Synthesis of benzosultams via an intramolecular sp2 C–H bond amination reaction of o-arylbenzenesulfonamides under metal-free conditions†
Yuewen
Li
a,
Qiuping
Ding
*a,
Guanyinsheng
Qiu
b and
Jie
Wu
*b
aKey Laboratory of Functional Small Organic Molecules, Ministry of Education and College of Chemistry & Chemical Engineering, Jiangxi Normal University, Nanchang, Jiangxi 330022, China. E-mail: dqpjxnu@gmail.com; Fax: +86 791 88120380; Tel: +86 791 88120380
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China. E-mail: jie_wu@fudan.edu.cn; Fax: +86 21 6564 1740; Tel: +86 21 6510 2412
First published on 29th October 2013
Abstract
A practical synthetic method for the generation of benzosultams via an intramolecular sp2 C–H bond amination reaction of o-arylbenzenesulfonamides with PhI(OAc)2–I2 under metal-free conditions is developed. A broad range of substrates are tolerated under mild reaction conditions, affording bioactive benzosultams in good to excellent yields. The resulting benzothiazines could be conveniently transformed into their corresponding iodinated derivatives via electrophilic substitution reactions.
Introduction
Benzosultams are important privileged structures ubiquitously found in drug discovery. Compounds with the core structure usually exhibit versatile inhibitory properties. For instance, benzothiazine dioxide derivatives have been found to show active inhibitory properties against a variety of enzymes such as 11β-HSD2,1 Calpain I,2 HIV integrand,3 and cyclooxygenase-2 (COX-2)4 (Fig. 1). Additionally, it was reported that members of the family also show powerful anti-inflammatory properties; some of the medicines like Meloxicam5 and Piroxicam6 have already been marketed widely.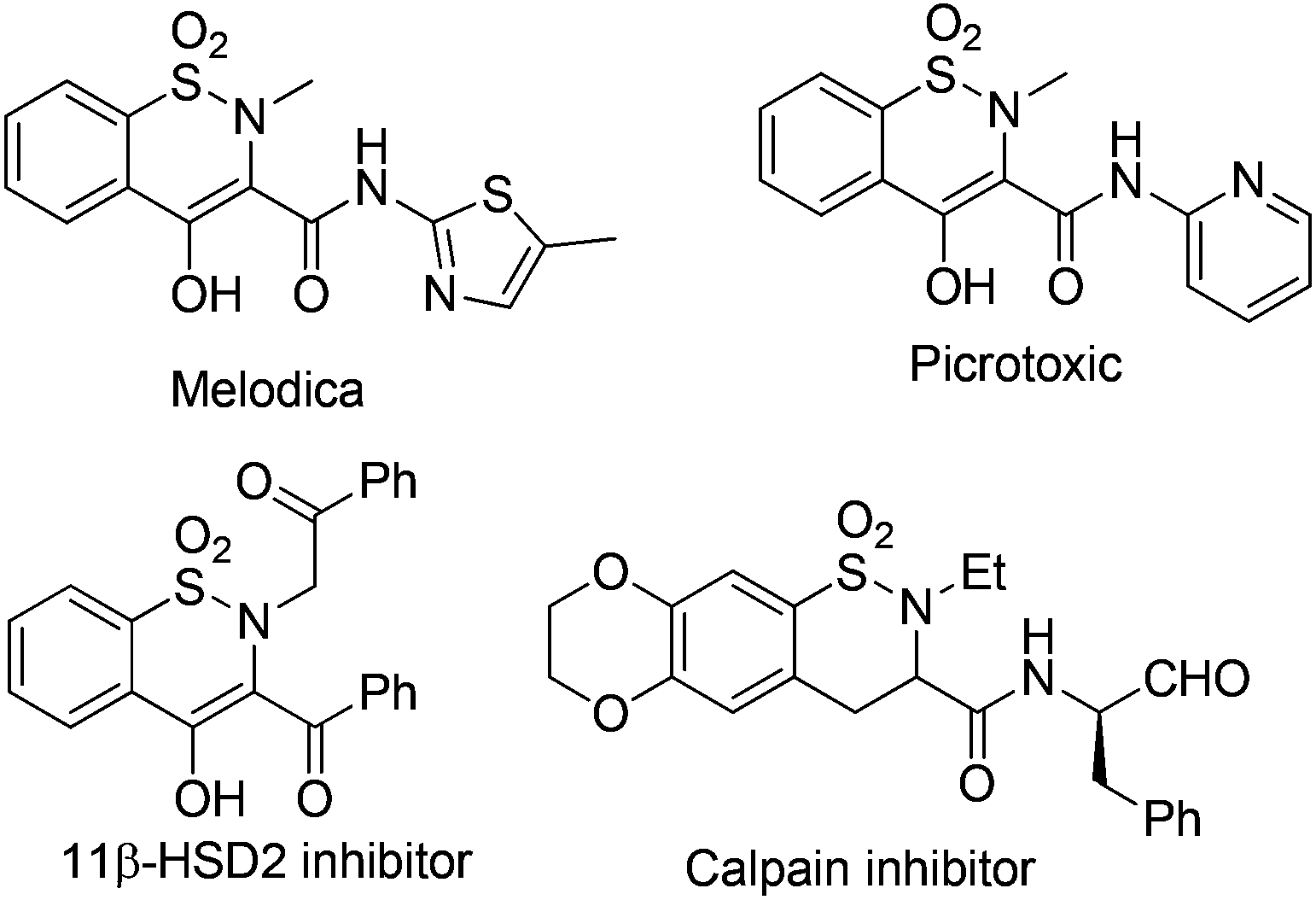 | ||
| Fig. 1 Structures of biologically active benzosultams. | ||
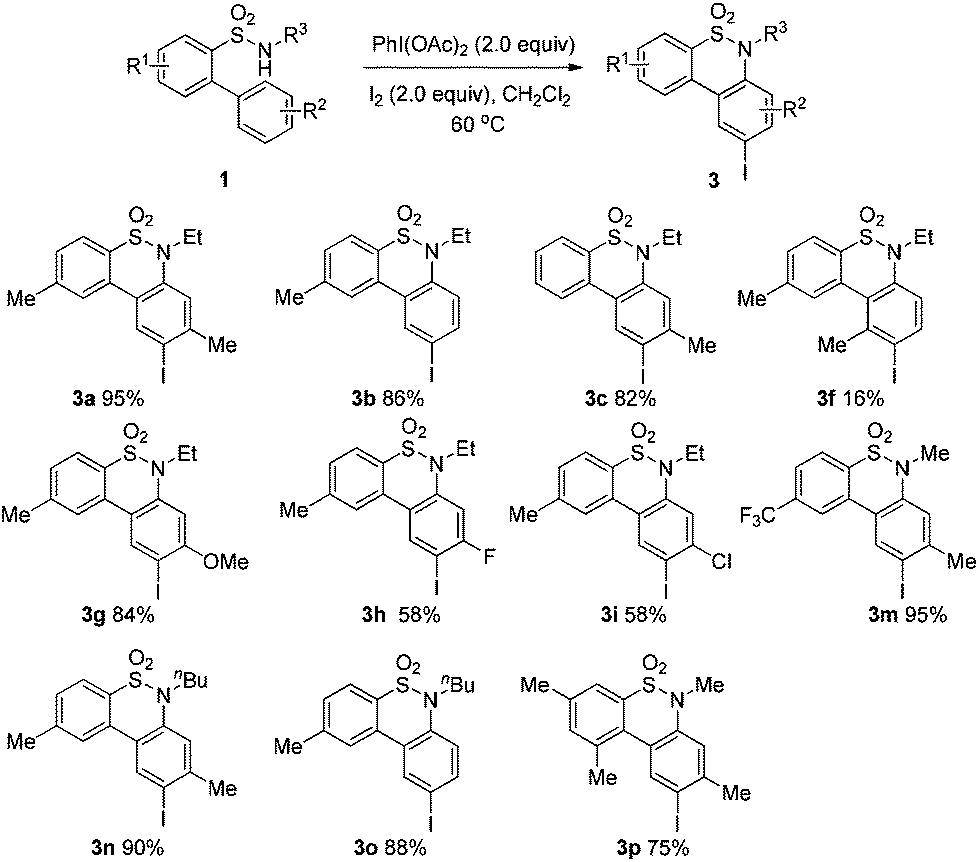
Consequently, there have been several routes successfully developed for the synthesis of benzothiazine derivatives, including the transition-metal or Lewis acid-catalyzed intramolecular cyclization,7 metal-free cyclization,8 as well as the intermolecular synthetic strategy.9 In 2007, Pal and co-workers7e reported a facile single-step Ag- or Cu-catalyzed intramolecular cyclization of o-(1-alkynyl)benzenesulfonamides to afford 3-substituted benzothiazines with high regioselectivity. Thibaudeau and co-workers8a described a novel method to access benzofused sultams starting from N-allylic sulfonamides through superelectrophilic activation by superacid HF–SbF5. Kanai and co-workers9a reported the synthesis of sultams via Cu-catalyzed intermolecular carboamination of alkenes with N-fluorobenzenesulfonimide. Recently, we have developed an efficient synthesis of 6H-benzo[f]cyclopean[d][1,2]thiazinepine 5,5-dioxides via a palladium-catalyzed tandem reaction of 2-(2-alkyl)benzene sulfonamides.10 Inspired by these results, and the recent advancement in C–H bond direct functionalizations,11 we are aware that it is of high urgency to develop a general catalytic system for the synthesis of benzothiazines based on the oxidative sp2 C–H amination strategy under mild and metal-free conditions. Herein we would like to report our recent success in the generation of 6H-dibenzo[c,e][1,2]thiazine 5,5-dioxides 2 from 1,1′-biphenyl-2-sulfonamides 1 with the PhI(OAc)2–I2 system12via an intramolecular sp2 C–H bond amination reaction. In addition, the resulting benzothiazines could be conveniently transformed into the corresponding iodinated derivatives 3via an electrophilic substitution reaction.
Results and discussion
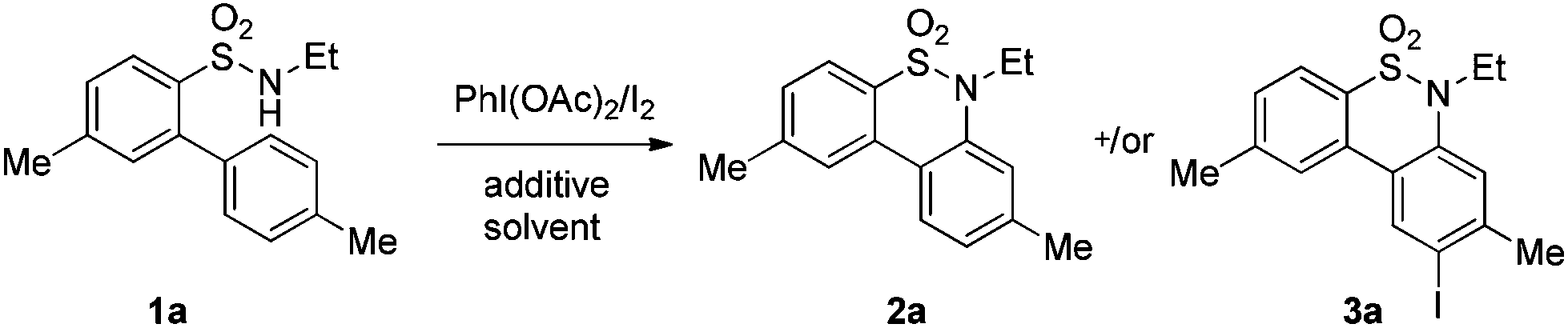
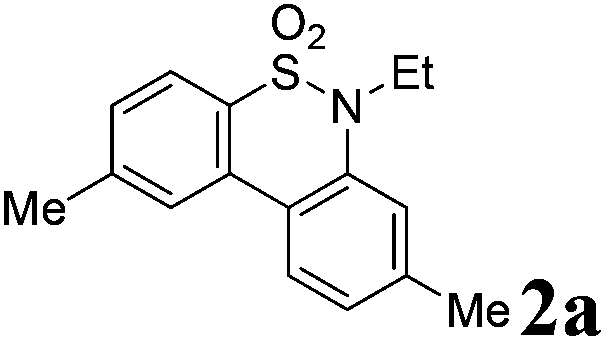
We began the intramolecular sp2 C–H bond amination reaction using N-ethyl-4′,5-dimethyl-[1,1′-biphenyl]-2-sulfonamide 1a as a model substrate. As shown in Table 1, the reaction conditions were initially screened with a combination of PhI(OAc)2 (1.1 equiv.) and I2 (1.1 equiv.) in different solvents at 60 °C. The cyclization reaction did not proceed at all in DMSO (Table 1, entry 1). However, we were pleased to isolate the corresponding product 3a in 39% yield when the reaction was performed in CH3CN (Table 1, entry 2), while the other solvents such as acetone, ethyl acetate, 1,2-dichloroethane, and 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) gave unsatisfactory results (Table 1, entries 3–6). An interesting solvent effect was observed on further screening. Under the same conditions, compound 2a was isolated as a major product in isopropanol, DMF, THF, dioxane, and CH2Cl2 (Table 1, entries 7–11), and the best results 2a and 3a were obtained in 64% and 15% yields in CH2Cl2, respectively (Table 1, entry 11). Further studies revealed that the yield of 2a could be improved when the reaction was carried out at 35 °C (71% yield, Table 1, entry 12). Lowering the amount of iodine to 10 mol% resulted in a drastic decrease of yield (24% yield of 2a, Table 1, entry 13). No product was detected when the reaction was carried out in the absence of PhI(OAc)2 or I2 (Table 1, entries 14 and 15), which indicates the important role of PhI(OAc)2 and I2 in the oxidative transformation. The structures of 2a and 3a were verified by 1H and 13C NMR, HRMS, as well as X-ray diffraction analysis (Fig. 2, also see ESI†). It was reported that potassium carbonate was used as an efficient promoter in the Hofmann–Fleer–Freytag reaction.8f Prompted by this result, we then optimized the reaction conditions using different bases [Na2CO3, KOH, KHCO3, and K2CO3] as additives (Table 1, entries 16–19). K2CO3 was found to be the most efficient one to yield a single product 2a in 94% yield (Table 1, entry 19). On the other hand, the desirable product 3a could be improved to 79% yield when 2.0 equivalents of PhI(OAc)2 were employed (Table 1, entry 20). While increasing the amount of PhI(OAc)2 did not facilitate the transformation, we could improve the yield of 3a to 95% by optimization of the reaction temperature (60 °C) and the amount of I2 (2.0 equivalents) (Table 1, entries 21–23).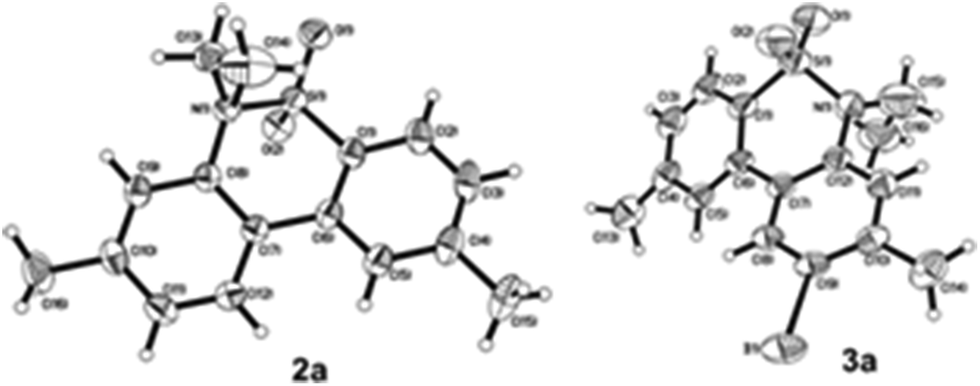 | ||
| Fig. 2 X-ray crystallographic structure of compounds 2a and 3a. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | PhI(OAc)2/I2 (equiv.) | Solvent | T (°C) | Yieldb (%) | |
| 2a | 3a | ||||
| a 1a (0.2 mmol), additive (1.5 equiv.), solvent (2.0 mL), 4 h. HFIP = 1,1,1,3,3,3-hexafluoropropan-2-ol. b Yield of isolated product; n.d. = not detected. c 1.5 Equivalents of Na2CO3 were added. d 1.5 Equivalents of KOH were added. e 1.5 Equivalents of KHCO3 were added. f 1.5 Equivalents of K2CO3 were added. | |||||
| 1 | 1.1/1.1 | DMSO | 60 | Trace | n.d. |
| 2 | 1.1/1.1 | CH3CN | 60 | n.d. | 39 |
| 3 | 1.1/1.1 | Acetone | 60 | n.d. | 12 |
| 4 | 1.1/1.1 | AcOEt | 60 | n.d. | 23 |
| 5 | 1.1/1.1 | DCE | 60 | n.d. | 28 |
| 6 | 1.1/1.1 | HFIP | 60 | n.d. | 37 |
| 7 | 1.1/1.1 | iPrOH | 60 | 40 | Trace |
| 8 | 1.1/1.1 | DMF | 60 | 31 | n.d. |
| 9 | 1.1/1.1 | THF | 60 | 51 | 9 |
| 10 | 1.1/1.1 | Dioxane | 60 | 48 | Trace |
| 11 | 1.1/1.1 | CH2Cl2 | 60 | 64 | 15 |
| 12 | 1.1/1.1 | CH2Cl2 | 35 | 71 | 14 |
| 13 | 1.1/0.1 | CH2Cl2 | 35 | 24 | Trace |
| 14 | 1.1/– | CH2Cl2 | 35 | n.d. | n.d. |
| 15 | –/1.1 | CH2Cl2 | 35 | n.d. | n.d. |
| 16c | 1.1/1.1 | CH2Cl2 | 35 | 78 | n.d. |
| 17d | 1.1/1.1 | CH2Cl2 | 35 | 10 | n.d. |
| 18e | 1.1/1.1 | CH2Cl2 | 35 | 52 | n.d. |
| 19f | 1.1/1.1 | CH2Cl2 | 35 | 94 | n.d. |
| 20 | 2.0/1.1 | CH2Cl2 | 35 | Trace | 79 |
| 21 | 3.0/1.1 | CH2Cl2 | 35 | Trace | 80 |
| 22 | 2.0/1.1 | CH2Cl2 | 60 | n.d. | 84 |
| 23 | 2.0/2.0 | CH2Cl2 | 60 | n.d. | 95 |
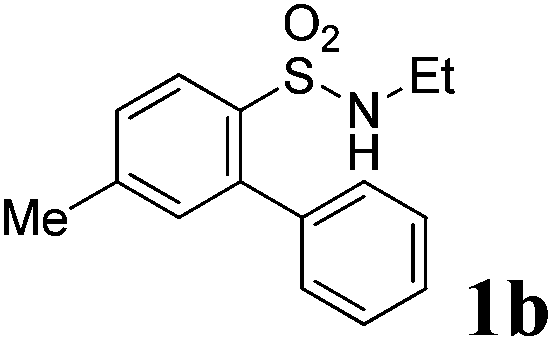
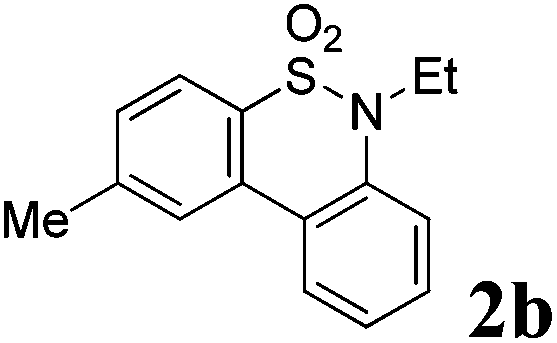
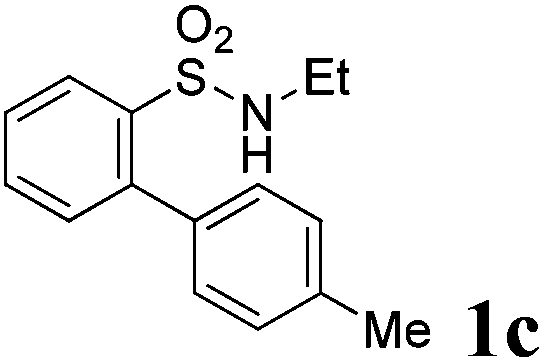
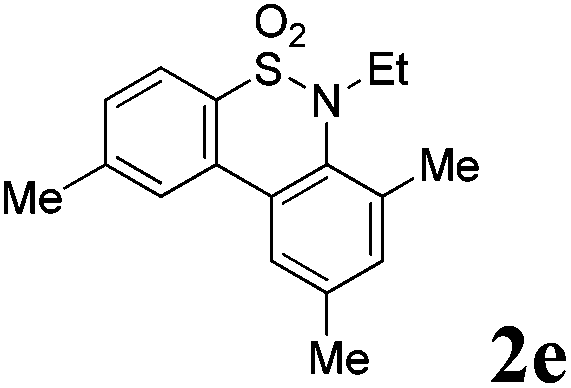
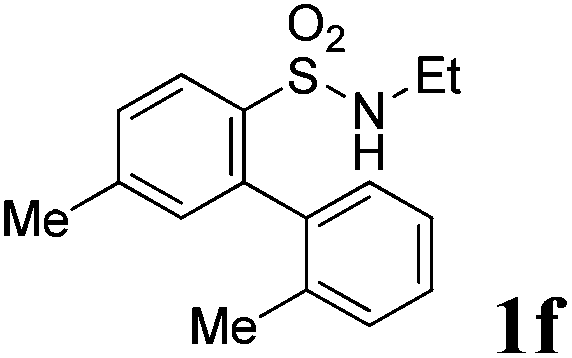
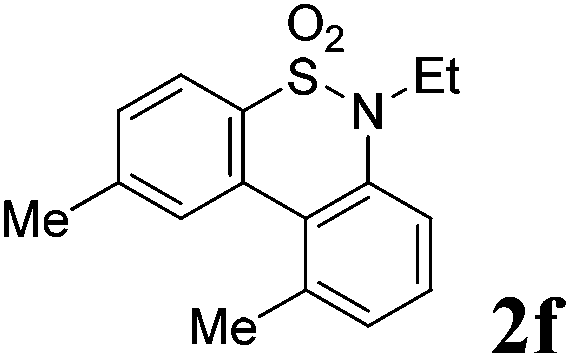
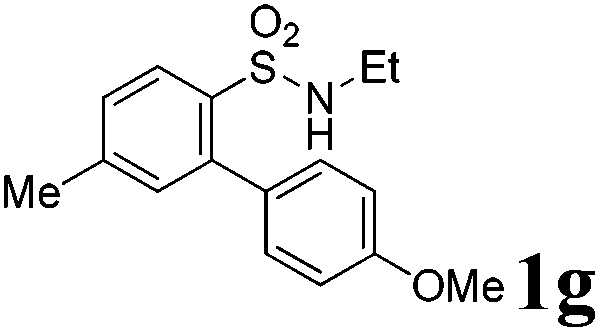
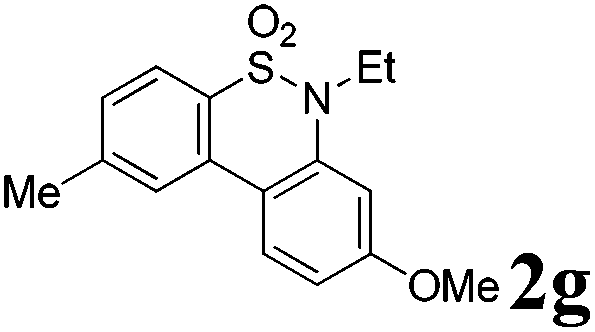
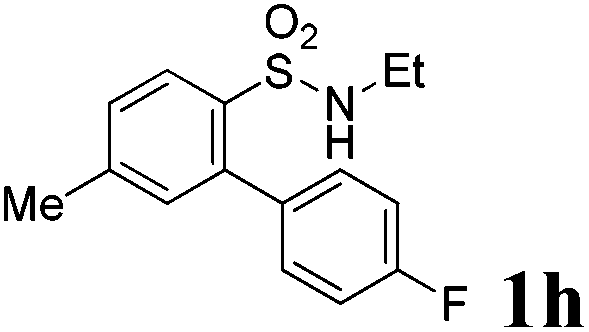
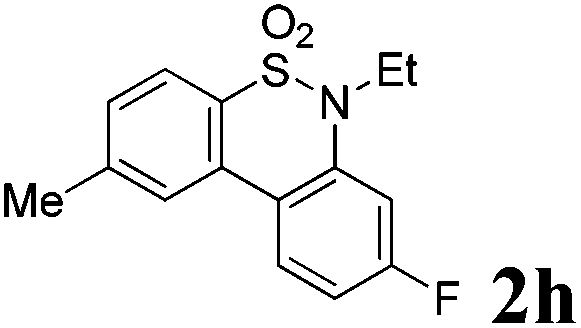
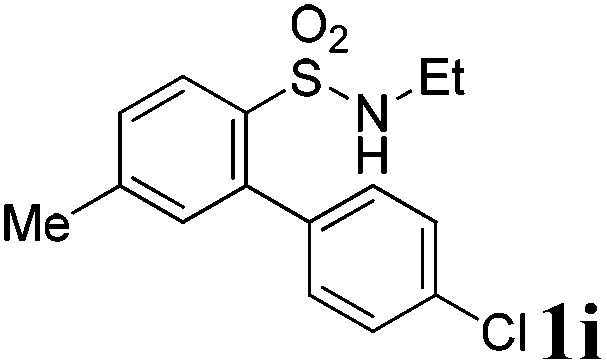
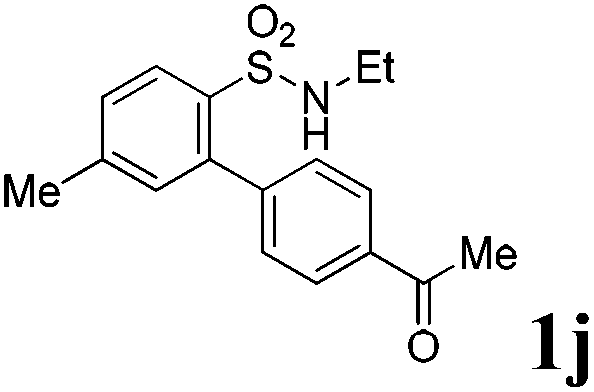
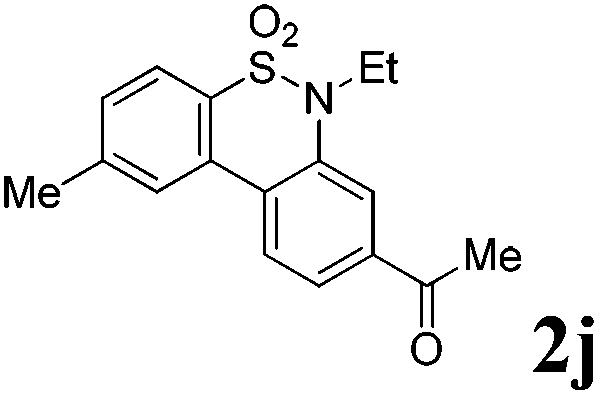
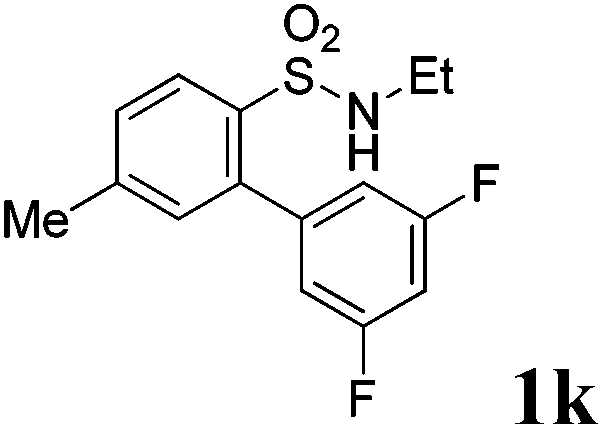
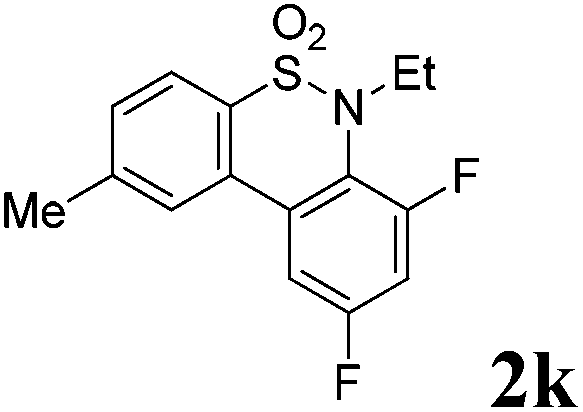
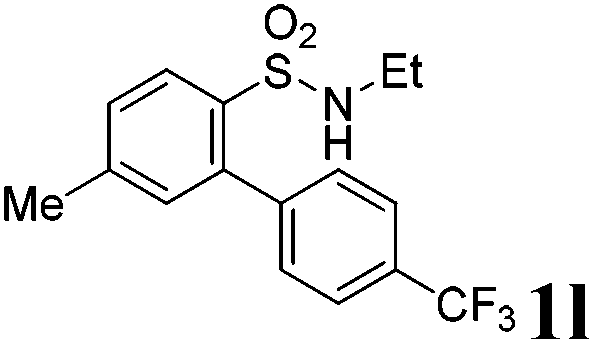
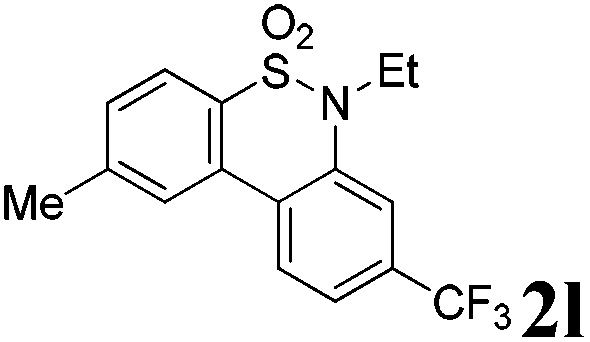
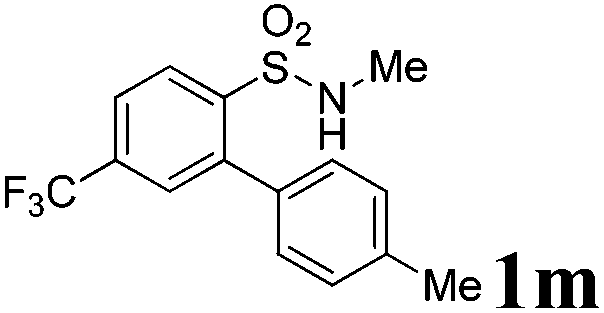
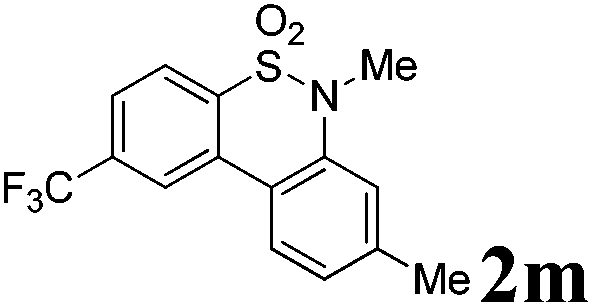
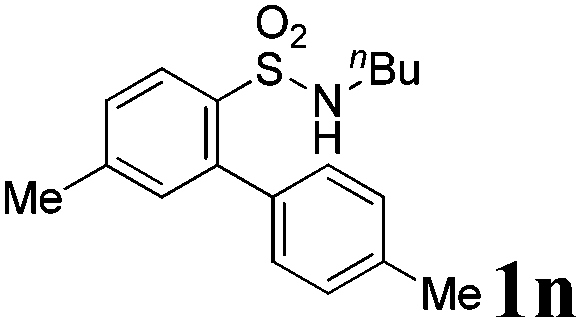
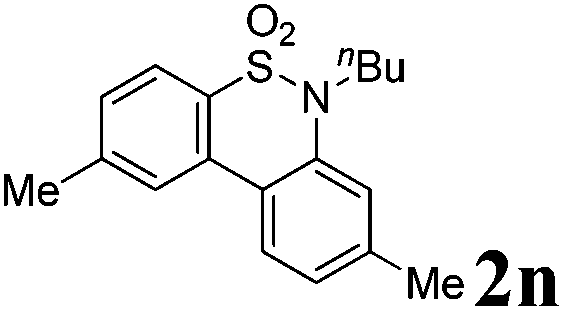
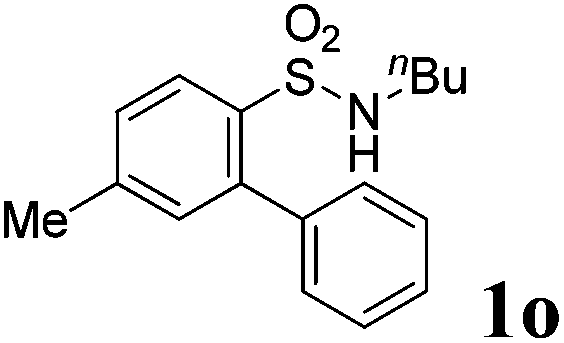
