Reaction site-driven regioselective synthesis of AChE inhibitors†
Emilia
Oueis
a,
Gianluca
Santoni
bcd,
Cyril
Ronco
a,
Olga
Syzgantseva
a,
Vincent
Tognetti
a,
Laurent
Joubert
a,
Anthony
Romieu
a,
Martin
Weik
bcd,
Ludovic
Jean
*a,
Cyrille
Sabot
*a,
Florian
Nachon
e and
Pierre-Yves
Renard
*a
aNormandie Univ, COBRA, UMR 6014 & FR 3038; Univ Rouen; INSA Rouen; CNRS, 1 rue Tesnière 76821 Mont-Saint-Aignan, Cedex, France. E-mail: pierre-yves.renard@univ-rouen.fr; Fax: (+) 33 2 35 52 29 71
bUniv Grenoble Alpes, IBS, F-38027 Grenoble, France
cCNRS, IBS, F-38027 Grenoble, France
dCEA, IBS, F-38027 Grenoble, France
eInstitut de Recherche Biomédicale des Armées BP73, F-91993 Brétigny-sur-Orge, France
First published on 28th October 2013
Abstract
The enzyme-directed synthesis is an emerging fragment-based lead discovery approach in which the biological target is able to assemble its own multidentate ligands from a pool of building blocks. Here, we report for the first time the use of the human acetylcholinesterase (AChE) as an enzyme for the design and synthesis of new potent heterodimeric huprine-based inhibitors. Both the specific click chemistry site within the protein and the regioselectivity of the Huisgen cycloaddition observed suggest promising alternatives in the design of efficient mono- and dimeric ligands of AChE. Finally, a detailed computational modelling of the click reaction was conducted to further understand the origin of this TGS selectivity.
Introduction
The target-guided synthesis (TGS) is a relatively unexplored but promising approach for lead discovery. More precisely, an enzyme acts as a template to autoassemble its own multidentate inhibitors from a pool of building blocks bearing complementary reacting fragments.1 This in situ ligation of building blocks is often achieved through the reliable alkyne–azide Huisgen cycloaddition (called “click chemistry”)2 to yield the corresponding 1,2,3-triazole-containing inhibitors. In addition, acetylcholinesterase that plays a key role in the treatment of Alzheimer's disease (AD)3 was the first enzyme used to validate the in situ click chemistry approach. It is well-known that the active site of AChE is composed of two distinct binding sites, named the peripheral (1) and the acylation sites (2), connected by a narrow hydrophobic channel, the AChE gorge (3) (Fig. 1A). Taking advantage of the enzyme structure, Sharpless and Kolb4 reported the capacity of mouse AChE (m-AChE) to catalyse exclusively the formation of highly potent syn-triazole heterodimeric inhibitors such as (±)-TZ2PIQA5 and (±)-TZ2PIQA6 (Fig. 1B). More precisely, the syn-triazole formation occurred from a mixture of tacrine azide (catalytic site binder) and phenyl tetrahydroisoquinoline alkyne (peripheral site binder) building blocks, in a particularly favourable constricted region composed of side chains of Tyr124, Phe297, Tyr337, and Phe338 at the bottom of the gorge (vide infra).5 The observed syn-regioselectivity was attributed to the parallel orientation of both azide and alkyne reactive groups that pointed toward the catalytic pocket.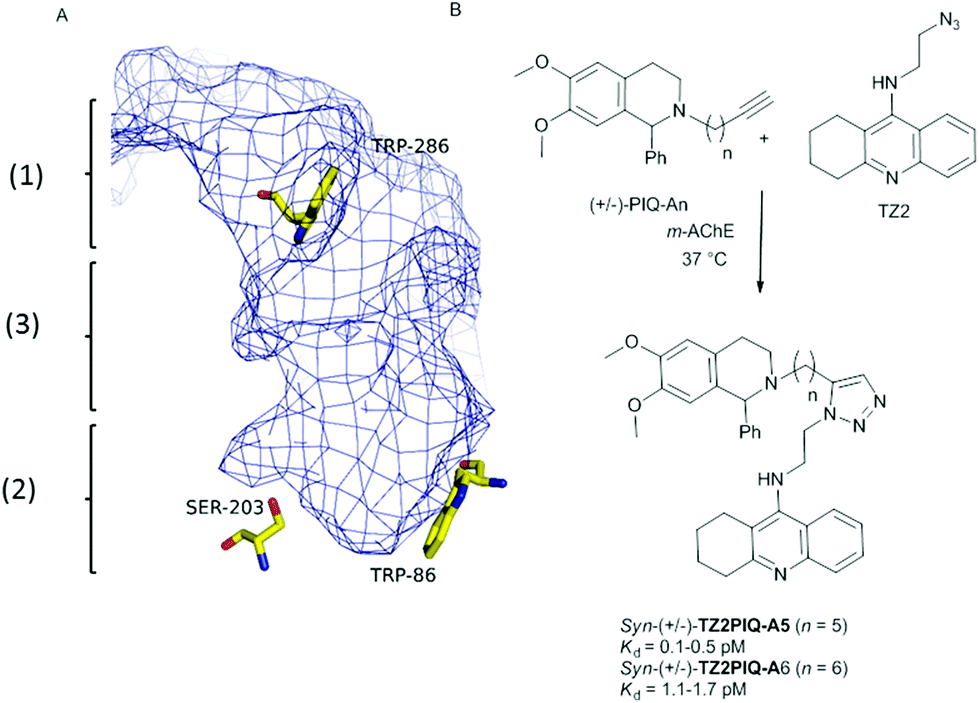 | ||
| Fig. 1 (A) Description of the active site of m-AChE: (1) peripheral anionic site; (2) acylation site; (3) hydrophobic channel. (B) Hits that emerged from in situ click chemistry experiments described by Sharpless and Kolb.4 | ||
Following these interesting results, the in situ click chemistry was performed in other constricted active sites of enzymes (HIV protease,6 carbonic anhydrase,7 or the transcriptional repressor EthR8) or at a presumably more flexible environment such as the subunit interfaces of the nicotinic acetylcholine receptor nAChR.9 To the best of our knowledge, only one click chemistry site was reported for each enzyme investigated to date, leading to one triazole regioisomer (either syn or anti). Indeed, not only the overall spacing between the two monomeric ligands but also the location of the triazole ring construction within the enzyme proved to be of crucial importance for the success of the in situ click chemistry approach.2b Consequently, in order to extend the scope of the TGS, we questioned whether the identification of a new reaction site within a protein would be possible. This should offer new opportunities in the rational design of new enzyme inhibitors by using various building block structures.
