Synthesis of crambescin B carboxylic acid, a potent inhibitor of voltage-gated sodium channels†
Atsuo
Nakazaki
a,
Yuki
Ishikawa
a,
Yusuke
Sawayama
a,
Mari
Yotsu-Yamashita
b and
Toshio
Nishikawa
*a
aGraduate School of Bioagricultural Sciences, Nagoya University, Chikusa, Nagoya 464-8601, Japan. E-mail: nisikawa@agr.nagoya-u.ac.jp
bGraduate School of Agricultural Science, Tohoku University, Aoba, Sendai 981-8555, Japan
First published on 30th October 2013
Abstract
The stereocontrolled synthesis of a racemic carboxylic acid of crambescin B, a marine alkaloid, is described. The synthesis features two highly stereoselective reactions: (i) palladium-catalyzed hydroxymethylation of an alkynyl aziridine having an N-guanidino group and (ii) cascade bromocyclization providing a spiro-hemiaminal structure. The cell-based colorimetric assay showed that the synthesized carboxylic acid exhibited a potent inhibitory activity on voltage-gated sodium channels.
Considerable attention is currently devoted to the synthesis and properties of guanidine-containing natural products.1 Well-known examples of such compounds include tetrodotoxin and saxitoxin isolated as toxic principles of pufferfish intoxication and paralytic shellfish poison, respectively. These compounds exhibit potent blockage of sodium ion influx through voltage-gated sodium channels.2 Recently, we have focused on developing a subtype selective blocker of the sodium channels on the basis of natural products such as tetrodotoxin3 and saxitoxin. In this context, we were intrigued by the biological activities of carboxylic acid 2 of an alkaloid crambescin B (1) isolated from the marine sponge Crambe crambe (Fig. 1),4,5 because of the similar zwitterion structure to tetrodotoxin; the spatial position of the guanidinium and carboxylate is similar to the guanidinium and orthoacid in tetrodotoxin (Fig. 2). Investigation of the biological activity of 2 has remained unexplored because crambescin B carboxylic acid 2 has not been isolated from natural sources and degradation of natural 1 into 2 has not been reported. Racemic synthesis of the twelve-carbon-chain homologue of carboxylic acid 2 has been reported by Snider et al.5b
