzed the coupling instead of the proposed method, so that repetition, scrutiny, and validation of these metal-free reactions must be made with extreme caution. While employing the aforementioned steps and precautions previously suggested, it is interesting to produce detailed mechanistic investigations on metal-free reactions. Since there are obviously overlapping reagents in both transition-metal catalyzed and metal free couplings, it is important to determine if they can possibly direct the coupling reaction in the total absence (or presence in extremely low concentrations) of a transition metal.
Conclusions and future prospects
The advent of green chemistry has stimulated scientists to develop innovative and increasingly environmentally benign synthetic strategies with comparable efficiencies to those employed in past years. With this, synthetic chemists have aimed to substitute established coupling methods with greener protocols, which improve traditional wasteful and hazardous methodologies. One of the issues of coupling chemistries, particularly for the synthesis of pharmaceuticals and biologically active compounds, deals with the disposal of waste and metal complexes that have deactivated after the reaction. Metal-free coupling methodologies have been feeding the minds of scientists for decades with the aim to advance organic synthetic protocols which overcome the aforementioned issues. However, a significant body of work in the past years has been devoted towards the previously mentioned Hamlet-type of duality “to be or not to be metal-free”. Moreover, from the seminal report from Leadbeater et al. on traces of Pd (as low as ppb) catalysing Suzuki-type chemistries, there has been a significant controversy in a range of reported protocols that even lead to some retraction of claimed metal-free processes or even more recently Fe-catalysed couplings.Away in principle from such controversy, this contribution has been focused on selecting useful synthetic examples of metal-free coupling reactions rather than emphasizing potential issues on traces of metals present (or not) in such protocols which could catalyse the proposed reactions. Many of the highlighted metal-free couplings have been demonstrated to be accomplished through a combination of approaches including microwave condition, base catalysis or even photocatalysis. Microwave reactions allow for substitution of organic solvents with water, while both basic and photocatalysis are highly dependent on substituent functional groups. Base catalyzed coupling reactions of substituted aryls mainly required electron withdrawing para-substituents, while light-mediated C–C forming reactions are better off with electron donating groups.
In line with the recent perspective from Leadbeater on when is metal free really free, we fully agree with the fact that all included synthetic transformations in this “metal free overview contribution” are synthetically useful, even many of them from a potential industrial standpoint. These transformations need obviously to be reproducible and with a wide applicability not only scientist-wise but also in terms of reagents, protocols and substrates. In some cases, regardless of the many tests and analysis conducted over metal-free coupling methodologies, several chemistries will be in the bullseye for metal-free claims and certainly challenging to fully prove the orgaNOmetallic (nice playword from Leadbeater and Muller) nature of the protocol.
In spite of many advances in the field in recent years and current extensive research to achieve a truly sustainable alternative method for coupling reactions (metal or not metal free), most methodologies included, as well as many others available in the literature, nicely complement their metal-catalysed versions in terms of substitution patterns, simplicity, and reaction conditions. What is more important, these may pave the way of future advanced synthetic coupling protocols that we all hope to enjoy in future years to come.
Acknowledgements
Rick Arneil D. Arancon wishes to thank the City University of Hong Kong (School of Energy and Environment) Graduate Teaching Assistant Scheme Category A for this wonderful academic exchange opportunity. Rafael Luque gratefully acknowledges Spanish MICINN for financial support via the concession of a RyC contract (ref: RYC-2009-04199) and funding under project CTQ2011-28954-C02-02 (MEC). Consejeria de Ciencia e Innovacion, Junta de Andalucia is also gratefully acknowledged for funding project P10-FQM-6711. The author Rafa Luque is also indebted to Prof. Guohua Chen, the Department of Chemical and Biomolecular Engineering (CBME) and HKUST for the provision of a Visiting Professorship at the CBME in 2013.References
- F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 2419 CAS.
- V. Farina, Adv. Synth. Catal., 2004, 346, 1553 CrossRef CAS.
- U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366 CrossRef CAS PubMed.
- D. Astruc, Inorg. Chem., 2007, 46, 1884–1894 CrossRef CAS PubMed.
- V. Calo, A. Nacci, A. Monopoli and F. Montingelli, J. Org. Chem., 2005, 70, 6040 CrossRef CAS PubMed.
- G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1698 CrossRef.
- I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS PubMed.
- V. P. Mehta and E. V. Van der Eycken, Chem. Soc. Rev., 2011, 40, 4925 RSC.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46 CrossRef CAS.
- J. W. Grissom, G. U. Gunawardena, D. Klingberg and D. Huang, Tetrahedron Lett., 1996, 52, 6453 CrossRef CAS.
- M. B. Nielsen and F. Diederich, Chem. Rev., 2005, 105, 1837 CrossRef CAS PubMed.
- C. Vargas, A. M. Balu, J. M. Campelo, C. González-Arellano, R. Luque and A. A. Romero, Curr. Org. Synth., 2010, 7, 568 CrossRef CAS.
