Silyl-protected dioxaborinanes: application in the Suzuki cross-coupling reaction†
Sean
Goggins
,
Eleanor
Rosevere
,
Clément
Bellini
,
Joseph C.
Allen
,
Barrie J.
Marsh
,
Mary F.
Mahon
and
Christopher G.
Frost
*
Department of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: c.g.frost@bath.ac.uk; Fax: +44 (0)1225 386231; Tel: +44 (0)1225 386142
First published on 7th November 2013
Abstract
The synthesis of a range of novel silyl-protected dioxaborinanes as a column- and bench-stable boron reagent were found to be advantageous to achieving good yields in palladium-catalysed cross-coupling reactions under standard conditions.
For Suzuki cross-coupling reactions, boronic acids are the coupling partners of choice in the majority of applications. However, boronic acids can be difficult to synthesise and there can often be issues with purification and manipulation. Also, an excess of the boronic acid has to be used due to the competing formation of trimeric cyclic anhydrides (boroxines) and protodeboronation processes, leading to difficulties in being able to accurately measure reaction stoichiometry.1 A number of elegant solutions to these problems have been presented involving the use of preformed borate and boronate reagents that can be isolated and stored prior to use (Fig. 1). These include tris(hydroxy)borates,2 lithium trimethoxyborate species,3 trifluoroborate salts4 and N-methyliminodiacetic acid (MIDA) boronates.5 An important addition to this range of donors is the cyclic triolborates synthesised by Miyaura.6 These borate reagents are reported to be stable in air and water and more soluble in organic solvents than potassium trifluoroborates. Recently, we described the synthesis of silyl-protected dioxaborinanes and exemplified their use as a coupling partner in the enantioselective synthesis of 4-arylchroman-2-ones.7 In this paper we describe an alternative procedure to the synthesis of silyl-protected dioxaborinanes and demonstrate their use as a boron reagent within the Suzuki cross-coupling reaction.