We then explored the substrate scope and generality of the method under the optimized reaction conditions A [o-arylbenzenesulfonamide 1 (0.2 mmol), PhI(OAc)2 (1.1 equiv.), I2 (1.1 equiv.), CH2Cl2 (2 mL), K2CO3 (1.5 equiv.), 35 °C, for 2 h], and the results are presented in Table 2. We first examined the substitution effect on the aryl moiety of compound 1. In general, the oxidative cyclization proceeded smoothly with substrates bearing electron-donating substituents (R2) at the nucleophilic phenyl part (Table 2, entries 3–5 and 7). Substrates with 4-methyl, 3,5-dimethyl, and 4-methoxyl groups were tolerated, providing the corresponding products 2 in yields ranging from 65 to 96%. For instance, when N-ethyl-3′,5,5′-trimethyl-[1,1′-biphenyl]-2-sulfonamide 1e was employed, product 2e was obtained in an excellent yield (96%, Table 2, entry 5). Except for the ortho-position substituted substrate 1f, the corresponding desired product 2f was obtained in poor yield (Table 2, entry 6), which indicated that the steric hindrance effect of the substrate was very obvious. In contrast, substrates with weak electron-withdrawing substituents (e.g. chloro, fluoro, and acetyl) exhibited good reactivity, giving the corresponding products in moderate to excellent yields (62–92%, Table 2, entries 8–10). 4′-Acetyl-N-ethyl-5-methyl-[1,1′-biphenyl]-2-sulfonamide 1j could be converted into the corresponding product 2j in 92% yield (Table 2, entry 10), whereas the reaction of substrates with strong electron-withdrawing substituents displayed lower reactivity. For instance, N-ethyl-5-methyl-4′-(trifluoromethyl)-[1,1′-biphenyl]-2-sulfonamide 1l under the standard reactions resulted in poor conversion (38% yield, Table 2, entry 12). For changing R1 from methyl to strong electron-withdrawing substituents (CF3), the reaction proceeded smoothly to furnish the desired product 2m in almost quantitative yield (99%, Table 2, entry 13). Subsequently, the effect of the nitrogen substituent was investigated. Under the standard reaction conditions A, substrates with R3 bearing alkyl or aryl substituents were studied next. Compounds 1n and 1o (R3 = nBu) exhibited good reactivity affording the expected products 2n and 2o in 85% and 93% yield, respectively (Table 2, entries 14 and 15). For comparison, the aryl-substituted substrates exhibited lower reactivity.