- Y. Liang, Y.-X. Xie and J.-H. Li, J. Org. Chem., 2006, 71, 379 CrossRef CAS PubMed.
- J. Mao, M. Wu, G. Xie and S. Jia, Adv. Synth. Catal., 2009, 351, 2101 CrossRef CAS.
- (a) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed; (b) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084 RSC.
- N. E. Leadbeater and B. J. Tominack, Tetrahedron Lett., 2003, 44, 8653 CrossRef CAS PubMed.
- B. Liang, M. Huang, Z. You, Z. Xiong, K. Lu, R. Fathi, J. Chen and Z. Yang, J. Org. Chem., 2005, 70, 6097 CrossRef CAS PubMed.
- T. Fukuyama, M. Shinmen, S. Nishitani, M. Sato and I. Ryu, Org. Lett., 2002, 4, 1691 CrossRef CAS PubMed.
- H. Cao, L. McNamee and H. Alper, Org. Lett., 2008, 10, 5281 CrossRef CAS PubMed.
- R. D. Tung and D. H. Rich, J. Am. Chem. Soc., 1985, 107, 4342 CrossRef CAS.
- L. A. Carpino and A. El-Faham, J. Org. Chem., 1994, 59, 695 CrossRef CAS.
- M. Hocek, A. Holý, I. Votruba and H. Dvořáková, J. Med. Chem., 2000, 43, 1817 CrossRef CAS PubMed.
- H. Plenio, Angew. Chem., Int. Ed., 2008, 47, 6954 CrossRef CAS PubMed.
- W. Rauf and J. M. Brown, Chem. Commun., 2013, 49, 8430 RSC.
- D. McCartney and P. J. Guiry, Chem. Soc. Rev., 2011, 40, 5122 RSC.
- N. A. Bumagin and V. V. Bykov, Tetrahedron Lett., 1997, 53, 14437 CrossRef CAS.
- H. Sakurai, T. Tsukuda and T. Hirao, J. Org. Chem., 2002, 67, 2721 CrossRef CAS PubMed.
- Y. Uozumi, H. Danjo and T. Hayashi, J. Org. Chem., 1999, 64, 3384 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 97, 2457 CrossRef.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed.
- A. S. Castanet, F. Colobert, J. R. Desmurs and S. Schlama, J. Mol. Catal., 2002, 481, 182 Search PubMed.
- R. K. Arvela, N. E. Leadbeater, M. S. Sangi, V. A. Williams, P. Granados and R. D. Singer, J. Org. Chem., 2004, 70, 161 CrossRef PubMed.
- F. Vallée, J. J. Mousseau and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 1514 CrossRef PubMed.
- W. Liu, H. Cao and A. Lei, Angew. Chem., Int. Ed., 2010, 122, 2048 CrossRef.
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J. M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC.
- J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2012, 46, 412 CrossRef PubMed.
- N. Leadbeater, Nat. Chem., 2010, 2, 1007 CrossRef CAS PubMed.
- N. Leadbeater, M. Marco and B. J. Tominack, Org. Lett., 2003, 21, 3919 CrossRef PubMed.
- N. E. Leadbeater and B. J. Tominack, Tetrahedron Lett., 2003, 44, 8653 CrossRef CAS PubMed.
- P. Appukkuttan, W. Dehaen and E. Van der Eycken, Eur. J. Org. Chem., 2003, 4713 CrossRef CAS.
- R. Luque and D. J. Macquarrie, Org. Biomol. Chem., 2009, 7, 1627 CAS.
- Z. Chen, H. Jiang, A. Wang and S. Yang, J. Org. Chem., 2010, 75, 6700 CrossRef CAS PubMed.
- J. Yan and L. Wang, Synth. Commun., 2005, 35, 2333 CrossRef CAS.
- V. P. Metha and B. Punji, RSC Adv., 2013, 3, 11957 RSC.
- S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673 CrossRef CAS PubMed.
- E. Shirakawa, K. I. Itoh, T. Higashino and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 15537 CrossRef CAS PubMed.
- K. Huang, S. F. Zheng, B. J. Li and Z. J. Shi, Nat. Chem., 2010, 2, 1044 CrossRef PubMed.
- W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 CrossRef CAS PubMed.
- (a) M. J. Mio, L. C. Kopel, J. B. Braun, T. L. Gadzikwa, K. L. Hull, R. G. Brisbois, C. J. Markworth and P. A. Grieco, Org. Lett., 2002, 4, 3199 CrossRef CAS PubMed; (b) A. Elangovan, Y. H. Wang and T. I. Ho, Org. Lett., 2003, 5, 1841 CrossRef CAS PubMed; (c) B. Liang, M. Dai, J. Chen and Z. Yang, J. Org. Chem., 2005, 70, 391 CrossRef CAS PubMed.