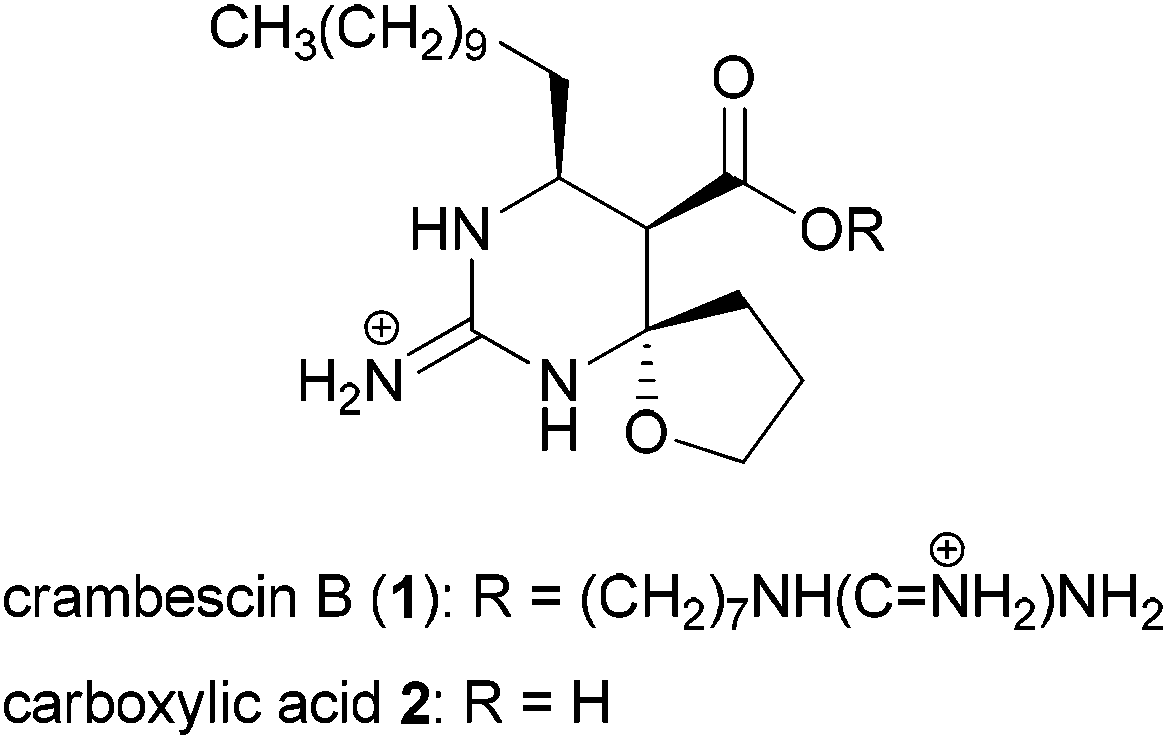 | ||
| Fig. 1 Structures of crambescin B (1) and its carboxylic acid 2. | ||
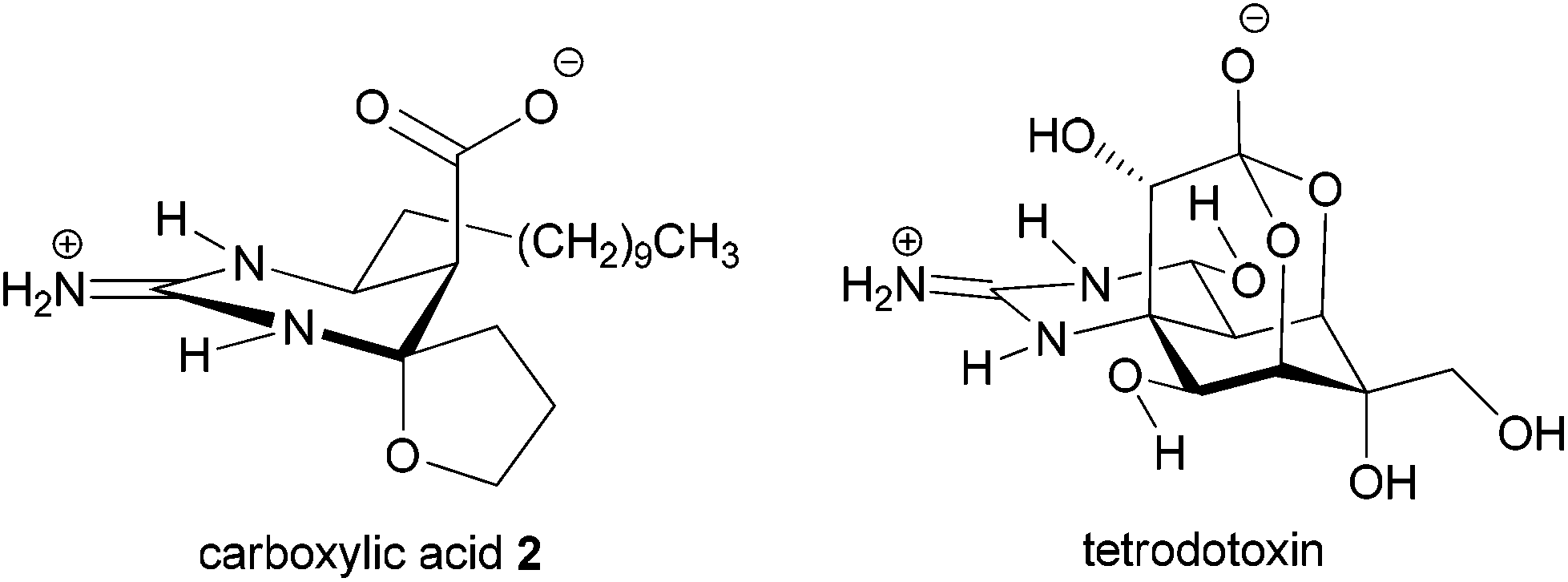 | ||
| Fig. 2 Structural comparison of crambescin B carboxylic acid 2 and tetrodotoxin. | ||
In this report, we describe the stereocontrolled synthesis of racemic carboxylic acid 2via cascade bromocyclization as a key reaction for the synthesis of a spiro-hemiaminal structure and preliminary results of sodium channel inhibitory activity of 2.
Our synthetic plan towards crambescin B carboxylic acid 2 is shown in Scheme 1. Carboxylic acid 2 would be synthesized from the corresponding alcohol A by oxidation of the primary alcohol and reduction of geminal dibromomethylene.6 The spirocyclic structure of A was envisaged to be constructed from (monocarbamoyl)guanidino-acetylene D through a cascade bromocyclization developed in this group.7 In this process, guanidino-acetylene D would undergo bromocation-induced cyclization to generate enamide B, which was further reacted with bromocations to form iminium ion intermediate C. And then the iminium ion in C would be trapped with the internal hydroxy group to furnish spiro-hemiaminal A. To synthesize guanidino-acetylene D bearing a hydroxymethyl group using cis-aziridine E, we planned to employ palladium-catalyzed stereoselective hydroxymethylation developed by Ohno et al.8cis-Aziridine E could be synthesized from the corresponding epoxide of cis-enyne 3 or by the direct aziridination of 3. cis-Enyne 3 was easily prepared by Sonogashira coupling between the known cis-1-iodotridecene and 3-butyn-1-ol. In principle, this synthetic plan allows us to synthesize 2 in an enantiomerically pure form from the corresponding enantio-enriched epoxide or aziridine prepared from cis-enyne 3.9 This plan posed two synthetic issues: stereoselective hydroxymethylation of cis-aziridine E and stereocontrol of the cascade bromocyclization of (monocarbamoyl)guanidino-acetylene D possessing a hydroxymethyl group at the propargylic position.