Thus, in this study we explored a novel AChE reaction site suitable for TGS, involving the use of bulky active site ligands such as huprine building blocks. Preliminary work from our laboratory suggests that the huprine moiety would be a suitable catalytic site binder for in situ click chemistry.10 Huprine derivatives, developed by Camps et al., are the best AChE active-site ligands reported to date, especially huprines X and Y that exhibit high affinity towards AChE's acylation site (IC50 = 0.75 nM and 0.78 nM respectively, 270-fold more potent than tacrine),11 and can be readily functionalized at position 9 or 12.12 Moreover, we selected the phenyl tetrahydroisoquinoline scaffold that already proved to be a convenient peripheral site binder in TGS.4 Not to mention the currently growing interest in developing multi-target-directed drugs and particularly new potent dual-binding site AChE inhibitors able to exert a dual action,13i.e. interacting simultaneously with the AChE active site (inhibition of cholinesterasic activity) and the peripheral site (inhibition of AChE-mediated Aβ deposition).14
From this investigation, the following important outcomes may be expected: (1) the design and synthesis of new potent dual-binding site inhibitors directed for the first time by the flexible recombinant human rh-AChE;15 (2) a better understanding of the relationship between the location of the click chemistry within the enzyme and the observed regioselectivity; and (3) a deeper insight into the fundamentals that govern the TGS strategy (Fig. 2).
Results and discussion
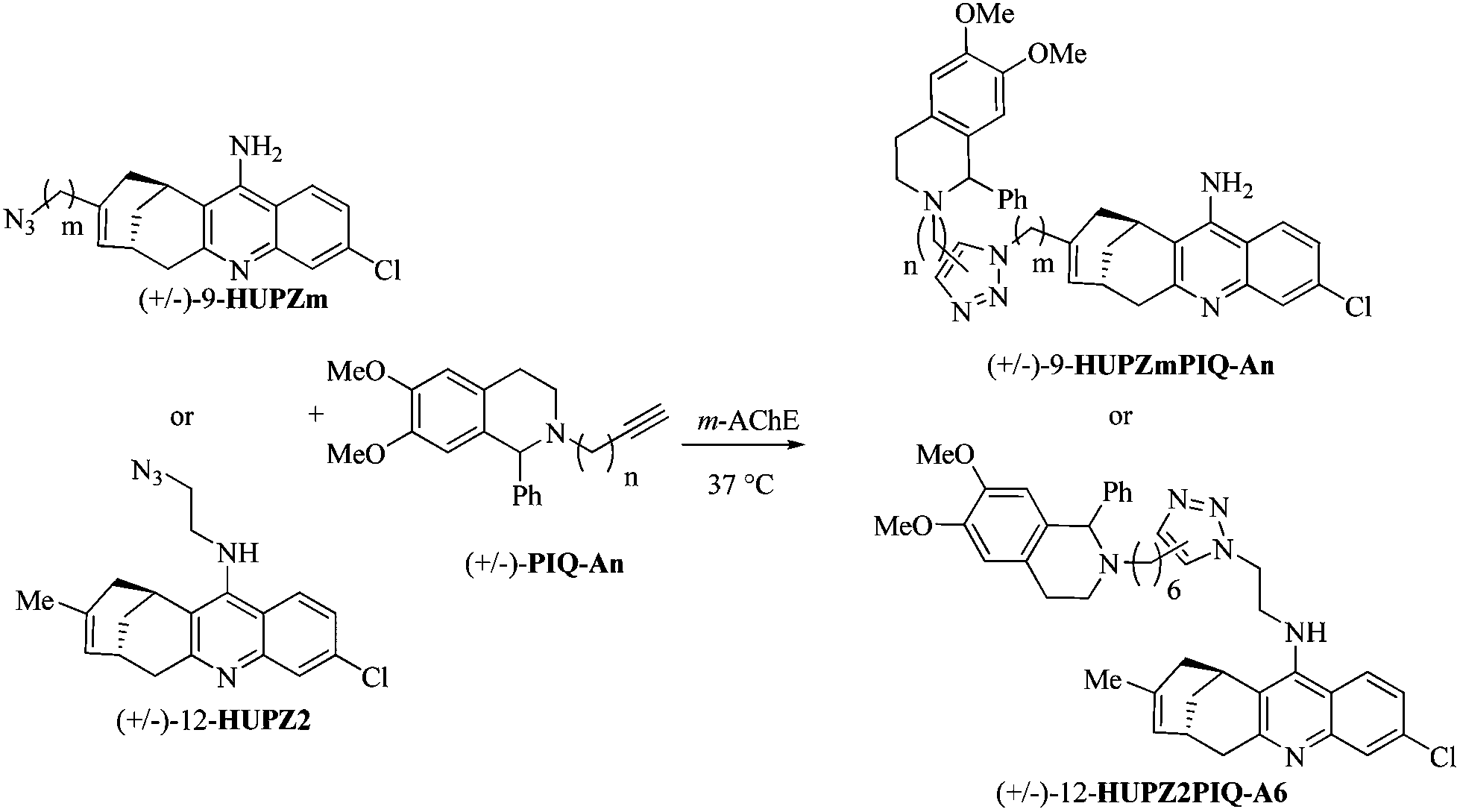
(±)-12-HUPZ2,16 functionalized by an amino ethyl azide group at position 12 on the quinoline ring, and (±)-PIQ-A6 (n = 6),17 bearing an alkyne group, were incubated in the presence of the model enzyme m-AChE (Table 1, entry 1). Interestingly, no trace of (±)-12-HUPZ2PIQ-A6 was observed using HPLC coupled with ESI/MS detection, whereas the incubation under similar conditions of (±)-PIQ-A6 and TZ2, the latter sharing important structural similarities with (±)-12-HUPZ2, afforded the corresponding dimeric inhibitor syn-(±)-TZ2PIQ-A6 (Fig. 1B) as reported by Sharpless and Kolb.4 Huprine and tacrine share similar binding zones within the active site of the enzyme, but the additional carbobicyclic ring system in the huprine scaffold hinders the access