- (a) J. H. Li, Y. Liang and Y. X. Xie, J. Org. Chem., 2005, 70, 4393 CrossRef CAS PubMed; (b) D. D. Hennings, T. Iwama and V. H. Rawal, Org. Lett., 1999, 1, 1205 CrossRef CAS; (c) J. Park, E. Park, A. Kim, S. A. Park, Y. Lee, K. W. Chi, Y. H. Jung and I. S. Kim, J. Org. Chem., 2011, 76, 2214 CrossRef CAS PubMed.
- D. Ramesh, U. Ramulu, S. Rajaram, P. Prabhakar and Y. Venkateswarlu, Tetrahedron Lett., 2010, 51, 4898 CrossRef CAS PubMed.
- O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC.
- J. Jin, Y. Li, Z. Wang, W. Qian and W. Bao, Eur. J. Org. Chem., 2010, 1235 CrossRef CAS.
- Y. Li and W. Bao, Adv. Synth. Catal., 2009, 351, 865 CrossRef CAS.
- D. Cheng and W. Bao, Adv. Synth. Catal., 2008, 350, 1263 CrossRef CAS.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 122, 5113 CrossRef.
- Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560 RSC.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494 CrossRef CAS PubMed.
- K. Matcha and A. P. Antonchick, Angew. Chem., 2013, 125, 2136 CrossRef.
- T. Dohi, K. Morimoto, C. Ogawa, H. Fujioka and Y. Kita, Chem. Pharm. Bull., 2009, 57, 710 CrossRef CAS.
- Y. Kita, K. Morimoto, M. Ito, C. Ogawa, A. Goto and T. Dohi, J. Am. Chem. Soc., 2009, 131, 1668 CrossRef CAS PubMed.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202 CrossRef CAS PubMed.
- S. Thakur and E. S. Tillman, J. Polym. Sci., 2007, 45, 3488 CAS.
- S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC.
- B. Prüger, G. E. Hofmeister, C. B. Jacobsen, D. G. Alberg, M. Nielsen and K. A. Jørgensen, Chem.–Eur. J., 2010, 16, 3783 CrossRef PubMed.
- M. Poirier, S. B. Goudreau, J. Poulin, J. Savoie and P. L. Beaulieu, Org. Lett., 2010, 12, 2334 CrossRef CAS PubMed.
- G. Deng, K. Ueda, S. Yanagisawa, K. Itami and C. J. Li, Chem.–Eur. J., 2009, 15, 333 CrossRef CAS PubMed.
- J. L. Bolliger and C. M. Frech, Tetrahedron Lett., 2009, 65, 1180 CrossRef CAS PubMed.
- F. R. Leroux, L. Bonnafoux, C. Heiss, F. Colobert and D. A. Lanfranchi, Adv. Synth. Catal., 2007, 349, 2705 CrossRef CAS.
- C. E. Knappke and A. J. von Wangelin, Chem. Soc. Rev., 2011, 40, 4948 RSC.
- T. Imamoto, N. Takiyama, K. Nakamura, T. Hatajima and Y. Kamiya, J. Am. Chem. Soc., 1989, 111, 4392 CrossRef CAS.
- J. Huang and S. P. Nolan, J. Am. Chem. Soc., 1999, 121, 9889 CrossRef CAS.
- K. Tamao, N. Ishida and M. Kumada, J. Org. Chem., 1983, 48, 2120 CrossRef CAS.
- R. A. Miller and R. S. Hoerrner, Org. Lett., 2003, 5, 285 CrossRef CAS PubMed.
- T. M. Hansen, G. J. Florence, P. Lugo-Mas, J. Chen, J. N. Abrams and C. J. Forsyth, Tetrahedron Lett., 2003, 44, 57 CrossRef CAS.
- P. L. Bragd, H. Van Bekkum and A. C. Besemer, Top. Catal., 2004, 27, 49 CrossRef CAS.
- M. S. Maji, S. Murarka and A. Studer, Org. Lett., 2010, 12, 3878 CrossRef CAS PubMed.
- A. Krasovskiy, A. Tishkov, V. del Amo, H. Mayr and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 5010 CrossRef CAS PubMed.
- S. Protti, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2005, 44, 5675 CrossRef CAS PubMed.
- M. De Carolis, S. Protti, M. Fagnoni and A. Albini, Angew. Chem., 2005, 117, 1258 CrossRef.
- D. P. Hari, P. Schroll and B. König, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed.
- D. A. Nagib and D. W. MacMillan, Nature, 2011, 480, 224 CrossRef CAS PubMed.
- G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521 CrossRef CAS PubMed.
- D. Becier and C. Darcel, Adv. Synth. Catal., 2009, 351, 1732 CrossRef.
- S. Buchwald and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5586 CrossRef CAS PubMed.
- T. Kylmala, A. Valkonen, K. Rissanen, Y. Xu and R. Franzen, Tetrahedron Lett., 2009, 50, 5692 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |