Chiral amides via copper-catalysed enantioselective conjugate addition
Anne K.
Schoonen
a,
M. Ángeles
Fernández-Ibáñez
ab,
Martín
Fañanás-Mastral
a,
Johannes F.
Teichert
ac and
Ben L.
Feringa
*a
aDepartment of Organic Chemistry and Molecular Inorganic Chemistry, Stratingh Institute, University of Groningen, Nijenborgh 4, 9747AG, Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl; Tel: +31 50 363 4278
bDepartamento de Química Orgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, Cantoblanco, 28049, Madrid, Spain
cInstitut für Chemie, Technische Universität Berlin, Straße des 17. Juni 115, D-10623 Berlin, Germany
First published on 29th October 2013
Abstract
A highly enantioselective one pot procedure for the synthesis of β-substituted amides was developed starting from the corresponding α,β-unsaturated esters. This new methodology is based on the copper-catalysed enantioselective conjugate addition of Grignard reagents to α,β-unsaturated esters and subsequent direct formation of amides by quenching the corresponding enolates with different amines. Various primary and secondary amines bearing alkyl or aryl substituents can be used giving rise to a large variety of β-substituted amides with excellent enantioselectivities.
Introduction
The catalytic asymmetric conjugate addition (CA) of organometallic reagents to α,β-unsaturated compounds is one of the most versatile methodologies for the formation of C–C bonds.1 In 2004, a highly enantioselective catalytic CA of Grignard reagents was reported from our laboratories.2 This paved the way for the efficient enantioselective CA of Grignard reagents to α,β-unsaturated ketones,3 esters4,5 and thioesters6 using catalysts based on Josiphos type ligands (L1 or L2)4 and various copper salts (Fig. 1). The group of Loh reported an alternative for the enantioselective CA to α,β-unsaturated esters with a catalyst consisting of Tol-BINAP (L3)5 and copper iodide (Fig. 1). Despite this progress, as far as we know only one method has been described for the enantioselective CA of Grignard reagents to α,β-unsaturated amides, with enantioselectivities ranging from 30 to 74%.7 This paucity in examples is probably due to the low intrinsic reactivity of α,β-unsaturated amides compared to esters and ketones.8 This limited the formation of chiral amides, one of the most common structural motifs in modern pharmaceuticals and biologically active compounds.9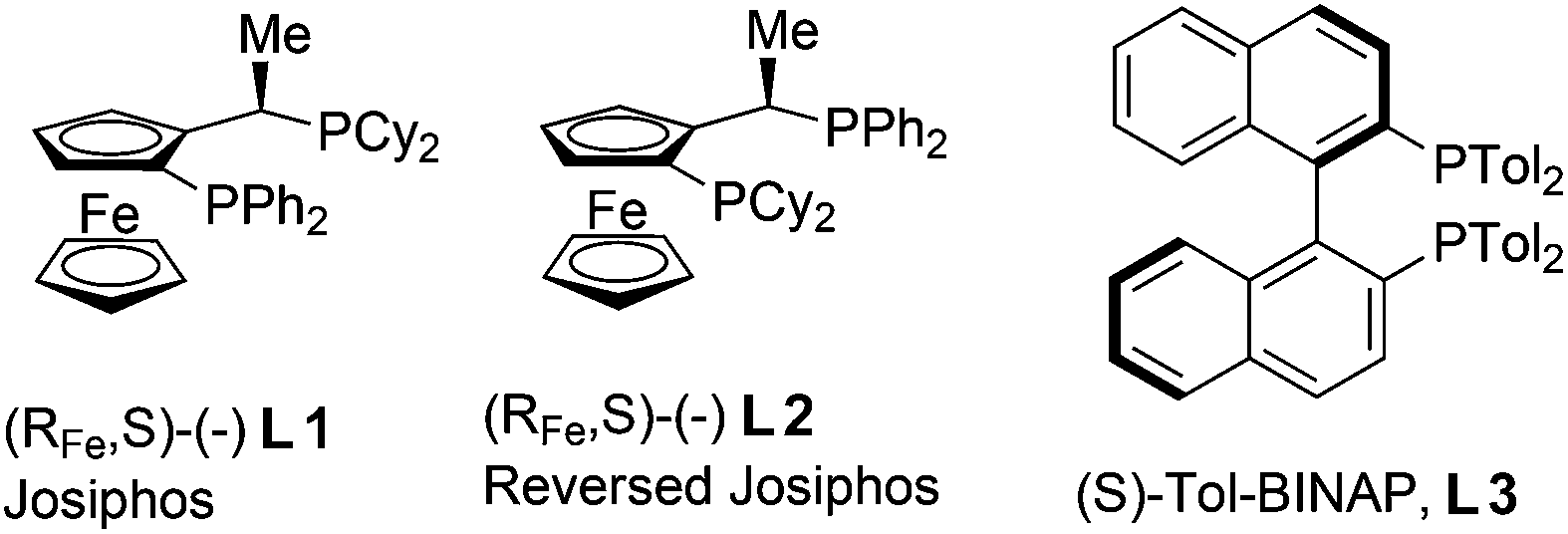 | ||
| Fig. 1 Diphosphine ligands used in Cu-catalysed asymmetric CA. | ||
Chiral β-substituted amides can be obtained with high stereoselectivities via the CA of Grignard reagents to α,β-unsaturated amides bearing (S,S)-pseudoephedrine as a chiral auxiliary.7,10,11 The addition proceeds both with7 and without10,11 copper as a catalyst. However, to obtain the desired chiral β-alkyl substituted amides via this route two additional steps are required: one to remove the auxiliary by hydrolysis and another to obtain the corresponding amide.
An alternative method to obtain β-alkyl substituted amides comprises the asymmetric CA of boron compounds to α,β-unsaturated amides (including 5,6-dihydro-2(1H)-pyridinones). This has been achieved via rhodium-catalysed addition of arylboron12 reagents and via copper-catalysed addition of diboron13 reagents. However, these methods do not allow the introduction of alkyl groups to furnish β-alkyl substituted amides. Therefore the formation of β-alkyl substituted chiral amides remains a challenging target as this structure is present in many biologically active compounds including cyclotheonamide E414 and orbiculamide A.15
In this context, the copper-catalysed enantioselective CA of dialkylzinc reagents to α,β-unsaturated N-acyloxazolidinones16 has been reported to provide the corresponding chiral β-alkyl compounds. The desired chiral β-alkyl substituted amide can be obtained from the corresponding oxazolidinones in one additional step. Furthermore, the copper-catalysed enantioselective CA to α,β-unsaturated lactams has been described with alkylzinc reagents17 and alkyl- and alkenylalananes17,18 giving rise to β-substituted lactams in good yields and selectivities. There is also one example of the asymmetric CA of Me3Al to an N-acylpyrrole derivative in which the corresponding methylated product is obtained in 54% yield and 96% ee.19
Recently, we discovered that β-alkyl substituted amides could be obtained directly by quenching a cyclic ester enolate with propylamine.20 This enolate was formed during the enantioselective CA of EtMgBr to coumarins; quenching with propylamine gave the corresponding amide in a good yield without compromising the ee of the enantioselective CA product. Inspired by this result, we envisioned a one pot procedure for the synthesis of β-alkyl substituted amides consisting of first the enantioselective CA of Grignard reagents to an acyclic α,β-unsaturated ester and subsequent in situ quenching with an amine to obtain the corresponding amide (Scheme 1). Herein, we present a new one pot method to obtain β-alkyl substituted amides in an asymmetric fashion, in which the amide formation occurs without the need for additional reagents or reaction steps.
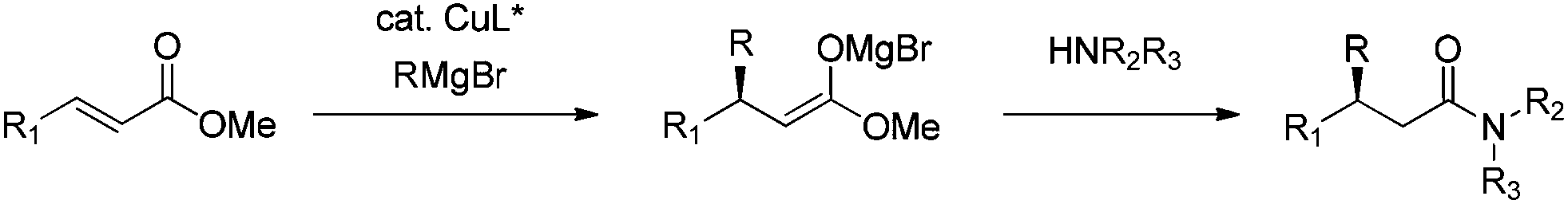 | ||
| Scheme 1 Catalytic asymmetric CA to esters and in situ formation of amides. | ||
Results and discussion
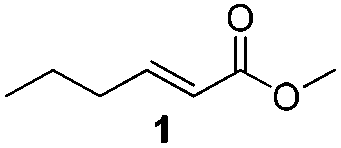
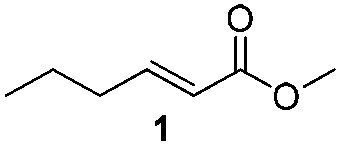
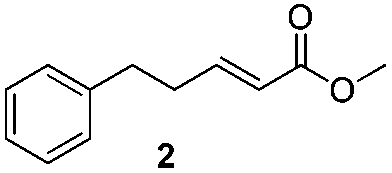
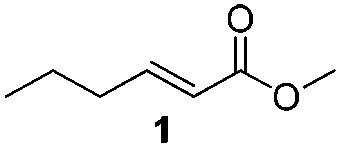
For this one pot procedure, we focused on the optimisation of the amide formation after the enantioselective CA step, which was carried out with ligand L1 and CuBr·SMe2 at −75 °C as previously reported.4 We optimised the reaction conditions and determined that the use of 3.0 equiv. of amine and stirring the reaction mixture at room temperature overnight (ca. 14 h) were the optimised conditions.For our investigations, we used α,β-unsaturated esters 1 and 2 as model substrates, because of their high enantioselectivities in the CA of Grignard reagents and as they represent substrates bearing alkyl or aryl substituents.21,22
The enantioselective CA to substrates 1 and 2 was performed, with EtMgBr (1.5 equiv.), CuBr·SMe2 (5 mol%) and L1 (7.5 mol%) in methyl tert-butyl ether (MTBE).4 Subsequently the intermediate enolate was quenched in situ with various amines and allowed to react under the optimised reaction conditions for amide formation (Table 1).