to the esteratic site, which would prevent the formation of the triazole of (±)-12-HUPZ2PIQ-A6 in this sequestered enzyme site. Moreover, the moderate IC50 value of (±)-12-HUPZ2PIQ-A6 (i.e. 7.0 nM) is in agreement with our previous studies showing that functionalization of huprines at the quinoline moiety (position 12) resulted in a significant decrease of their inhibitory potency. Indeed, the free amino group on the quinoline ring is involved in key hydrogen bonds with the nearby structural water molecule.12d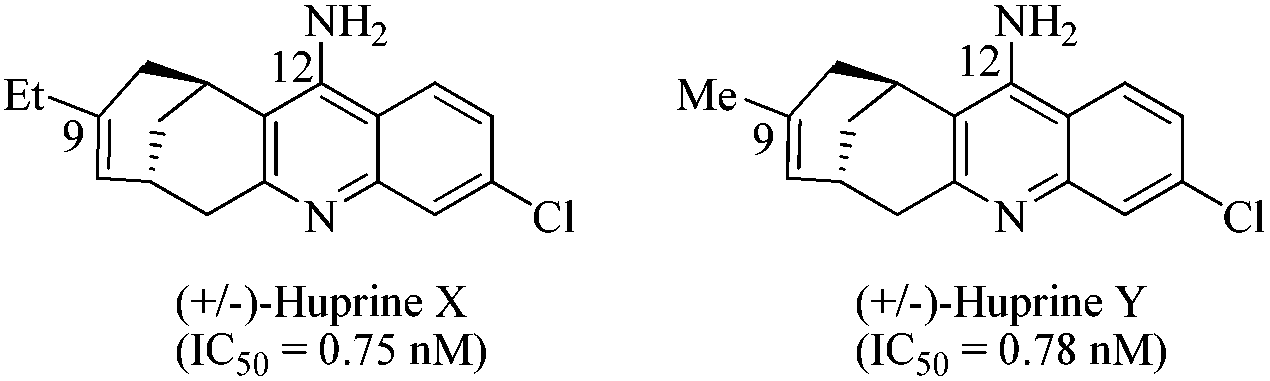 | ||
| Fig. 2 (±)-Huprine X, Y. | ||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Ligand | m | n | Click in situ | IC50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (nM) a (nM) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Corresponding to the anti isomer. b Not applicable. c Detected after a two-day incubation period. d Detected after a seven-day incubation period. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | (±)-12-HUPZ2PIQ-A6 | n.a.b | 6 | — | 7.0 ± 1.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | (±)-9-HUPZ4PIQ-A1 | 4 | 1 | — | 1.5 ± 0.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | (±)-9-HUPZ4PIQ-A2 | 4 | 2 | anti | 0.4 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | (±)-9-HUPZ4PIQ-A3 | 4 | 3 | syn/antid | 1.2 ± 0.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | (±)-9-HUPZ4PIQ-A4 | 4 | 4 | — | 3.1 ± 0.6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | (±)-9-HUPZ2PIQ-A4 | 2 | 4 | — | 13.0 ± 2.5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
This preliminary result emphasizes the importance of finding alternative reaction sites within an enzyme, especially to widen the scope of bivalent inhibitors designed via in situ click chemistry. Furthermore, we recently reported that suitable modifications at position 9 of the huprine derivative leave the water network unchanged, and could increase their affinity toward AChE. Interestingly, our previous docking studies suggested that the ligation to a peripheral site binder could be carried out at position 9.10 Consequently, the enzymatic triazole synthesis could be envisioned in the a priori less favorable mid-gorge of m-AChE, i.e. closer to the peripheral anionic site. Yet, the higher degree of enzyme flexibility expected at this location may disadvantage the click chemistry process.