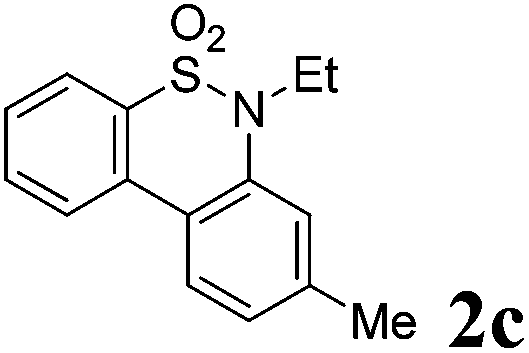
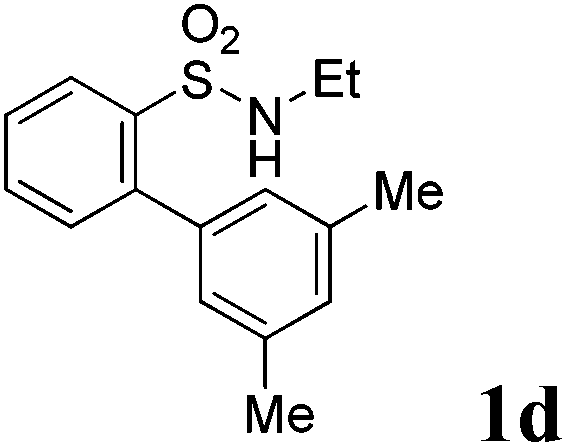
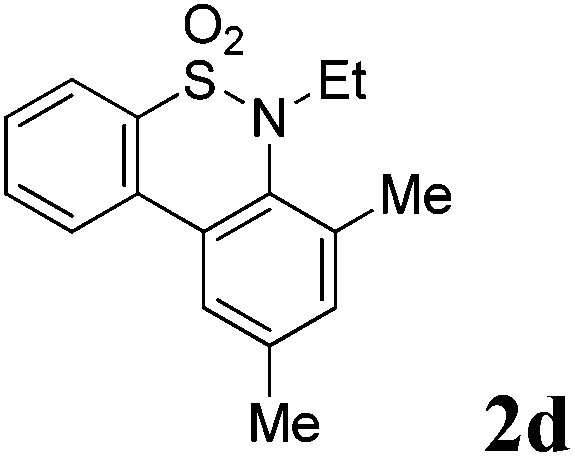
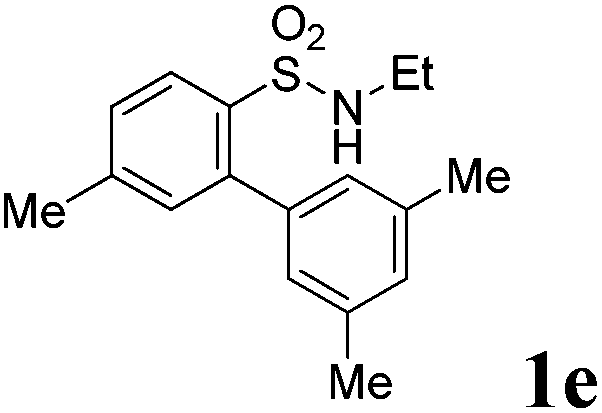
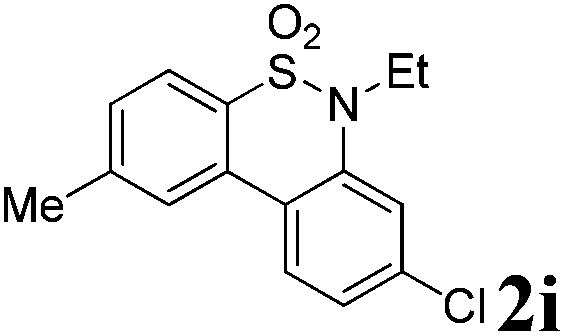
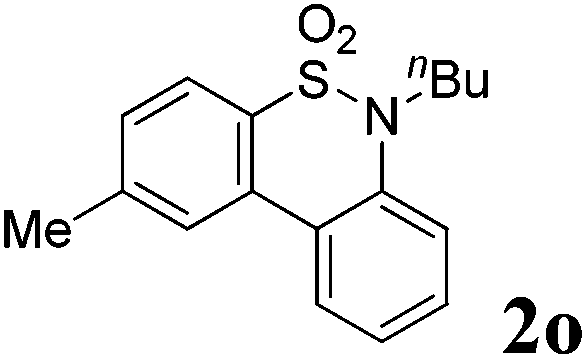
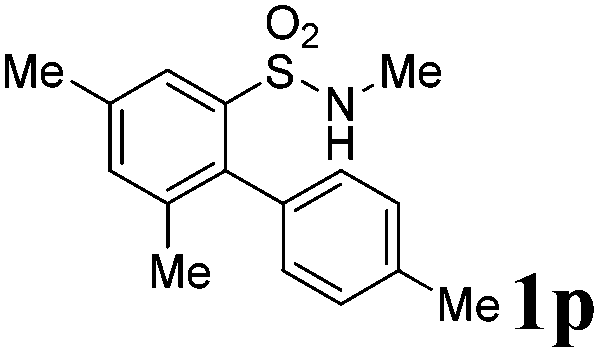
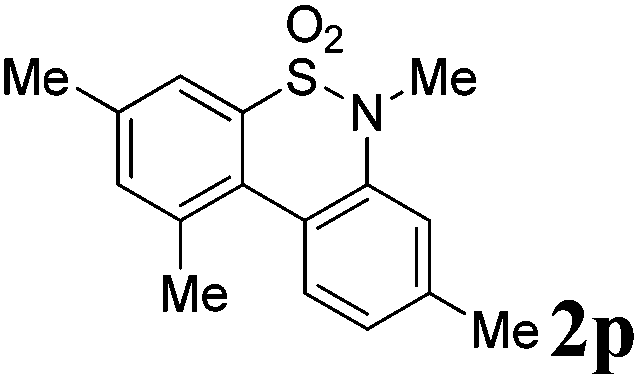
After refining the oxidative intramolecular sp2 C–H bond amination for the synthesis of benzosultams 2 under the reaction conditions A, we next switched our attention to the cascade electrophilic substitution reaction for the synthesis of iodinated benzosultams 3 under the optimized reaction conditions B [o-arylbenzenesulfonamide 1 (0.2 mmol), PhI(OAc)2 (2.0 equiv.), I2 (2.0 equiv.), CH2Cl2 (2 mL), 60 °C, for 2 h] and the results are summarized in Table 3. In general, the reactivity of the cascade reaction under conditions B is similar to the former C–H amination reaction under conditions A, which tolerates a wide range of substituents such as methyl, methoxy, halogen (F and Cl), and trifluoromethyl groups. When substrates 1d and 1e (R2 = 3,5-dimethyl) were subjected to conditions B, 2d and 2e were obtained in high yields (86% and 94%, respectively) (data not shown in Table 2). This might be the reason that the active site of the electrophilic substitution reaction was occupied by the methyl group. The same treatment with substrates 1j–1l (containing 4-acetyl, 3,5-difluoro, and 4-trifluoromethyl groups) afforded amination products 2j–2l only, instead of the electrophilic substituted products 3 (data not shown in Table 2), which was due to the reduction of electron density by electron-withdrawing substituents (R2) on the aromatic ring toward the iodonium species. Substrates 1m–1p were also examined in this reaction; the corresponding expected products 3m–3p were obtained in good to excellent yields ranging from 75 to 95%.
A plausible reaction mechanism based on the PhI(OAc)2–I2 system12 is illustrated in Scheme 1. Firstly, a reaction of PhI(OAc)2 and iodine would occur to produce a reactive electrophile IOAc,12d which reacts with substrate 1 to form the N-iodinated compound I. The homolytic cleavage of N–I bonds in compound I would occur to give a reactive radical species II. Intramolecular radical recombination of intermediate II followed by oxidative aromatization generated the amination product 2. The iodinated product 3 would be formed via an electrophilic substitution reaction of compound 2. To verify the possible pathway of the proposed mechanism, 2.0 equivalents of TEMPO serving as a radical-trapping agent were added to the model reaction; only a trace of the desired product 2a was observed. This result suggested that free radical intermediates were possibly involved in the reaction. In addition, when compound 2a was employed as a test substrate, the electrophilic substitution reaction took place smoothly under the standard conditions B to generate product 3a in 95% yield (Scheme 2).