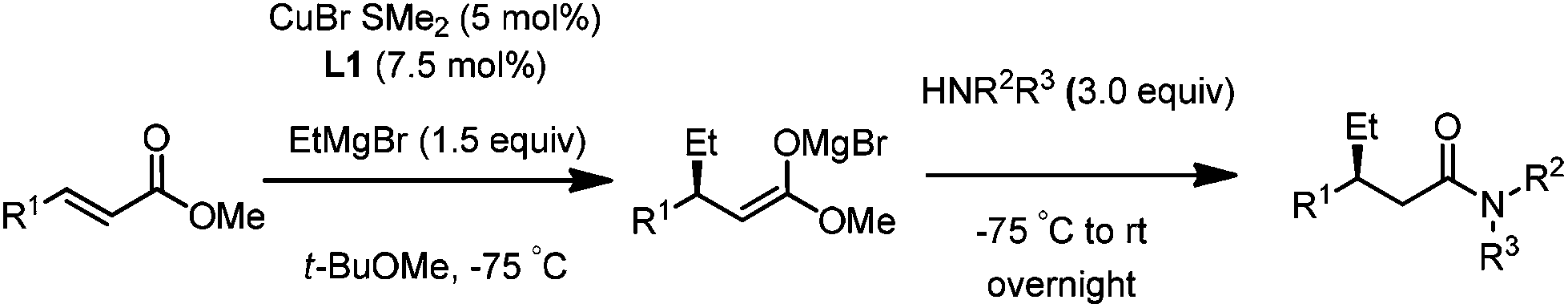
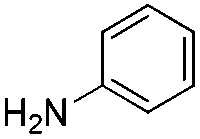
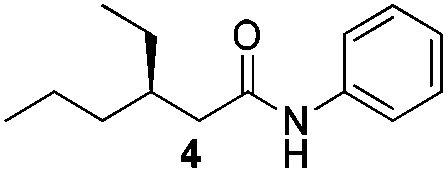
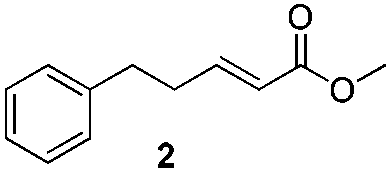
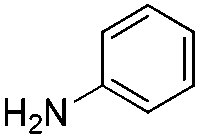
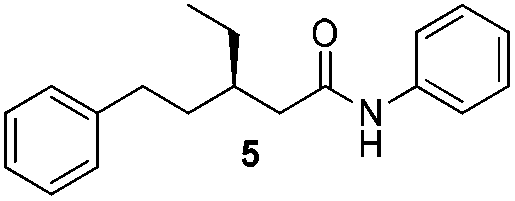
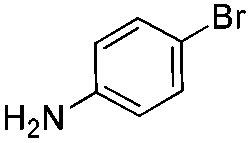
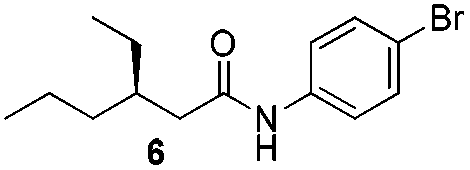
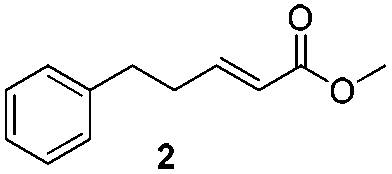
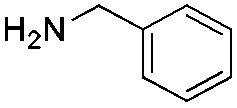

We found that the reaction of α,β-unsaturated esters 1 and 2 with 3.0 equiv. of aniline provided the corresponding amides 4 and 5 with good yields (65% and 73%, respectively) and excellent enantioselectivities, revealing that no racemisation occurs during the process (entries 1 and 2). The reaction using p-bromoaniline furnished amide 6 with good yield (77%) without compromising the ee (entry 3). We also carried out the reaction employing benzylamine and similar results were obtained (entry 4).
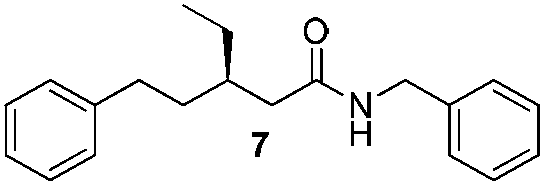
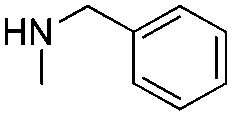
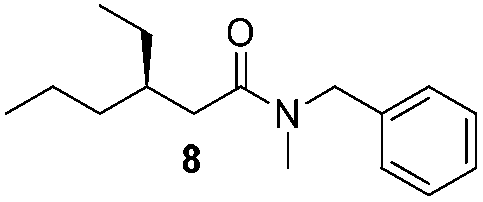
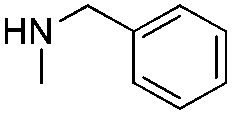
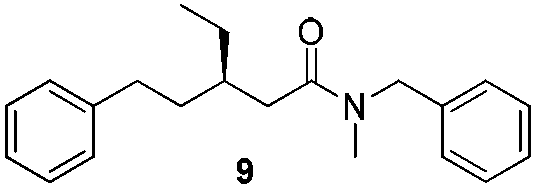
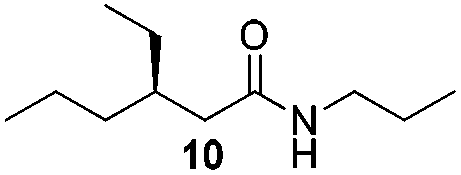
In order to broaden the amine scope, the reaction was carried out with the secondary N-methylbenzylamine giving rise to the corresponding amides 8 and 9 with synthetically useful yields (entries 5 and 6). Finally, the reaction was performed with aliphatic propylamine, which yields amide 10 in moderate yield and excellent ee (entry 7).23 In addition, this method has been employed to obtain the biologically active amide 1124 with 40% yield and 98% ee in a one pot two step synthesis starting from commercially available materials (entry 8).
To underscore the synthetic utility of this method, we performed the one pot synthesis of amide 6 on a large scale (entry 3) starting from 1.0 g (7.8 mmol) of the α,β-unsaturated ester 1 and we obtained 1.8 g (75% yield) of amide 6 with 92% ee.
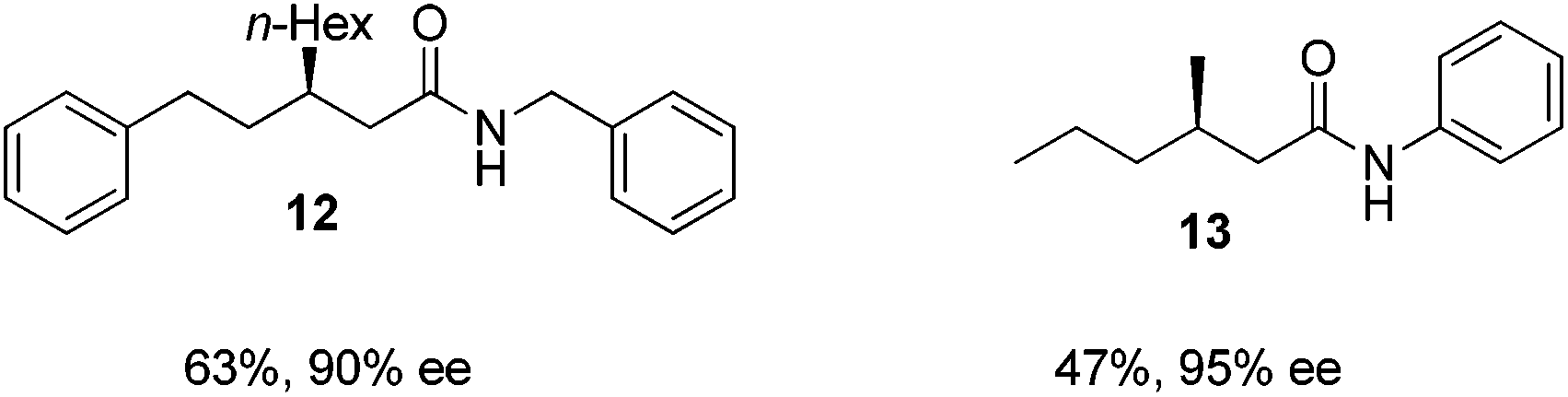
Finally, we employed different Grignard reagents including the less reactive MeMgBr (Fig. 2). The reaction using n-HexMgBr and benzylamine furnished the amide 12 with good yield (63%) and excellent ee. In addition, the Me group is introduced employing the conditions of Loh et al.5b followed by the addition of aniline, which afforded the amide 13 in moderate yield (47%) but excellent enantioselectivity.
 | ||
| Fig. 2 Different alkyl Grignard reagents. | ||
Conclusions
In conclusion, we have developed a highly enantioselective one pot procedure for the synthesis of β-substituted amides starting from the corresponding α,β-unsaturated esters. This new methodology is based on the previously developed copper-catalysed asymmetric CA of Grignard reagents to α,β-unsaturated esters and the subsequent direct formation of the corresponding amides by quenching the enolate with an amine without the need of extra reagents. This methodology is compatible with a range of α,β-unsaturated esters, Grignard reagents and amines, including primary and secondary amines bearing alkyl or aryl substituents. Importantly, this one pot two-step process occurs without racemisation. Furthermore, the robustness of the process is demonstrated by performing the reaction on a 1.0 g scale. Further synthetic applications are in progress.Experimental
General methods
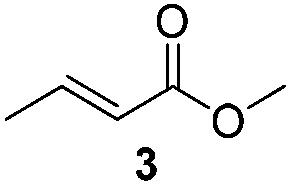
Chromatography: Merck silica gel type 9385 230–400 mesh, TLC: Merck silica gel 60, 0.25 mm. Components were visualized by UV and cerium/molybdenum staining. Mass spectra were recorded on a LTQ Orbitrap XL (ESI+). 1H- and 13C-NMR were recorded on a Varian AMX400 (400 and 100 MHz, respectively) or a Varian Mercury Plus (200 and 50 MHz, respectively) using CDCl3 as a solvent. Chemical shift values are reported in ppm with the solvent resonance as the internal standard (CHCl3: δ 7.26 for 1H, δ 77.16 for 13C). Optical rotations were measured using a Schmidt+Haensch polarimeter (Polartronic MH8) with a 10 cm cell (c given in g per 100 mL). Enantiomeric excesses were determined by HPLC analysis using a Shimadzu LC-10ADVP HPLC equipped with a Shimadzu SPD-M10AVP diode array detector or by capillary GC analysis (Chiraldex B-PM (30 m × 0.25 mm × 0.25 μm)) using a flame ionization detector. All reactions were carried out under a nitrogen atmosphere using flame-dried glassware and using standard Schlenk techniques. Dichloromethane and MTBE were dried using the solvent purification system SPS 800 from MBraun. Substrates 2 and 3 were synthesised according to literature procedures.20,21 Methyl trans-cinnamate (4), copper salts (CuI and copper(I) bromide dimethylsulfide complex (CuBr)), propylamine and N-benzylmethylamine, all Grignard reagents (MeMgBr (3.0 M in Et2O), EtMgBr (3.0 M in Et2O), n-HexMgBr (2.0 M in Et2O)), ligands L2 and L3 were purchased from Aldrich. The benzylamine was purchased from Acros and aniline from Merck; all amines were distilled before use.General procedure for the amide synthesis
In a Schlenk tube equipped with a septum and a stirring bar, L2 (6.7 mg, 10.5 μmol) and CuBr·SMe2 (1.85 mg, 9.0 μmol) were dissolved in MTBE (1.0 mL) and the mixture was stirred under nitrogen at r.t. for 20 min. The mixture was then cooled to −75 °C and the Grignard reagent (0.45 mmol in solution) was added. After stirring for 5 min, a solution of the substrate (0.3 mmol) in MTBE (0.20 mL) was added dropwise over 1 h. After stirring at −75 °C for two additional hours, the corresponding amine (0.9 mmol) was added and the reaction mixture was stirred overnight while being slowly warmed to r.t. Subsequently a saturated aqueous solution of NH4Cl (2 mL) was added. After extraction with CH2Cl2 (3 × 10 mL) the combined organic phases were dried over MgSO4 and concentrated under reduced pressure. The crude product was purified using flash chromatography on silica gel (pentane–EtOAc as an eluent) to yield the pure product.25![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 138.0 (C), 128.93 (CH), 124.1 (CH), 119.8 (CH), 42.5 (CH2), 36.6 (CH), 35.6 (CH2), 26.2 (CH2), 19.8 (CH2), 14.4 (CH3), 10.8 (CH3); HRMS (APCI+, m/z): calculated for C14H22NO [M + H+]: 220.1696, found: 220.1696; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 95
O), 138.0 (C), 128.93 (CH), 124.1 (CH), 119.8 (CH), 42.5 (CH2), 36.6 (CH), 35.6 (CH2), 26.2 (CH2), 19.8 (CH2), 14.4 (CH3), 10.8 (CH3); HRMS (APCI+, m/z): calculated for C14H22NO [M + H+]: 220.1696, found: 220.1696; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 40 °C, 0.5 mL min−1, 213 nm, retention times (min) t1: 34.74 (minor) and t2: 38.93 (major).
O), 142.5 (C), 138.0 (C), 128.9 (CH), 128.4 (CH), 128.3 (CH), 125.8 (CH), 124.2 (CH), 120.0 (CH), 42.3 (CH2), 36.6 (CH), 35.2 (CH2), 33.0 (CH2), 26.1 (CH2), 10.7. (CH3); HRMS (APCI+, m/z): calculated for C16H26NO [M + H+]: 281.1852, found: 282.1853; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 90:10, 40 °C, 0.5 mL min−1, 213 nm, retention times (min) t1: 12.8 (minor) and t2: 14.10 (major).
O), 136.9 (C), 132.1 (CH), 121.6 (CH), 118.7 (CBr), 42.8 (CH2), 36.8 (CH), 35.9 (CH2), 26.5 (CH2), 20.0 (CH2), 14.5 (CH3), 11.0 (CH3); HRMS (APCI+, m/z): calculated for C14H21BrNO [M + H+]: 298.0801, found: 298.0803; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OJ-H column, n-heptane–i-PrOH 98:2, 40 °C, 0.5 mL min−1, 249 nm, retention times (min) t1: 47.7 (major) and t2: 49.6 (minor).
O), 142.6 (C), 138.4 (C), 128.7 (CH), 128.3 (CH), 128.3 (CH), 127.9 (CH), 127.5 (CH), 125.7 (CH), 43.6 (CH2N), 41.3 (CH2), 36.5 (CH), 35.2 (CH2), 33.0 (CH2), 26.1 (CH2), 10.7 (CH3); HRMS (APCI+, m/z): calculated for C20H26NO [M + H+]: 296.2009, found: 296.2008; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 90:10, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 25.12 (minor) and t2: 28.53 (major).
:3 mixture of conformers A and B). [α]20D = +39 (c 1.0 in CHCl3); 1H NMR (400 MHz) δ 7.45–7.20 (m, 4H), 7.16 (d, J = 7.4 Hz, 1H), 4.60 (s, NCH2Ph, conformer A), 4.54 (s, NCH2Ph, conformer B) 2.94 (s, NCH3, conformer B), 2.92 (s, NCH3, conformer A), 2.34–2.25 (m, 2H), 2.05–1.78 (m, 1H), 1.45–1.20 (m, 6H), 1.01–0.77 (m, 6H); 13C NMR (50 MHz) δ 173.4 (CO), 173.0 (CO), 137.7 (C), 128.9 (CH), 128.5 (CH), 128.0 (CH), 127.5 (CH), 127.2 (CH), 126.3 (CH), 53.4 (CH2), 50.8 (CH2), 37.8 (CH2), 37.5 (CH2), 36.1 (CH3), 36.0 (CH3), 35.8 (CH), 35.7 (CH), 26.3 (CH2), 26.2 (CH2), 19.8 (CH2), 14.4 (CH3), 10.9 (CH3); HRMS (APCI+, m/z): calculated for C16H26NO [M + H+]: 248.2009, found: 248.2015; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OJ-H column, n-heptane–i-PrOH 98:02, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 13.12 (minor) and t2: 13.87 (major).
:5 mixture of conformers A and B). [α]20D = −10 (c 1.0 in CHCl3); 1H NMR (200 MHz) δ 7.31 (m, 10H), 4.60 (s, NCH2Ph, conformer B), 4.52 (s, NCH2Ph, conformer A) 2.94 (s, NCH3, conformer A), 2.89 (s, NCH3, conformer B), 2.61 (m, 2H), 2.35 (m, 2H), 2.16–1.92 (m, 1H), 1.77–1.54 (m, 2H), 1.54–1.35 (m, 2H), 0.94 (t, J = 5.9 Hz, 3H, conformer B), 0.87 (t, J = 5.8 Hz, 3H, conformer A); 13C NMR (50 MHz) δ 173.3.0 (CO), 172.7 (CO), 142.7 (C), 137.6 (C), 136.8 (C), 129.6 (CH), 128.9 (CH), 128.8 (CH), 128.5 (CH), 128.4 (CH), 128.3 (CH), 128.0 (CH), 127.5 (CH), 127.3 (CH), 126.3 (CH), 125.6 (CH), 53.9 (CH2), 50.9 (CH2), 37.7 (CH2), 37.4 (CH2) 36.0 (CH), 35.4 (CH3), 35.3 (CH2), 33.1 (CH2), 26.3 (CH2), 26.2 (CH2), 10.8 (CH3); HRMS (APCI+, m/z): calculated for C21H28NO [M + H+]: 310.2165, found: 310.2167; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OJ-H column, n-heptane–i-PrOH 95:5, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 24.97 (minor) and t2: 26.45 (major).