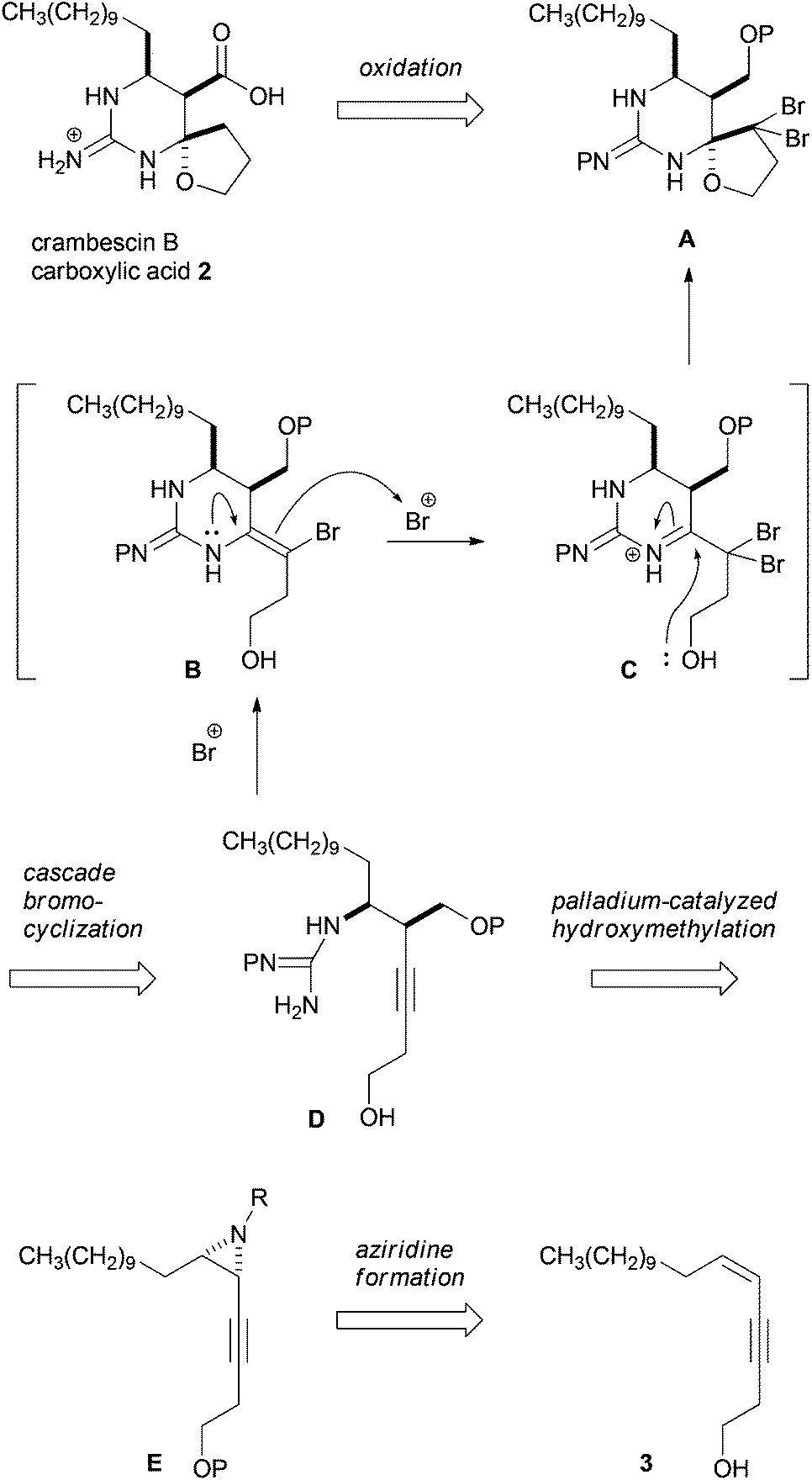 | ||
| Scheme 1 Synthetic plan of crambescin B carboxylic acid 2. | ||
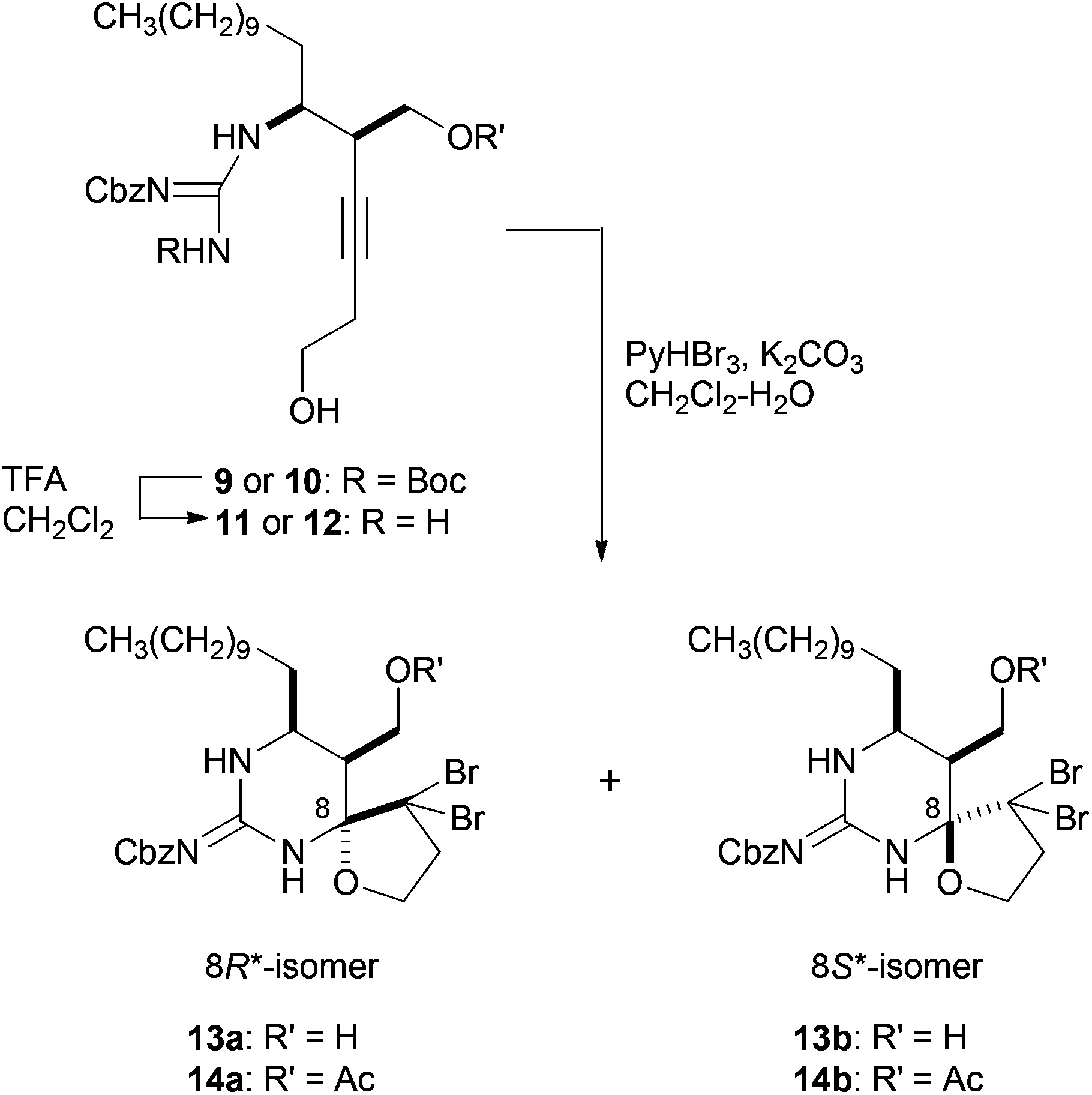
The synthesis of guanidino-acetylenes 9 and 10, precursors of the bromocyclization, is shown in Scheme 2. Enyne 3 was treated with mCPBA to produce epoxide 4 in 71% yield, which was then transformed into cis-aziridine 5 in two steps including regioselective addition of azide to epoxide 4 and sequential reductive cyclization of the resulting azide alcohol.10,11 The protection of the primary alcohol provided TBDPS ether 6 in 56% overall yield from 4. Aziridine 6 was guanylated with Cbz,Boc-protected methylisothiourea7a in the presence of HgCl2 and Et3N to afford guanidino-aziridine 7. Stereoselective hydroxymethylation of the ethynylaziridine developed by Ohno et al. was next applied.8 Thus, guanidino-aziridine 7 was subjected to formalin in the presence of indium iodide and Pd(PPh3)4 to produce the requisite alcohol 8 in 75% yield as a single diastereomer. The stereochemistry of the newly generated stereogenic center of the resulting 8 was confirmed after the cascade reaction (see ESI† for details). The high regio- and stereoselectivity can be rationalized by an analogous cyclic transition state involving an allenylindium intermediate and formaldehyde, reported by Ohno et al.8 It is noteworthy that in addition to carbamoyl and sulfonyl groups previously reported, a (biscarbamoyl)guanidino group can serve as an activating group of aziridine in the hydroxymethylation. Bromocyclization precursors 9 and 10 were obtained by simple protective group manipulations.
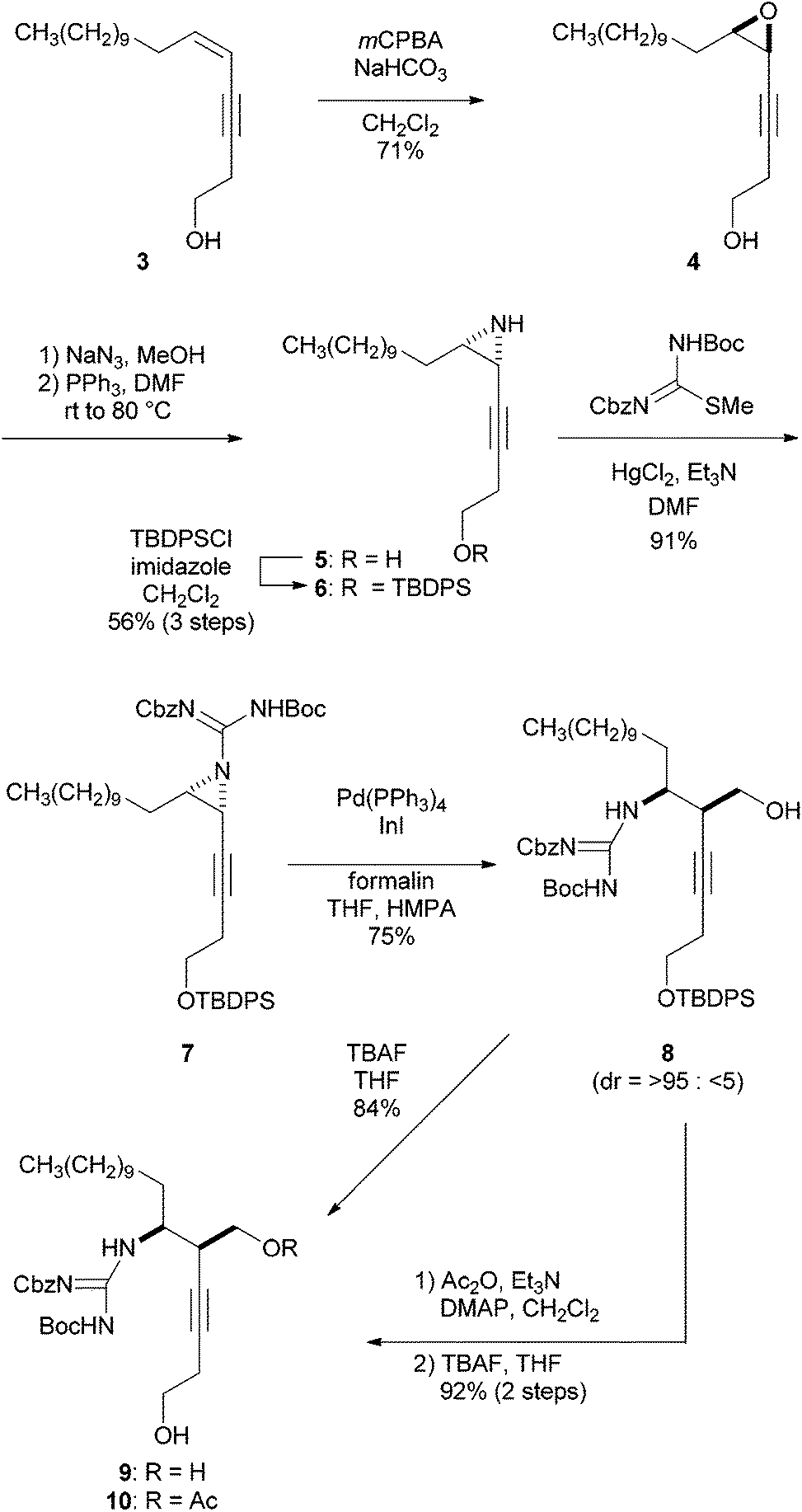 | ||
| Scheme 2 Synthesis of guanidino alcohols 9 and 10. | ||
With the precursors 9 and 10 in hand, key cascade bromocyclization was next investigated (Table 1). The Boc group of 9 was deprotected and then the product was exposed to the conditions for the bromocyclization,7 providing a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of spiro-hemiaminals 13a and 13b as C8-epimers (Table 1, entry 1).12 In contrast, the corresponding acetate 12 underwent bromocyclization under the same conditions to give the desired 14a exclusively (Table 1, entry 2). The observed high diasteroselectivity could be rationalized by the neighboring group participation of the acetyl group to shield the β-face of an iminium ion intermediate (Scheme 3).
:1 mixture of spiro-hemiaminals 13a and 13b as C8-epimers (Table 1, entry 1).12 In contrast, the corresponding acetate 12 underwent bromocyclization under the same conditions to give the desired 14a exclusively (Table 1, entry 2). The observed high diasteroselectivity could be rationalized by the neighboring group participation of the acetyl group to shield the β-face of an iminium ion intermediate (Scheme 3).
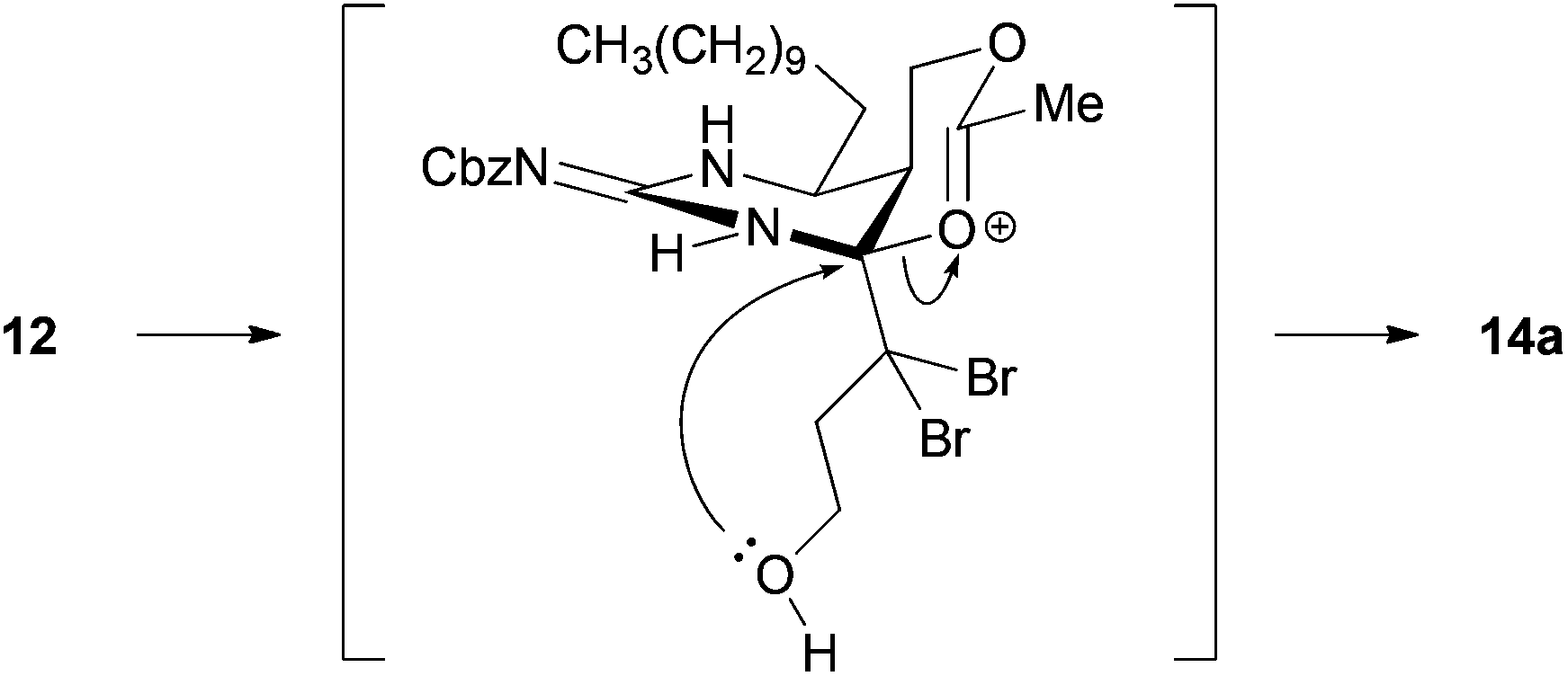 | ||
| Scheme 3 Possible rationalization of high diastereocontrol for the formation of 14a. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | R′ | Two step yield | |
| 8R*-isomer | 8S*-isomer | |||
| 1 | 11 | H | 13a: 27% | 13b: 26% |
| 2 | 12 | Ac | 14a: 73% | 14b: 0% |
The final task was to oxidize a primary alcohol in 14a to carboxylic acid (Scheme 4). The geminal dibromomethylene of spiro-hemiaminal 14a was reduced with Bu3SnH and Et3B to afford 15, whose acetate was reductively deprotected with DIBAL leading to alcohol 16a in good yield. However, the oxidation of 16a to carboxylic acid proved to be difficult. In this event, we found that the Jones oxidation of guanidinium compound 1713 prepared by the conventional hydrogenolytic deprotection of the Cbz group of 16a furnished carboxylic acid 2 in 32% yield for the two steps.14 It is worth noting that the use of the unprotected guanidinium compound as an oxidation precursor would be crucial for this oxidation. The 1H and 13C NMR spectra of the hydrochloric acid salt of 2 were in good agreement with those reported by Snider et al.5b,15
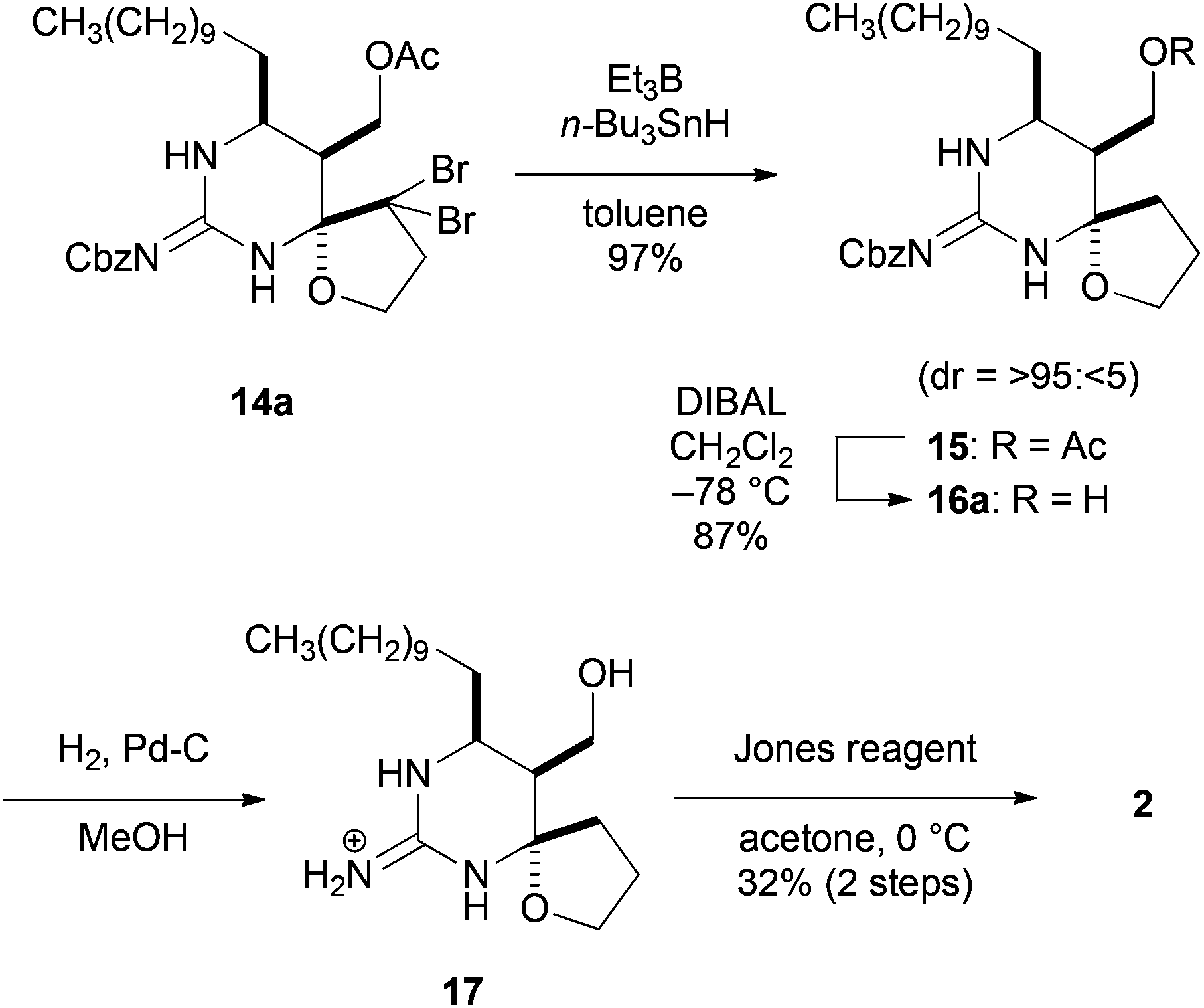 | ||
| Scheme 4 Synthesis of crambescin B carboxylic acid 2. | ||
Inhibitory activity of 2 on voltage-gated sodium channels was investigated by cytotoxicity of ouabain and veratridine to mouse neuroblastoma cells (Neuro-2a), as we previously estimated the activity of tetrodotoxin and its analogues.16 The ED50 for carboxylic acid 2 was estimated to be 42 ± 25 nM (mean, s.d.) (n = 5), which was nine fold larger than that of tetrodotoxin (ED50 = 4.8 ± 2.6 nM, n = 4). This result suggests that carboxylic acid 2 exhibited a potent inhibitory activity on voltage-gated sodium channels.17
Conclusions
In conclusion, we have developed the stereocontrolled synthesis of crambescin B carboxylic acid 2 by means of two highly stereoselective reactions as key steps: (i) palladium-catalyzed hydroxymethylation with (biscarbamoyl)guanidino-aziridine and (ii) the cascade bromocyclization of guanidino-acetylenes bearing a branched hydroxymethyl group, and we demonstrated the utility of these reactions. This study should provide a facile synthetic route for not only enantio-enriched crambescin B carboxylic acid which probably could not be obtained by Snider's route but also its naturally occurring analogues such as crambescins A and C. Further synthetic studies of the related compounds as well as detailed investigations on the biological activities of 2 are in progress.Acknowledgements
This work was financially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Chemical Biology of Natural Products” and a Grant-in-Aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology, Japan; Grant for Basic Science Research Projects from the Sumitomo Foundation; and the Daiichi-Sankyo Foundation of Life Science. We thank Prof. B. B. Snider (Brandeis University) for discussions regarding the NMR data of 2.Notes and references
- R. G. S. Berlinck, A. E. Trindade-Silva and M. F. C. Santos, Nat. Prod. Rep., 2012, 29, 1382–1406 RSC.
- (a) T. Narahashi, J. Toxicol., Toxin Rev., 2001, 20, 67–84 CrossRef CAS PubMed; (b) T. Narahashi, Proc. Jpn. Acad., Ser. B, 2008, 84, 147–154 CrossRef CAS; (c) L. E. Llewellyn, Nat. Prod. Rep., 2006, 23, 200–222 RSC.
- For recent synthetic studies on tetrodotoxin from this laboratory, see: (a) M. Adachi, T. Imazu, M. Isobe and T. Nishikawa, J. Org. Chem., 2013, 78, 1699–1705 CrossRef CAS PubMed , and references cited therein; (b) T. Nishikawa and M. Isobe, Chem. Rec., 2013, 13, 286–302 CrossRef CAS PubMed.