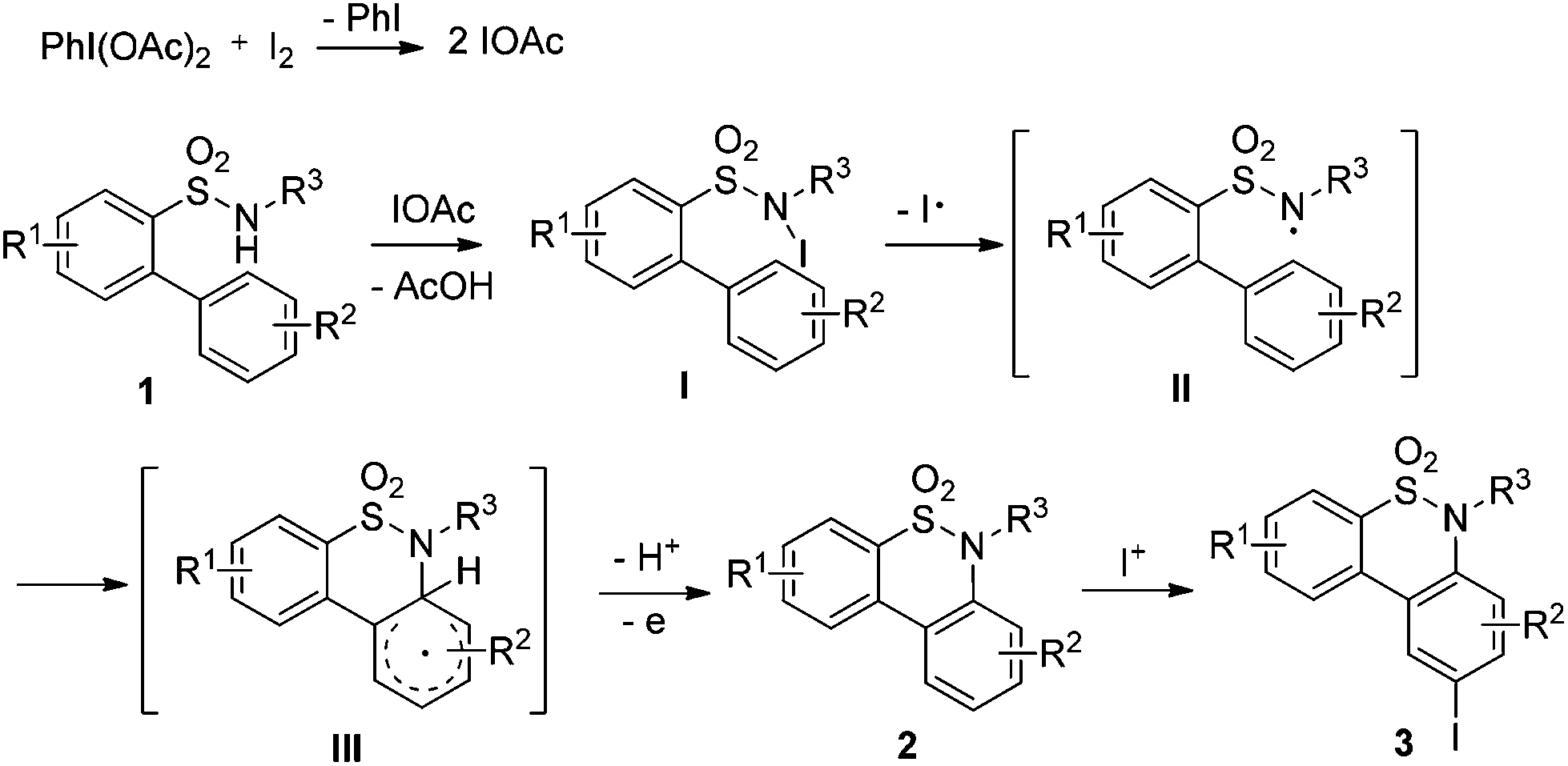 | ||
| Scheme 1 Plausible reaction mechanism. | ||
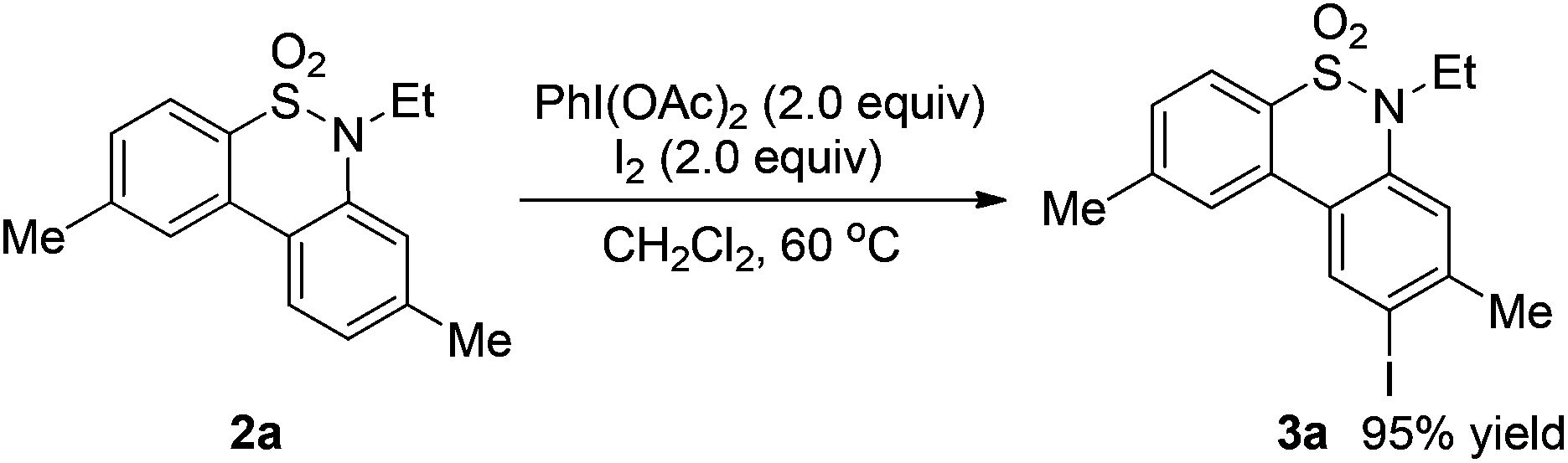 | ||
| Scheme 2 The reaction of 2a under conditions B. | ||
Conclusion
In summary, we have demonstrated an efficient intramolecular sp2 C–H bond amination reaction of aryl sulfonamides with PhI(OAc)2–I2 to give benzosultams in good to excellent yields. The cyclization was proposed to proceed via a radical mechanism under metal-free conditions with a good functional group tolerance. The resulting benzothiazines could be conveniently transformed into the corresponding iodinated derivatives in high yields. Further screening of biological activities of these molecules is under investigation in our laboratory.Experimental
General procedure for the synthesis of 6H-dibenzo[c,e][1,2]thiazine 5,5-dioxide derivatives
Conditions A: a solution of o-arylbenzenesulfonamide 1 (0.2 mmol), PhI(OAc)2 (70.8 mg, 0.22 mmol), I2 (55.9 mg, 0.22 mmol), and K2CO3 (41.4 mg, 0.3 mmol) in DCM (2.0 mL) was stirred at 35 °C.Conditions B: a solution of o-arylbenzenesulfonamide 1 (0.2 mmol), PhI(OAc)2 (128.8 mg, 0.4 mmol), and I2 (101.6 mg, 0.4 mmol) in DCM (2.0 mL) was stirred at 60 °C.
After completion of the reaction as detected by TLC, the reaction mixture was cooled to room temperature, and purified directly by silica gel chromatography (EtOAc–hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 v/v) to provide the title compound 2 or 3 as an amorphous solid.
:20 v/v) to provide the title compound 2 or 3 as an amorphous solid.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21002042), the Project of Jiangxi Youth Scientist (20122BCB23012), Jiangxi Educational Committee (GJJ12169), and Open Project Program of Key Laboratory of Functional Small Organic Molecule, Ministry of Education, Jiangxi Normal University (no. KLFS-KF-201203 and 201217) is gratefully acknowledged.Notes and references
- S. H. Kim, R. Amu, S. W. Kwon, S.-H. Lee, C. H. Kim, Se. K. Kang, D. S. Rhee, M. A. Bae, S. H. Ahn and D. C. Ha, Bioorg. Med. Chem. Lett., 2010, 20, 1065 CrossRef CAS PubMed.
- (a) R. Bihovsky, M. Tao, J. P. Mallamo and G. J. Wells, Bioorg. Med. Chem. Lett., 2004, 14, 1035 CrossRef CAS PubMed; (b) G. J. Wells, M. Tao, K. A. Josef and R. Bihovsky, J. Med. Chem., 2001, 44, 3488 CrossRef CAS PubMed.