O), 41.6 (CH2), 41.1 (CH2), 36.5 (CH), 35.6 (CH2), 26.0 (CH2), 22.9 (CH2), 19.7 (CH2), 14.3 (CH3), 11.3 (CH3), 10.7 (CH3); HRMS (APCI+, m/z): calculated for C11H24NO [M + H+]: 186.1852, found: 186.1850; enantiomeric excess was determined by chiral GC analysis, Chiraldex B-PM column, T 110 (5 min), 120 (5 min), 130 (5 min), 135 (15 min), ΔT 5 °C min−1, retention times (min) t1: 27.54 (major) and t2: 28.83 (minor).
O), 138.9 (C), 128.7 (CH), 128.6 (CH), 126.4 (CH), 44.2 (CH2), 40.4 (CH2), 35.8 (CH2), 32.3 (CH), 29.3 (CH2), 19.1 (CH3), 11.3 (CH3); HRMS (APCI+, m/z): calculated for C14H22NO [M + H+]: 220.1697, found: 220.1696; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 98:2, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 28.60 (major) and t2: 35.59 (minor).
O), 142.6 (C), 138.4 (C), 128.7 (CH), 128.3 (CH), 128.3 (CH), 127.9 (CH), 127.5 (CH), 125.7 (CH), 43.6 (CH2), 41.8 (CH2), 35.7 (CH), 35.3 (CH2), 33.7 (CH2), 33.0 (CH2), 31.9 (CH2), 29.0 (CH2), 26.4 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (APCI+, m/z): calculated for C24H34NO [M + H+]: 352.2635, found: 352.2632; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 90:10, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 21.26 (minor) and t2: 22.52 (major).
O), 137.9 (C), 128.9 (CH), 124.1 (CH), 119.8 (CH), 45.6 (CH2), 39.1 (CH), 30.6 (CH2), 20.1 (CH2), 19.6 (CH3), 14.2 (CH3); HRMS (APCI+, m/z): calculated for C13H20NO [M + H+]: 206.1539, found: 206.1540; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 95:5, 40 °C, 0.5 mL min−1, 254 nm, retention times (min) t1: 35.38 (minor) and t2: 43.70 (major).
:5, 40 °C, 0.5 mL min−1, 213 nm, retention times (min) t1: 34.74 (minor) and t2: 38.93 (major).
O), 142.5 (C), 138.0 (C), 128.9 (CH), 128.4 (CH), 128.3 (CH), 125.8 (CH), 124.2 (CH), 120.0 (CH), 42.3 (CH2), 36.6 (CH), 35.2 (CH2), 33.0 (CH2), 26.1 (CH2), 10.7. (CH3); HRMS (APCI+, m/z): calculated for C16H26NO [M + H+]: 281.1852, found: 282.1853; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 90:10, 40 °C, 0.5 mL min−1, 213 nm, retention times (min) t1: 12.8 (minor) and t2: 14.10 (major).
O), 136.9 (C), 132.1 (CH), 121.6 (CH), 118.7 (CBr), 42.8 (CH2), 36.8 (CH), 35.9 (CH2), 26.5 (CH2), 20.0 (CH2), 14.5 (CH3), 11.0 (CH3); HRMS (APCI+, m/z): calculated for C14H21BrNO [M + H+]: 298.0801, found: 298.0803; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OJ-H column, n-heptane–i-PrOH 98:2, 40 °C, 0.5 mL min−1, 249 nm, retention times (min) t1: 47.7 (major) and t2: 49.6 (minor).
O), 142.6 (C), 138.4 (C), 128.7 (CH), 128.3 (CH), 128.3 (CH), 127.9 (CH), 127.5 (CH), 125.7 (CH), 43.6 (CH2N), 41.3 (CH2), 36.5 (CH), 35.2 (CH2), 33.0 (CH2), 26.1 (CH2), 10.7 (CH3); HRMS (APCI+, m/z): calculated for C20H26NO [M + H+]: 296.2009, found: 296.2008; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 90:10, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 25.12 (minor) and t2: 28.53 (major).
:3 mixture of conformers A and B). [α]20D = +39 (c 1.0 in CHCl3); 1H NMR (400 MHz) δ 7.45–7.20 (m, 4H), 7.16 (d, J = 7.4 Hz, 1H), 4.60 (s, NCH2Ph, conformer A), 4.54 (s, NCH2Ph, conformer B) 2.94 (s, NCH3, conformer B), 2.92 (s, NCH3, conformer A), 2.34–2.25 (m, 2H), 2.05–1.78 (m, 1H), 1.45–1.20 (m, 6H), 1.01–0.77 (m, 6H); 13C NMR (50 MHz) δ 173.4 (CO), 173.0 (CO), 137.7 (C), 128.9 (CH), 128.5 (CH), 128.0 (CH), 127.5 (CH), 127.2 (CH), 126.3 (CH), 53.4 (CH2), 50.8 (CH2), 37.8 (CH2), 37.5 (CH2), 36.1 (CH3), 36.0 (CH3), 35.8 (CH), 35.7 (CH), 26.3 (CH2), 26.2 (CH2), 19.8 (CH2), 14.4 (CH3), 10.9 (CH3); HRMS (APCI+, m/z): calculated for C16H26NO [M + H+]: 248.2009, found: 248.2015; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OJ-H column, n-heptane–i-PrOH 98:02, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 13.12 (minor) and t2: 13.87 (major).