Thus, huprines bearing alkyl azide (m = 2 and 4), and phenyl tetrahydroisoquinolines decorated with alkyl acetylene of varying chain lengths (n = 1–4), were incubated in the presence of m-AChE. Control experiments were also performed without an enzyme (or with BSA protein). The reaction mixtures were next analysed by RP-HPLC through electrospray ionisation mass spectrometry in the positive selected ion monitoring mode (SIM) (Fig. 3), and compared to authentic samples obtained by thermal or copper-catalysed 1,3-dipolar cycloaddition.18
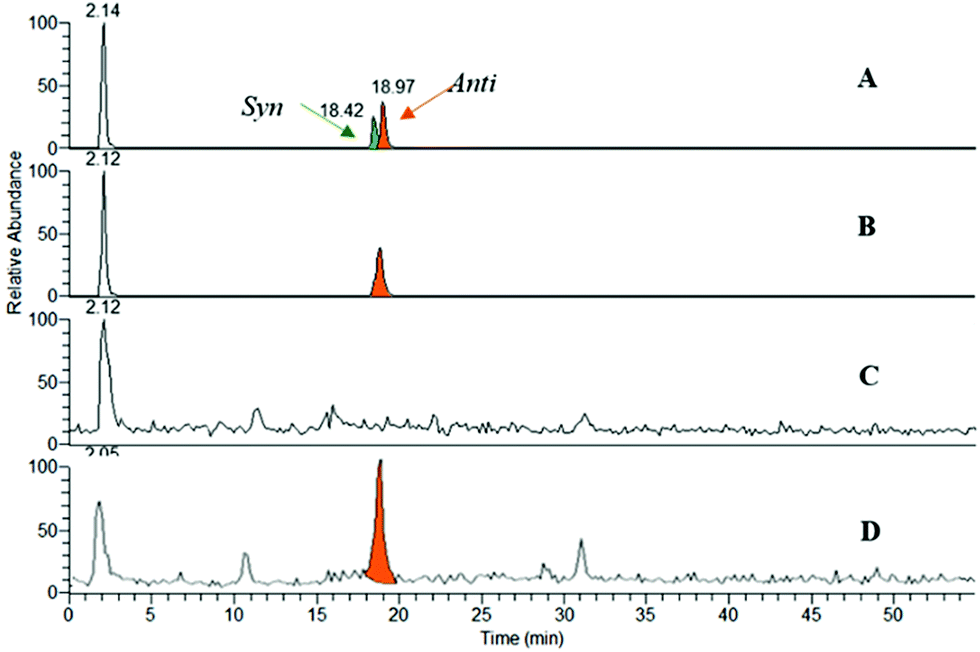 | ||
| Fig. 3 LCMS/SIM traces of (±)-9-HUPZ4PIQ-A2: (A) a mixture of anti and syn-(±)-9-HUPZ4PIQ-A2 obtained under the thermal Huisgen process; (B) anti-(±)-9-HUPZ4PIQ-A2 formed by catalytic CuAAC; (C) trace of the blank experiment without m-AChE; and (D) trace of the click experiment with m-AChE. | ||
From ten heterodimers that could theoretically be formed in situ (Table 1, entries 2–6), only the anti-9-HUPZ4PIQ-A2 and the regioisomeric mixture of syn- and anti-9-HUPZ4PIQ-A3 (≈ 1:2) were observed (Table 1, entries 3 and 4). These results show that the acetylene moiety of the peripheral site ligand (±)-PIQ-A2, with a two carbon-atom chain length, would be located at an optimal distance from the azide unit of (±)-9-HUPZ4 to adopt a specific conformation leading to a unique regioisomer (i.e. anti). However, the presence of an additional methylene unit in (±)-PIQ-A3 would enable the alkyne chain to fold, and lead to the subsequent syn-triazole isomer as well. Interestingly, no trace of (±)-9-HUPZ2PIQ-A4 was formed (entry 6), even though it has the same overall spacing between the peripheral and catalytic site binders as (±)-9-HUPZ4PIQ-A2. In this case, the triazole unit would be unfavourably located at the bottleneck of the active site gorge.
It is noteworthy that this study, together with Sharpless and Kolb's results, shows that AChE is able to template either the anti isomer of (±)-9-HUPZ4PIQ-A2 (entry 3) or the syn isomer of (±)-TZ2PIQ-A6, depending on the location of the click chemistry within the active site of the enzyme. Indeed, as depicted in Fig. 4, these dual-binding site inhibitors are templated at two clearly distinct sites of m-AChE, located on either side of the enzyme bottleneck. syn-(±)-TZ2PIQ-A6 is assembled in the constricted region located in proximity to the acylation site of the enzyme, resulting from reacting functionalities extended in a parallel orientation. anti-(±)-9-HUPZ4PIQ-A2 is ligated closer to the rim of the gorge, an unprecedented reaction site of AChE, from complementary functional groups lying in an antiparallel orientation.19
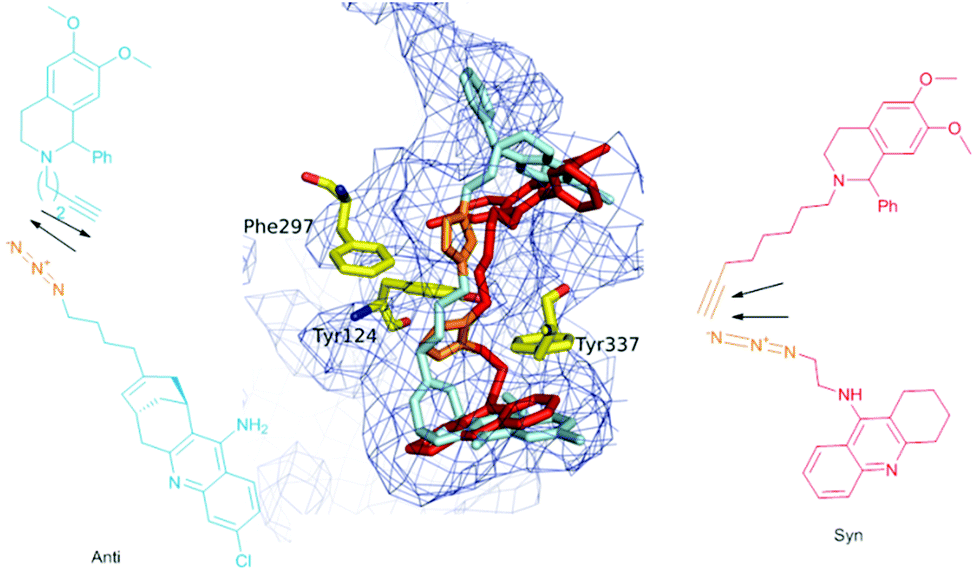 | ||
| Fig. 4 Superimposition of in situ auto-assembled ligand syn-(±)-TZ2PIQ-A6 (in red) and anti-(±)-9-HUPZ4PIQ-A2 (in blue) docked in the active site of m-AChE (pdb code 1Q83). The triazole unit of each ligand is colored in orange. | ||
We next examined whether these promising results could be extrapolated to the more flexible rh-AChE. In fact, the symptomatic approach to treat neurological disorders such as AD consists of controlling the enzymatic activity of human AChE. It is noteworthy that switching from m- to rh-AChE template is not a trivial task as both enzymes have marked differences in dynamic properties despite fully conserved active site residues.15b The same series of binary mixtures was incubated in the presence of rh-AChE to direct the synthesis of heterodimeric huprine-based inhibitors (Table 2). Gratifyingly, in situ click chemistry was observed and similar results were obtained with the clinically relevant enzyme. Note that longer reaction times were necessary before detecting the triazole-adducts (±)-9-HUPZ4PIQ-A2 and (±)-9-HUPZ4PIQ-A3, probably due to the greater flexibility of the rh-AChE active site compared with the m-AChE one.