- (a) R. G. S. Berlinck, J. C. Braekman, D. Daloze, K. Hallenga, R. Ottinger, I. Bruno and R. Riccio, Tetrahedron Lett., 1990, 31, 6531–6534 CrossRef CAS; (b) E. A. Jares-Erijman, A. A. Ingrum, F. Sun and K. L. Rinehart, J. Nat. Prod., 1993, 56, 2186–2188 CrossRef CAS; (c) S. Bondu, G. Genta-Jouve, M. Leiròs, C. Vale, J. Guigonis, L. M. Botana and O. P. Thomas, RSC Adv., 2012, 2, 2828–2835 RSC . The crambescins are classified into three major groups, A–C, according to the structural features of the central core scaffold. Although Thomas et al. proposed using a more specific name, crambescin B2, for analogues having an upper alkyl side chain whose carbon number is higher than eleven, we used the major classified name crambescin B for compound 1 for clarity.
- The structural revision and total synthesis of racemic crambescin B analogues were reported. (a) B. B. Snider and Z. Shi, J. Org. Chem., 1992, 57, 2526–2528 CrossRef CAS; (b) B. B. Snider and Z. Shi, J. Org. Chem., 1993, 58, 3828–3839 CrossRef CAS.
- In our initial studies, the addition of cyanide to aziridine E (R = Ns, P = H) was attempted in order to introduce a carboxylic acid moiety; however, the desired product could not be obtained, probably because of the instability of the adduct. Elimination of the N-Ns amide was observed in the reaction of N-Ns aziridine with KCN, indicating its β-elimination.
- (a) Y. Sawayama and T. Nishikawa, Synlett, 2011, 651–654 CAS; (b) Y. Sawayama and T. Nishikawa, Angew. Chem., Int. Ed., 2011, 50, 7176–7178 CrossRef CAS PubMed; (c) Y. Sawayama and T. Nishikawa, J. Synth. Org. Chem., Jpn., 2012, 70, 1178–1186 CrossRef CAS.
- (a) H. Ohno, H. Hamaguchi and T. Tanaka, Org. Lett., 2000, 2, 2161–2163 CrossRef CAS PubMed; (b) H. Ohno, H. Hamaguchi and T. Tanaka, J. Org. Chem., 2001, 66, 1867–1875 CrossRef CAS PubMed; (c) J. Inuki, A. Iwata, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2011, 76, 2072–2083 CrossRef PubMed.
- A number of synthetic methods for enantio-enriched epoxides or aziridines from cis-enyne compounds have been reported. For reviews on asymmetric epoxidation, see: (a) T. Katsuki, in Catalytic Asymmetric Synthesis, ed. I. Ojima, VCH, New York, 2000, ch. 6B Search PubMed; (b) O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958–3987 CrossRef CAS PubMed and references therein. For a review on asymmetric aziridination, see: ; (c) H. Pellissier, Tetrahedron, 2010, 66, 1509–1555 CrossRef CAS PubMed.
- Y. Ittah, Y. Sasson, I. Shahak, S. Tsaroom and J. Blum, J. Org. Chem., 1978, 43, 4271–4273 CrossRef.
- This azide alcohol was obtained as a single regioisomer through the addition of azide at the propargylic position of 4.
- The stereochemistries of the C8 stereogenic centers of the resulting spiro-hemiaminals 13a and 13b were determined by the NOESY correlations of the reduced 16a and 16b, respectively, prepared from 13a and 13b by treatment with n-Bu3SnH and Et3B. See ESI† for details.
- The stereochemistry of the C8 stereogenic centers of 17 was confirmed by the NOESY correlations.
- Interestingly, the 16b (8R*-isomer) was converted not into the corresponding deprotected 8R*-isomer but into its epimer 17 (8S*-isomer) under the deprotection conditions [Pd–C, H2 (1 atm), MeOH], which indicates that epimerization under the hydrogenation conditions occurred leading to a thermodynamically more stable isomer 17.
- The spectral data of the hydrochloric acid salt of carboxylic acid 2 were identical to those of the related carboxylic acid with a twelve-carbon alkyl side chain reported by Snider et al., with the exception of one extra methylene.
- For details of the cell culture assay, see: M. Yotsu-Yamashita, D. Urabe, M. Asai, T. Nishikawa and M. Isobe, Toxicon, 2003, 42, 557–560 CrossRef CAS PubMed.
- During the preparation of this manuscript, Botana et al. reported that crambescin C1, an open form analogue of the spiro-hemiaminal structure of crambescin B, exhibited a weak inhibitory activity of total Na+ current. V. Martín, C. Vale, S. Bondu, O. P. Thomas, M. R. Vieytes and L. M. Botana, Chem. Res. Toxicol., 2013, 26, 169–178 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data and 1H and 13C NMR spectra. See DOI: 10.1039/c3ob42017e |
| This journal is © The Royal Society of Chemistry 2014 |