:5 mixture of conformers A and B). [α]20D = −10 (c 1.0 in CHCl3); 1H NMR (200 MHz) δ 7.31 (m, 10H), 4.60 (s, NCH2Ph, conformer B), 4.52 (s, NCH2Ph, conformer A) 2.94 (s, NCH3, conformer A), 2.89 (s, NCH3, conformer B), 2.61 (m, 2H), 2.35 (m, 2H), 2.16–1.92 (m, 1H), 1.77–1.54 (m, 2H), 1.54–1.35 (m, 2H), 0.94 (t, J = 5.9 Hz, 3H, conformer B), 0.87 (t, J = 5.8 Hz, 3H, conformer A); 13C NMR (50 MHz) δ 173.3.0 (CO), 172.7 (CO), 142.7 (C), 137.6 (C), 136.8 (C), 129.6 (CH), 128.9 (CH), 128.8 (CH), 128.5 (CH), 128.4 (CH), 128.3 (CH), 128.0 (CH), 127.5 (CH), 127.3 (CH), 126.3 (CH), 125.6 (CH), 53.9 (CH2), 50.9 (CH2), 37.7 (CH2), 37.4 (CH2) 36.0 (CH), 35.4 (CH3), 35.3 (CH2), 33.1 (CH2), 26.3 (CH2), 26.2 (CH2), 10.8 (CH3); HRMS (APCI+, m/z): calculated for C21H28NO [M + H+]: 310.2165, found: 310.2167; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OJ-H column, n-heptane–i-PrOH 95:5, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 24.97 (minor) and t2: 26.45 (major).
O), 41.6 (CH2), 41.1 (CH2), 36.5 (CH), 35.6 (CH2), 26.0 (CH2), 22.9 (CH2), 19.7 (CH2), 14.3 (CH3), 11.3 (CH3), 10.7 (CH3); HRMS (APCI+, m/z): calculated for C11H24NO [M + H+]: 186.1852, found: 186.1850; enantiomeric excess was determined by chiral GC analysis, Chiraldex B-PM column, T 110 (5 min), 120 (5 min), 130 (5 min), 135 (15 min), ΔT 5 °C min−1, retention times (min) t1: 27.54 (major) and t2: 28.83 (minor).
O), 138.9 (C), 128.7 (CH), 128.6 (CH), 126.4 (CH), 44.2 (CH2), 40.4 (CH2), 35.8 (CH2), 32.3 (CH), 29.3 (CH2), 19.1 (CH3), 11.3 (CH3); HRMS (APCI+, m/z): calculated for C14H22NO [M + H+]: 220.1697, found: 220.1696; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 98:2, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 28.60 (major) and t2: 35.59 (minor).
O), 142.6 (C), 138.4 (C), 128.7 (CH), 128.3 (CH), 128.3 (CH), 127.9 (CH), 127.5 (CH), 125.7 (CH), 43.6 (CH2), 41.8 (CH2), 35.7 (CH), 35.3 (CH2), 33.7 (CH2), 33.0 (CH2), 31.9 (CH2), 29.0 (CH2), 26.4 (CH2), 22.7 (CH2), 14.1 (CH3); HRMS (APCI+, m/z): calculated for C24H34NO [M + H+]: 352.2635, found: 352.2632; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 90:10, 40 °C, 0.5 mL min−1, 212 nm, retention times (min) t1: 21.26 (minor) and t2: 22.52 (major).
O), 137.9 (C), 128.9 (CH), 124.1 (CH), 119.8 (CH), 45.6 (CH2), 39.1 (CH), 30.6 (CH2), 20.1 (CH2), 19.6 (CH3), 14.2 (CH3); HRMS (APCI+, m/z): calculated for C13H20NO [M + H+]: 206.1539, found: 206.1540; enantiomeric excess was determined by chiral HPLC analysis, Chiralcel OD-H column, n-heptane–i-PrOH 95:5, 40 °C, 0.5 mL min−1, 254 nm, retention times (min) t1: 35.38 (minor) and t2: 43.70 (major).
Acknowledgements
The financial support of the NWO astrochemistry program the National Research School of Catalysis (NRSC-C), and the Ministry of Education, Culture and Science (Gravity Program 024.001.035) is gratefully acknowledged.Notes and references
- (a) P. Knochel, M. C. P. Yeh, S. C. Berk and J. Talbert, J. Org. Chem., 1988, 53, 2390–2392 CrossRef CAS; (b) G. van Koten, J. Organomet. Chem., 1990, 400, 283–301 CrossRef CAS; (c) J. E. Bäckvall and P. G. Andersson, J. Am. Chem. Soc., 1990, 112, 3683 CrossRef; (d) M. Van Klaveren, E. S. M. Persson, A. Del Villar, D. M. Grove, J. E. Bäckvall and G. Van Koten, Tetrahedron Lett., 1995, 36, 3059–3062 CrossRef CAS; (e) K. Tomioka and Y. Nagaoka, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, New York, 1999, vol. III, pp. 1105–1120 Search PubMed; (f) B. L. Feringa, Acc. Chem. Res., 2000, 33, 346–353 CrossRef CAS PubMed; (g) M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033–8061 CrossRef CAS; (h) N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 171–196 CrossRef CAS PubMed; (i) B. L. Feringa, R. Naasz, R. Imbos and L. A. Arnold, in Modern Organocopper Chemistry, ed. N. Krause, VCH, Weinheim, 2002, pp. 224–258 Search PubMed; (j) A. Alexakis and C. Benhaim, Eur. J. Org. Chem., 2002, 3221–3236 CrossRef CAS; (k) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed; (l) P. K. Fraser and S. Woodward, Chem.–Eur. J., 2003, 9, 776–783 CrossRef CAS PubMed; (m) A. H. Hoveyda, A. W. Hird and M. A. Kacprzynski, Chem. Commun., 2004, 1779–1785 RSC; (n) S. Woodward, Angew. Chem., Int. Ed., 2005, 44, 5560–5562 CrossRef CAS PubMed; (o) S. Sulzer-Mosse and A. Alexakis, Chem. Commun., 2007, 3123–3135 RSC; (p) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824–2852 CrossRef CAS PubMed; (q) A. Alexakis, J. E. Bäckvall, N. Krause, O. Pàmies and M. Diéguez, Chem. Rev., 2008, 108, 2796–2823 CrossRef CAS PubMed; (r) T. Jerphagnon, M. G. Pizzuti, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2009, 38, 1039–1075 RSC.