| Entry | Ligand | m | n | Click in situ | IC50a (nM) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Corresponding to the anti isomer. b Not applicable. c Detected after a nine-day incubation period. d Detected after a thirteen-day incubation period. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | (±)-12-HUPZ2PIQ-A6 | n.a.b | 6 | — | 6.1 ± 0.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | (±)-9-HUPZ4PIQ-A1 | 4 | 1 | — | 3.3 ± 0.2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | (±)-9-HUPZ4PIQ-A2 | 4 | 2 | anti | 0.6 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | (±)-9-HUPZ4PIQ-A3 | 4 | 3 | syn/antid | 0.6 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | (±)-9-HUPZ4PIQ-A4 | 4 | 4 | — | 0.8 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | (±)-9-HUPZ2PIQ-A4 | 2 | 4 | — | 5.3 ± 0.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Consequently, these in situ click chemistry results highlighted that the functionalization of the huprine scaffold at position 9 was an appropriate strategy to access the peripheral anionic site of AChE (Table 2, entry 1 vs. 3 and 4). As expected, the overall chain length between the catalytic and the peripheral site ligand (entries 2 and 5 vs. 3 and 4), as well as the location of the triazole ring into the gorge, should be considered (entry 3 vs. 6). The latter information would be particularly valuable to build efficient triazole-containing dimeric and monomeric inhibitors. Indeed, the present TGS approach confirmed our previous observations10 showing that ligands bearing a triazole unit at four carbon atoms away from the huprine scaffold, such as compounds 3 and 4, were much more potent than their two carbon atom chain length analogue 1 or 2, due to better accommodation midway in the active site gorge, and notably favourable interactions of the triazole with Tyr341 and the main chain of Phe295 (Fig. 5).20 Compound 4 constitutes one of the best rh-AChE active-site ligands reported to date (IC50 = 0.43 nM).
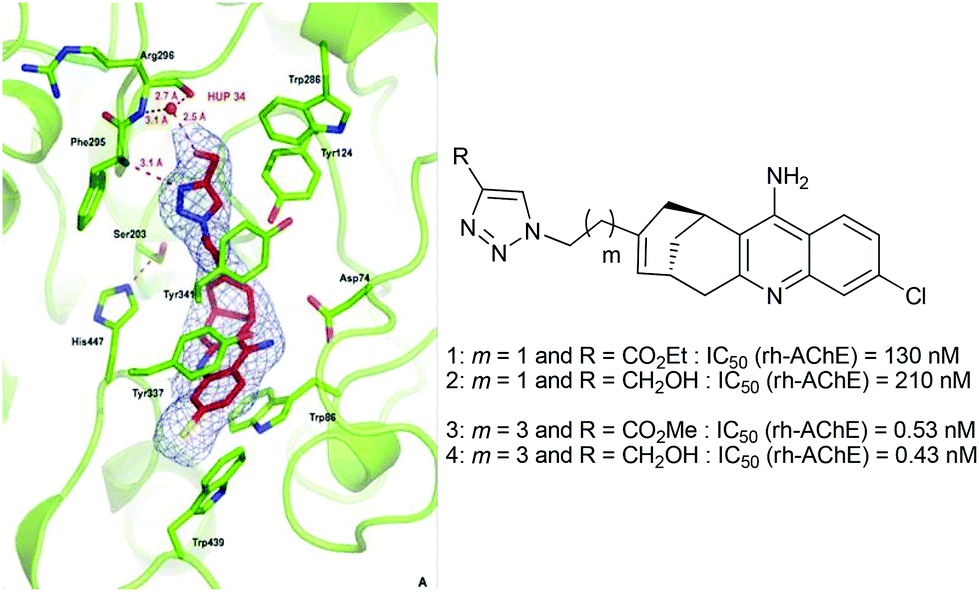 | ||
| Fig. 5 X-ray crystal structure of the 4-m-AChE complex (pdb code 4A16) and comparative IC50 values of huprines bearing a triazole unit separated by a two- and four-carbon chain length. | ||
In order to investigate the potential reasons for the regioselective formation of the anti-(±)-9-HUPZ4PIQ-A2 (i.e. the role of the residues in close proximity to the reaction site, the reaction barriers and the total energy of the protein–substrate complex), additional theoretical studies were undertaken. To this aim, the structures of protein–ligand complexes for syn- and anti-molecules were obtained by docking and MD equilibration. The structure of the complexes was used as the basis to model the click reaction between substrates, using the combined quantum mechanics/molecular mechanics approach (QM/MM). A detailed computational procedure is given in ESI.†
First, pre-reactive complexes of syn-(±)-9-HUPZ4PIQ-A2 and anti-(±)-9-HUPZ4PIQ-A2 in the active site of rh-AChE were analysed (Fig. 6). The huprine moiety is firmly maintained by aromatic stacking in between Trp86 and Tyr337, while the azido group is confined in an aromatic pocket formed by Phe295, Phe297, Tyr337 and Phe338.