- F. Brzozowski, F. Saczewski and N. Neamati, Bioorg. Med. Chem. Lett., 2006, 16, 5298 CrossRef PubMed.
- (a) J. Wang, D. Limburg, J. Ctarter, G. Mbalaviele, J. Gierse and M. Vazquez, Bioorg. Med. Chem. Lett., 2010, 20, 1604 CrossRef CAS PubMed; (b) L. Churchill, A. G. Graham, C. K. Shih, D. Pauletti, P. R. Farina and P. M. Grob, Inflammopharmacology, 1996, 4, 125 CrossRef CAS.
- (a) T. Mezei, N. Mesterházy, T. Bakó, M. Porcs-Makkay, G. Simig and B. Volk, Org. Process Res. Dev., 2009, 13, 567 CrossRef CAS; (b) R. C. Obach, A. S. Kalgutkar, T. F. Ryder and G. S. Walker, Chem. Res. Toxicol., 2008, 21, 1890 CrossRef CAS PubMed; (c) T. Hakan, M. Z. Berkman, T. Ersoy, I. Karatas, T. San, S. Arbak and J. Lin, Neuroscience, 2008, 15, 55 CAS.
- (a) I. Fujita, T. Okumura, A. Sakakibara and Y. Kita, J. Pharm. Pharmacol., 2012, 64, 747 CrossRef CAS PubMed; (b) C. Knottenbelt, G. Gault, G. Chambers and D. L. Argyle, J. Small Anim. Pract., 2006, 47, 14 CrossRef CAS PubMed; (c) J. G. Lombardino and E. H. Wiseman, Med. Res. Rev., 1982, 2, 127 CrossRef CAS; (d) J. G. Lombardino, J. Med. Chem., 1981, 24, 39 CrossRef CAS.
- (a) C. B. Bheeter, J. K. Bera and H. Doucet, Adv. Synth. Catal., 2012, 354, 3533 CrossRef CAS; (b) S. Rousseaux, S. I. Gorelsky, B. K. W. Chung and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 10692 CrossRef CAS PubMed; (c) K. C. Majumdar, M. Shovan and D. Nirupam, Synthesis, 2009, 3127 CrossRef PubMed; (d) J. Minville, J. Poulin, C. Dufresne and C. F. Sturino, Tetrahedron Lett., 2008, 49, 3677 CrossRef CAS PubMed; (e) D. K. Barange, T. C. Nishad, N. Swamy, K. V. Bandameedi, D. Kumar, B. R. Sreekanth, K. Vyas and M. Pal, J. Org. Chem., 2007, 72, 8547 CrossRef CAS PubMed; (f) W. Zeng and S. R. Chemler, J. Am. Chem. Soc., 2007, 129, 12948 CrossRef CAS PubMed; (g) P. Dauban and R. H. Dodd, Org. Lett., 2000, 2, 2327 CrossRef CAS PubMed; (h) R. A. Abramovitch and K. Miyashita, J. Chem. Soc., Perkin Trans. 1, 1975, 2413 RSC.
- (a) F. Liu, A. Martin-Mingot, M.-P. Jouannetaud, F. Zunino and S. Thibaudeau, Org. Lett., 2010, 12, 868 CrossRef CAS PubMed; (b) Y. Ishiwata and H. Togo, Tetrahedron Lett., 2009, 50, 5354 CrossRef CAS PubMed; (c) D. K. Barange, V. R. Batchu, D. Gorja, V. R. Pattabiraman, L. K. Tatini, J. M. Babu and M. Pal, Tetrahedron, 2007, 63, 1775 CrossRef CAS PubMed; (d) Y. Misu and H. Togo, Org. Biomol. Chem., 2003, 1, 1342 RSC; (e) H. Togo and A. Ryokawa, Tetrahedron, 2001, 57, 5915 CrossRef; (f) H. Togo, Y. Harada and M. Yokoyama, J. Org. Chem., 2000, 65, 926 CrossRef CAS; (g) D. Blondet and J.-C. Pascal, Tetrahedron Lett., 1994, 35, 2911 CrossRef CAS; (h) S. A. Glover, A. Goosen, C. W. McCleland and J. L. Schoonraad, J. Chem. Soc., Perkin Trans. 2, 1986, 645 RSC; (i) B. Loev, M. F. Kormendy and K. M. Snader, J. Org. Chem., 1966, 31, 3531 CrossRef CAS; (j) B. Loev and M. F. Kormendy, J. Org. Chem., 1965, 30, 3163 CrossRef CAS.
- (a) K. Kaneko, T. Yoshino, S. Matsunaga and M. Kanai, Org. Lett., 2013, 15, 2502 CrossRef CAS PubMed; (b) M. Yamagishi, K. Nishigai, A. Ishii, T. Hata and H. Urabe, Angew. Chem., Int. Ed., 2012, 51, 6471 CrossRef CAS PubMed; (c) D. K. Rayabarapu, A. Zhou, K. O. Jeon, T. Samarakoon, A. Rolfe, H. Siddiqui and P. R. Hanson, Tetrahedron, 2009, 65, 3180 CrossRef CAS PubMed; (d) W. J. Layman, T. D. Greenwood, A. L. Downey and J. F. Wolfe, J. Org. Chem., 2005, 70, 9147 CrossRef CAS PubMed.
- (a) Q. Xiao, Y. Luo, Y. Yan, D. Ding and J. Wu, RSC Adv., 2013, 3, 5779 RSC; (b) H. Wang, Y. Luo, X. Hou and J. Wu, Dalton Trans., 2013, 42, 4410 RSC.
- (a) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (b) Y. Park, J. Park and C. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed; (c) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013 CrossRef CAS PubMed; (d) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed; (e) D. Colby, R. Bergman and J. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (f) C. Copéret, Chem. Rev., 2010, 110, 656 CrossRef PubMed; (g) S. Conejero, M. Paneque, M. Poveda, L. Santos and E. Carmona, Acc. Chem. Res., 2010, 43, 572 CrossRef CAS PubMed; (h) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (i) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (j) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (k) A. N. Verdernikov, Acc. Chem. Res., 2012, 45, 803 CrossRef PubMed; (l) D.-G. Yu, B.-J. Li and Z.-J. Shi, Tetrahedron, 2012, 68, 5130 CrossRef CAS PubMed.
- (a) V. P. Mehta and B. Punji, RSC Adv., 2013, 3, 11957 RSC; (b) A. A. Lamar and K. M. Nicholas, J. Org. Chem., 2010, 75, 7644 CrossRef CAS PubMed; (c) H. J. Lu, Q. Chen and C. Z. Li, J. Org. Chem., 2007, 72, 2564 CrossRef CAS PubMed; (d) R. Fan, D. Pu, F. Wen and J. Wu, J. Org. Chem., 2007, 72, 8994 CrossRef CAS PubMed; (e) H. Togo and M. Kotohgi, Synlett, 2001, 565 CAS; (f) M. Katohgi and H. Togo, Tetrahedron, 2001, 57, 7481 CrossRef CAS; (g) J. Madsen, C. Viuf and M. Bols, Chem.–Eur. J., 2000, 6, 1140 CrossRef CAS; (h) C. G. Francisco, M. C. Gonzalez and E. Suarez, J. Org. Chem., 1998, 63, 8092 CrossRef CAS; (i) H. Togo, Y. Hoshina, T. Muraki, H. Nakayama and M. Yokoyama, J. Org. Chem., 1998, 63, 5193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all new compounds. CCDC 951230 and 951231. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob41810c |
| This journal is © The Royal Society of Chemistry 2014 |