- B. L. Feringa, R. Badorrey, D. Peña, S. R. Harutyunyan and A. J. Minnaard, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5834–5838 CrossRef CAS PubMed.
- (a) G. M. Villacorta, C. P. Rao and S. J. Lippard, J. Am. Chem. Soc., 1988, 110, 3175–3182 CrossRef CAS; (b) K. H. Ahn, R. B. Klassen and S. J. Lippard, Organometallics, 1990, 9, 3178–3181 CrossRef CAS; (c) F. López, S. R. Harutyunyan, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2004, 126, 12784–12785 CrossRef PubMed; (d) B. Maciá, M. Á. Fernández-Ibáñez, N. Mršić, A. J. Minnaard and B. L. Feringa, Tetrahedron Lett., 2008, 49, 1877–1880 CrossRef PubMed.
- F. López, S. Harutyunyan, A. Meetsma, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2005, 44, 2752–2756 CrossRef PubMed.
- (a) S. Wang, S. Ji and T. Loh, J. Am. Chem. Soc., 2007, 129, 276–277 CrossRef CAS PubMed; (b) S. Wang, T. Lum, S. Ji and T. Loh, Adv. Synth. Catal., 2008, 350, 673–677 CrossRef CAS; (c) S. Wang and T. Loh, Chem. Commun., 2010, 46, 8694–8703 RSC.
- (a) B. M. Ruíz, K. Geurts, M. Á. Fernández-Ibáñez, B. ter Horst, A. J. Minnaard and B. L. Feringa, Org. Lett., 2007, 9, 5123–5126 CrossRef PubMed; (b) R. Des Mazery, M. Pullez, F. López, S. R. Harutyunyan, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 9966–9967 CrossRef CAS PubMed.
- K. Biswas and S. Woodward, Tetrahedron: Asymmetry, 2008, 19, 1702–1708 CrossRef CAS PubMed.
- For an explanation of the reactivity of α,β-unsaturated carboxylic acid derivatives relative to that of enones on the basis of their respective LUMO energies, see: S. Matsunaga, T. Kinoshita, S. Okada, S. Harada and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 7559–7570 CrossRef CAS PubMed.
- (a) B. L. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 CrossRef CAS PubMed; (b) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (c) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (d) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- T. Mukaiyama and N. Iwasawa, Chem. Lett., 1981, 913–916 CrossRef CAS.
- E. Reyes, J. L. Vicario, L. Carrillo, D. Badía, U. Uria and A. Iza, J. Org. Chem., 2006, 71, 7763–7772 CrossRef CAS PubMed.
- (a) T. Senda, M. Ogasawara and T. Hayashi, J. Org. Chem., 2001, 66, 6852–6856 CrossRef CAS PubMed; (b) S. Sakuma and N. Miyaura, J. Org. Chem., 2001, 66, 8944–8946 CrossRef CAS PubMed; (c) M. Pucheault, V. Michaut, S. Darses and J. Genet, Tetrahedron Lett., 2004, 45, 4729–4732 CrossRef CAS PubMed.
- H. Chea, H. Sim and J. Yun, Adv. Synth. Catal., 2009, 351, 855–858 CrossRef CAS.
- Y. Murakami, M. Takei, K. Shindo, C. Kitazume, J. Tanaka, T. Higa and H. Fukamachi, J. Nat. Prod., 2002, 65, 259–261 CrossRef CAS PubMed.
- N. Fusetani, T. Sugawara, S. Matsunaga and H. Hirota, J. Am. Chem. Soc., 1991, 113, 7811–7812 CrossRef CAS.
- A. W. Hird and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 1276–1279 CrossRef CAS PubMed.
- M. Pineschi, F. Del Moro, F. Gini, A. J. Minnaard and B. L. Feringa, Chem. Commun., 2004, 1244–1245 RSC.
- P. Cottet, D. Müler and A. Alexakis, Org. Lett., 2013, 15, 828–831 CrossRef CAS PubMed.
- K. Endo, D. Hamada, S. Yakeishi and T. Shibata, Angew. Chem., Int. Ed., 2013, 52, 606–610 CrossRef CAS PubMed.
- J. F. Teichert and B. L. Feringa, Chem. Commun., 2011, 47, 2679–2681 RSC.
- S. G. Alcock, J. E. Baldwin, R. Bohlmann, L. M. Harwood and J. I. Seeman, J. Org. Chem., 1985, 50, 3526–3535 CrossRef CAS.
- R. A. Barrow, T. Hemscheidt, J. Liang, S. Paik, R. E. Moore and M. A. Tius, J. Am. Chem. Soc., 1995, 117, 2479–2490 CrossRef CAS.
- The analysis of the crude mixture by 1H NMR shows that the corresponding β-disubstituted-ester is still present (∼50%), indicating that the second step to obtain the amide does not go to full conversion. Increasing the temperature or prolonging the reaction time did not improve the results.
- S. Schulz, J. S. Dickschat, B. Kunze, I. Wagner-Dobler, R. Diestel and F. Sasse, Mar. Drugs, 2010, 8, 2976–2987 CrossRef CAS PubMed.
- The absolute stereochemistry is assumed to be the same as for the β-substituted esters, see ref. 4 and 5.
| This journal is © The Royal Society of Chemistry 2014 |