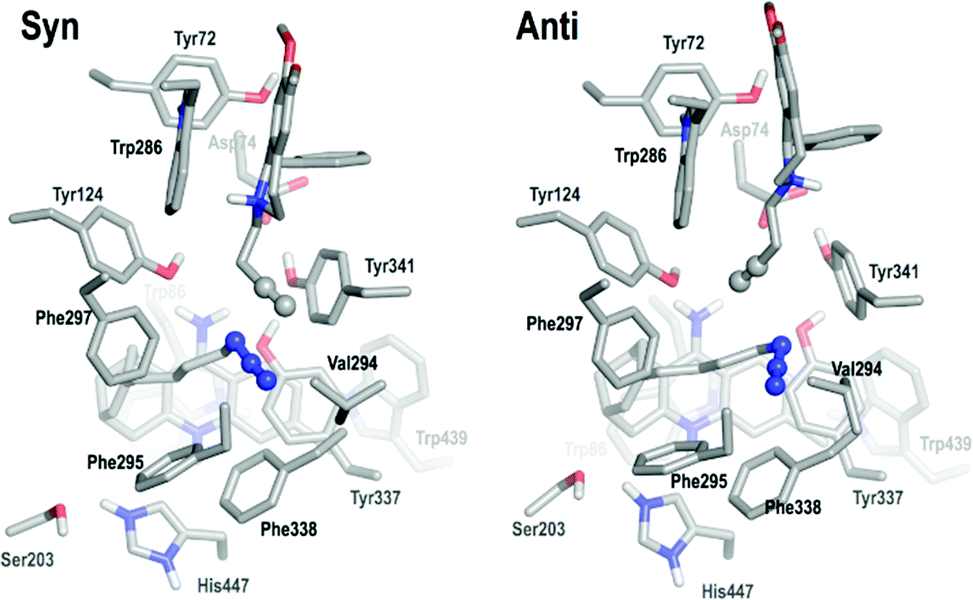 | ||
| Fig. 6 Pre-reactive complexes of syn-(±)-9-HUPZ4PIQ-A2 and anti-(±)-9-HUPZ4PIQ-A2 in the active site of rh-AChE. The main chain of active site residues is represented as grey sticks, with oxygen in red and nitrogen in blue. Atoms of the reactive azido and ethyne groups are represented as spheres. | ||
The tetrahydroisoquinoline substrate is bound to the peripheral site, stacking parallel to Trp286 with additional T-stacking involving its phenyl ring and Tyr341. In the pre-syn complex, the alkynyl group lies away from Trp286 (4.6 Å) parallel to the azido group and makes favorable π–π interaction with Tyr341 (3.4 Å). By contrast, the alkynyl group lies orthogonally to the azido group in the pre-anti complex, at closer distance from Trp286 than Tyr341 (3.4 Å vs. 4.0 Å). However, no clear conclusion could be drawn at this stage from this preliminary analysis.
Consequently, reaction barriers for syn and anti substrates in the protein environment were next investigated from a quantum chemistry viewpoint by fully optimizing the corresponding transition states, and were found to be 16.3 kcal mol−1 and 20.5 kcal mol−1, respectively. These values (both accessible at ambient temperature) suggest a slight but not definitive kinetic preference for the syn product. It can be noticed that Senapati and co-workers21 reported a similar difference (4 kcal mol−1) between the activation barriers in favour of the experimentally non-observed product. The strong catalytic role of the enzyme can also be enlightened by remarking that the activation energies calculated using the same computational protocol for the formation of the heterodimeric inhibitors without AChE are about twice (35 kcal mol−1) the ones calculated in the presence of the enzyme.
From a thermodynamic point of view, a direct comparison of the total energies of protein–substrate complexes shows that the anti-complex is more stable than the syn-complex by 173 kcal mol−1 at the MM level and 144 kcal mol−1 at the QM/MM level. This means that once the system is trapped in the anti conformation, it cannot be converted to the syn isomer. Thus, the selective formation of anti-9-HUPZ4PIQ-A2 is mainly the result of accumulation of protein–substrate complexes and interactions in the anti configuration. It should be mentioned that similar findings were reported earlier by Senapati and co-workers21 on the selective formation of the observed product in m-AChE. It thus seems reasonable to conclude that the physicochemical factors driving the selectivity of the process in rh-AChE are the same as those in m-AChE.
Finally, the total energy difference (i.e. 144 kcal mol−1) between syn and anti products can be decomposed into three contributions: substrate and protein internal energy differences and binding interaction (protein–substrate) differences between both conformations. It was found that 9.6 kcal mol−1 is brought about by the stabilization of the anti-substrate with respect to the syn-substrate, 17.3 kcal mol−1 is due to a higher binding interaction energy of the anti-isomer with the protein and the larger contribution of 117.5 kcal mol−1 comes from the protein internal energy difference between both conformations. Interestingly, these three contributions have cumulative effects, favouring the formation of the anti-complex. The main contribution comes clearly from the protein internal energy differences, and thus highlights the induced fit contribution to the triazole formation. These results, which are in agreement with experiments, also highlight the relevance of such a QM/MM approach.
Conclusions
Herein, we have reported the first use of human AChE as the template for in situ synthesis of subnanomolar dual binding site inhibitors. To achieve this outcome, the identification of a novel click chemistry site within the AChE gorge that enables the use of bulky huprine building blocks as an acylation site binder was required. Whilst the syn-triazole isomer was previously reported by Sharpless and Kolb to be preferentially formed in the sequestered region within the AChE gorge, interestingly, we observed a reversal of regioselectivity moving the reaction toward the upper part of the gorge, in the mid-gorge site of AChE. This selectivity is thermodynamically driven and is principally related to the protein internal energy difference between both conformations, and shows for the first time that an enzyme could govern the absolute regioselectivity of the Huisgen reaction (either syn or anti isomer), depending on the reaction site selected and the overall spacing between the two monomeric ligands.Importantly, this study shows that the TGS, sustained by docking experiments, is a flexible tool capable of highlighting different click chemistry sites within an enzyme, depending on the combination of building blocks selected. Consequently, the identification of a broader diversity of inhibitors may be expected.
Acknowledgements
This work was supported by Agence Nationale pour la Recherche (through ANR_06_BLAN_0163 DETOXNEURO and ANR_09_BLAN_0192 ReAChE programs), and the Région Haute Normandie (the CRUNCh program, and a PhD fellowship for EO). C. Thiot Sabot is acknowledged for helpful discussions.Notes and references
- (a) D. Rideout, T. Calogeropoulou, J. Jaworski and M. Mccarthy, Biopolymers, 1990, 29, 247–262 CrossRef CAS PubMed; (b) G. E. Boldt, T. J. Dicherson and K. D. Janda, Drug Discovery Today, 2004, 9, 143–148 Search PubMed; (c) A. V. Eliseev, Drug Discovery Today, 2004, 9, 348–349 CrossRef; (d) K. B. Sharpless and R. Manetsh, Expert Opin. Drug Discovery, 2006, 1, 525–538 CrossRef CAS PubMed.
- (a) W. G. Lewis, L. G. Green, F. Grynszpan, Z. Radič, P. R. Carlier, P. Taylor, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 1053–1057 CAS; (b) R. Manetsch, A. Krasinski, Z. Radič, J. Raushel, P. Taylor, K. B. Sharpless and H. C. Kolb, J. Am. Chem. Soc., 2004, 126, 12809–12818 CrossRef CAS PubMed; (c) T. Suzuki, Y. Ota, Y. Kasuya, M. Mutsuga, Y. Kawamura, H. Tsumoto, H. Nakagawa, M. G. Finn and N. Miyata, Angew. Chem., Int. Ed., 2010, 49, 6817–6820 CrossRef CAS PubMed; (d) T. Suzuki, Y. Ota, Y. Kasuya, M. Mutsuga, Y. Kawamura, H. Tsumoto, H. Nakagawa, M. G. Finn and N. Miyata, Angew. Chem., Int. Ed., 2010, 122, 6969–6972 CrossRef; (e) S. K. Mamidyala and M. G. Finn, Chem. Soc. Rev., 2010, 39, 1252–1261 RSC; (f) X. Hu and R. Manetsch, Chem. Soc. Rev., 2010, 39, 1316–1324 RSC; (g) S. W. Millward, R. K. Henning, G. A. Kwong, S. M. Pitram, H. D. Agnew, K. M. Deyle, A. Nag, J. E. Hein, S. S. Lee, J. Lim, J. A. Pfeilsticker, K. B. Sharpless and J. R. Heath, J. Am. Chem. Soc., 2011, 133, 18280–18288 CrossRef CAS PubMed; (h) C. Peruzzotti, S. Borrelli, M. Ventura, R. Pantano, G. Fumagalli, M. S. Christodoulou, D. Monticelli, M. Luzzani, A. L. Fallacara, C. Tintori, M. Botta and D. Passarella, ACS Med. Chem. Lett., 2013, 4, 274–277 CrossRef CAS; (i) W. Tieu, T. P. Soares da Costa, M. Y. Yap, K. L. Keeling, M. C. J. Wilce, J. C. Wallace, G. W. Booker, S. W. Polyak and A. D. Abell, Chem. Sci., 2013, 4, 3533–3537 RSC.
- P. Anand and B. Singh, Arch. Pharmacal. Res., 2013, 36, 375–399 CrossRef CAS PubMed.
- A. Krasinski, Z. Radič, R. Manetsch, J. Raushel, P. Taylor, K. B. Sharpless and H. C. Kolb, J. Am. Chem. Soc., 2005, 127, 6686–6692 CrossRef CAS PubMed.
- For the crystal structure of the syn-(±)-TZ2PIQA6-mouse AChE complex, see: Y. Bourne, H. C. Kolbe, Z. Radic, K. B. Sharpless, P. Taylor and P. Marchot, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1449–1454 CrossRef CAS PubMed.
- M. Whiting, J. Muldoon, Y.-C. Lin, S. M. Silverman, W. Lindstrom, A. J. Olson, H. C. Kolb, M. G. Finn, K. B. Sharpless, J. H. Elder and V. V. Fokin, Angew. Chem., Int. Ed., 2006, 45, 1435–1439 CrossRef CAS PubMed.
- V. P. Mocharla, B. Colasson, L. V. Lee, S. Röper, K. B. Sharpless, C.-H. Wong and H. C. Kolb, Angew. Chem., Int. Ed., 2005, 44, 116–120 CrossRef CAS PubMed.
- N. Willand, M. Desroses, P. Toto, B. Dirié, Z. Lens, V. Villeret, P. Rucktooa, C. Locht, A. Baulard and B. Deprez, ACS Chem. Biol., 2010, 5, 1007–1013 CrossRef CAS PubMed.
- N. P. Grimster, B. Stump, J. R. Fotsing, T. Weide, T. T. Talley, J. G. Yamauchi, Á. Nemecz, C. Kim, K.-Y. Ho, K. B. Sharpless, P. Taylor and V. V. Fokin, J. Am. Chem. Soc., 2012, 134, 6732–6740 CrossRef CAS PubMed.
- C. Ronco, E. Carletti, J.-P. Colletier, M. Weik, F. Nachon, L. Jean and P.-Y. Renard, ChemMedChem, 2012, 7, 400–405 CrossRef CAS PubMed.
- (a) P. Camps, R. El Achab, D. M. Görbig, J. Morral, D. Muñoz-Torrero, A. Badia, J. E. Baños, N. M. Vivas, X. Barril, M. Orozco and F. J. Luque, J. Med. Chem., 1999, 42, 3227–3242 CrossRef CAS PubMed; (b) P. Camps, R. El Achab, J. Morral, D. Muñoz-Torrero, A. Badia, J. E. Baños, N. M. Vivas, X. Barril, M. Orozco and F. J. Luque, J. Med. Chem., 2000, 43, 4657–4666 CrossRef CAS PubMed.
- (a) C. Ronco, G. Sorin, F. Nachon, R. Foucault, L. Jean, A. Romieu and P.-Y. Renard, Bioorg. Med. Chem., 2009, 17, 4523–4536 CrossRef CAS PubMed; (b) C. Ronco, L. Jean and P.-Y. Renard, Tetrahedron, 2010, 66, 7399–7404 CrossRef CAS PubMed; (c) C. Ronco, L. Jean, H. Outaabout and P.-Y. Renard, Eur. J. Org. Chem., 2011, 302–310 CrossRef CAS; (d) C. Ronco, R. Foucault, E. Gillon, P. Bohn, F. Nachon, L. Jean and P.-Y. Renard, ChemMedChem, 2011, 6, 876–888 CrossRef CAS PubMed; (e) F. Nachon, E. Carletti, C. Ronco, M. Trovaslet, Y. Nicolet, L. Jean and P.-Y. Renard, Biochem. J., 2013, 453, 393–399 CrossRef CAS PubMed.
- A. Cavalli, M. L. Bolognesi, A. Minarini, M. Rosini, V. Tumiatti, M. Recanatini and C. Melchiorre, J. Med. Chem., 2008, 51, 347–371 CrossRef CAS PubMed.
- (a) P. Camps, X. Formosa, D. Muñoz-Torrero, J. Petrignet, A. Badia and M. V. Clos, J. Med. Chem., 2005, 48, 1701–1704 CrossRef CAS PubMed; (b) P. Camps, X. Formosa, C. Galdeano, T. Gómez, D. Muñoz-Torrero, M. Scarpellini, E. Viayna, A. Badia, M. V. Clos, A. Camins, M. Pallàs, M. Bartolini, F. Mancini, V. Andrisano, J. Estelrich, M. Lizondo, A. Bidon-Chanal and F. J. Luque, J. Med. Chem., 2008, 51, 3588–3598 CrossRef CAS PubMed; (c) E. Viayna, T. Gómez, C. Galdeano, L. Ramírez, M. Ratia, A. Badia, M. V. Clos, E. Verdaguer, F. Junyent, A. Camins, M. Pallàs, M. Bartolini, F. Mancini, V. Andrisano, M. P. Arce, M. I. Rodríguez-Franco, A. Bidon-Chanal, F. J. Luque, P. Camps and D. Muñoz-Torrero, ChemMedChem, 2010, 5, 1855–1870 CrossRef CAS PubMed.
- (a) J. Peters, M. Trovaslet, M. Trapp, F. Nachon, F. Hill, E. Royer, F. Gabel, L. van Eijck, P. Masson and M. Tehei, Phys. Chem. Chem. Phys., 2012, 19, 6764–6770 RSC; (b) M. Trovaslet, M. Trapp, M. Weik, F. Nachon, P. Masson, M. Tehei and J. Peters, Chem.–Biol. Interact., 2013, 1, 14–18 CrossRef PubMed.
- (−)-Huprine derivatives are about 2 orders of magnitude more active than their racemic mixtures (cf.ref. 11b).
- The configuration of the chiral center of PIQ has little effect on the binding affinity of the corresponding heterodimers (cf.ref. 4).
- We failed to prepare regioselectively the syn-triazole heterodimers under ruthenium-catalyzed 1,3-dipolar cycloaddition.
- For information, the most potent ligands of Drosophila melanogaster were anti substituted triazoles: Z. Radič, R. Manetsch, D. Fournier, K. B. Sharpless and P. Taylor, Chem. Biol. Interact., 2008, 175, 161–165 CrossRef PubMed.
- Crystallization of the m or rh-AChE and anti-(±)-9-HUPZ4PIQ-A2 proved unsuccessful.
- S. Senapati, Y. Cheng and J. A. McCammon, J. Med. Chem., 2006, 49, 6222–6230 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ob42109k |
| This journal is © The Royal Society of Chemistry 2014 |