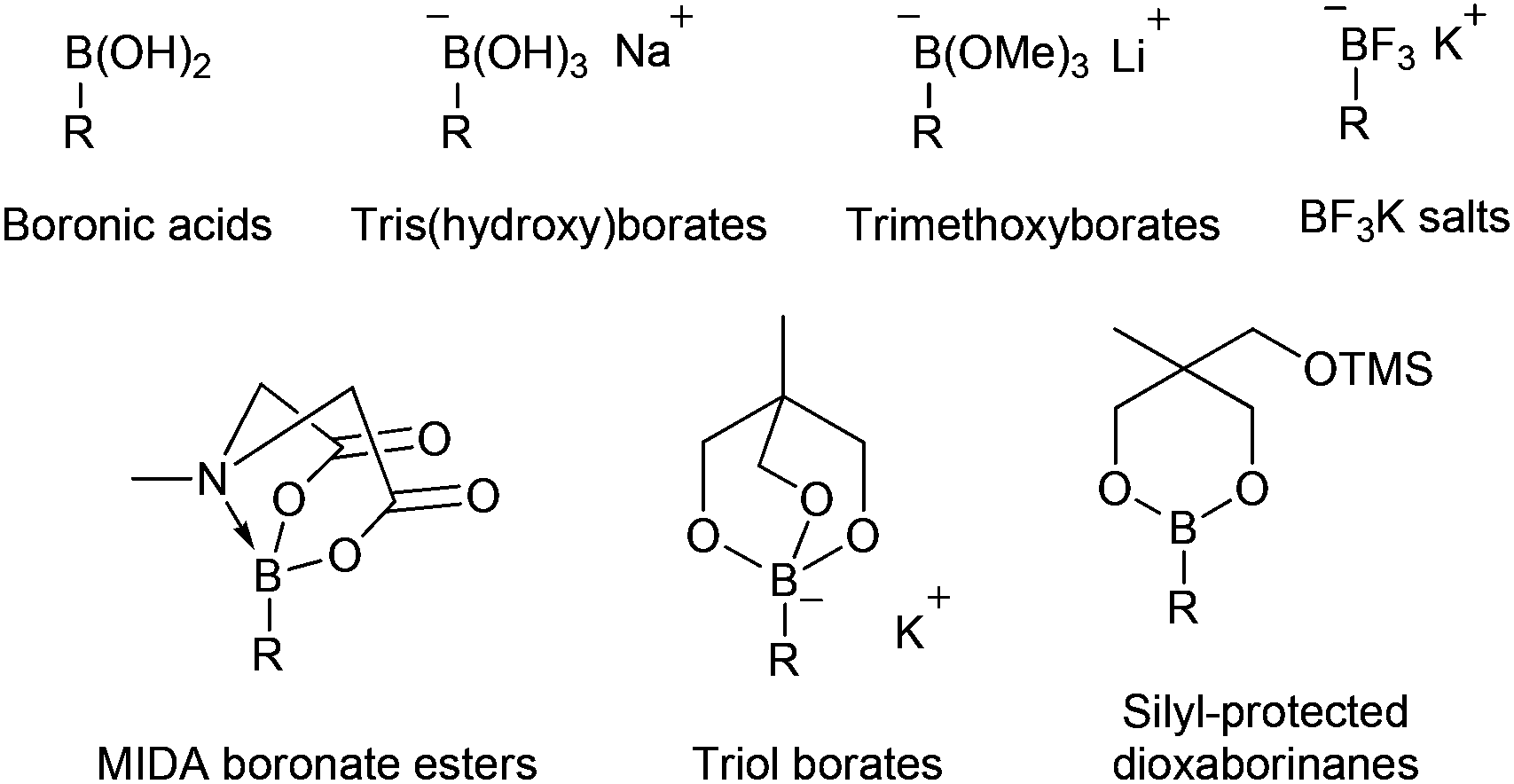 | ||
| Fig. 1 Organoboron reagents. | ||
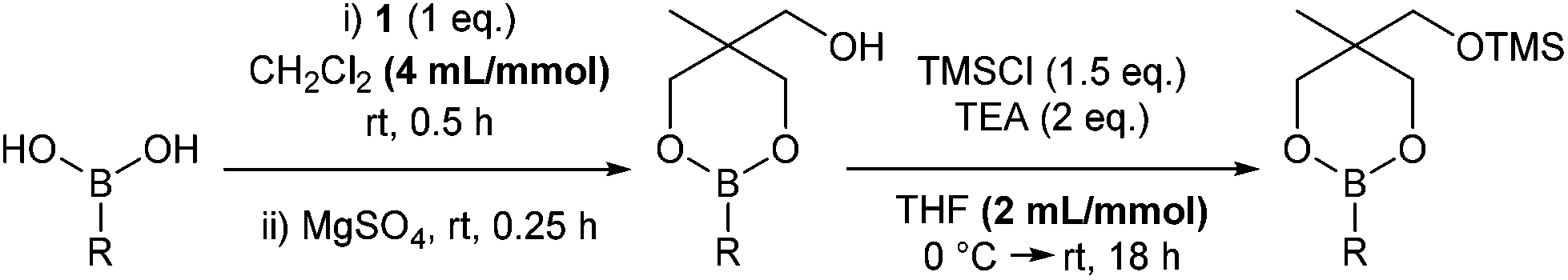
Previously, we reported that heating boronic acids with 1,1,1-tris(hydroxymethyl)ethane 1, under Dean-Stark conditions gives the dioxaborinane intermediate prior to silylation.7 Alternatively, stirring the boronic acid with the triol at room temperature in dichloromethane leads to the formation of the same intermediate without losing any boronic acid to boroxine formation. Following work-up, treatment with chlorotrimethylsilane in the presence of triethylamine gives the silyl-protected dioxaborinane in improved yields (Scheme 1). Also, the concentration of the reaction mixture for both reactions was found to be important in order to gain optimum yields via the telescoped procedure. Purification can be achieved through standard column chromatography to afford the desired protected boron species. Once synthesised and purified, silyl-protected dioxaborinanes are stable on a bench-top for several months without any decomposition being observed.
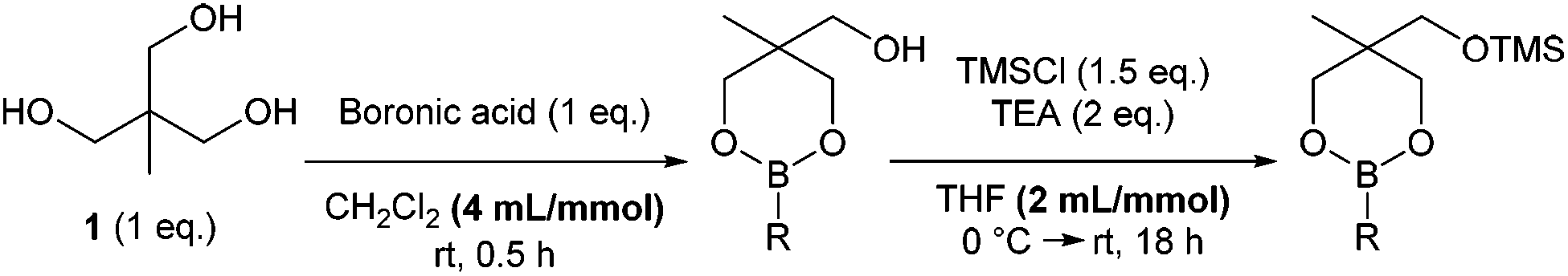 | ||
| Scheme 1 General procedure for the synthesis of silyl-protected dioxaborinanes. | ||
The synthesis was then applied to a wide range of commercially available boronic acids and the corresponding silyl-protected dioxaborinanes were obtained in all cases (Table 1). Simple aryl boronic acids gave excellent yields over the two steps but yields decreased somewhat for both electron-withdrawing aryl boronic acids as well as sterically demanding substrates. Pleasingly, heteroaromatics were also tolerated under the reaction conditions. The structure of silyl-protected dioxaborinane 2k was confirmed by X-ray crystallography (Fig. 2).
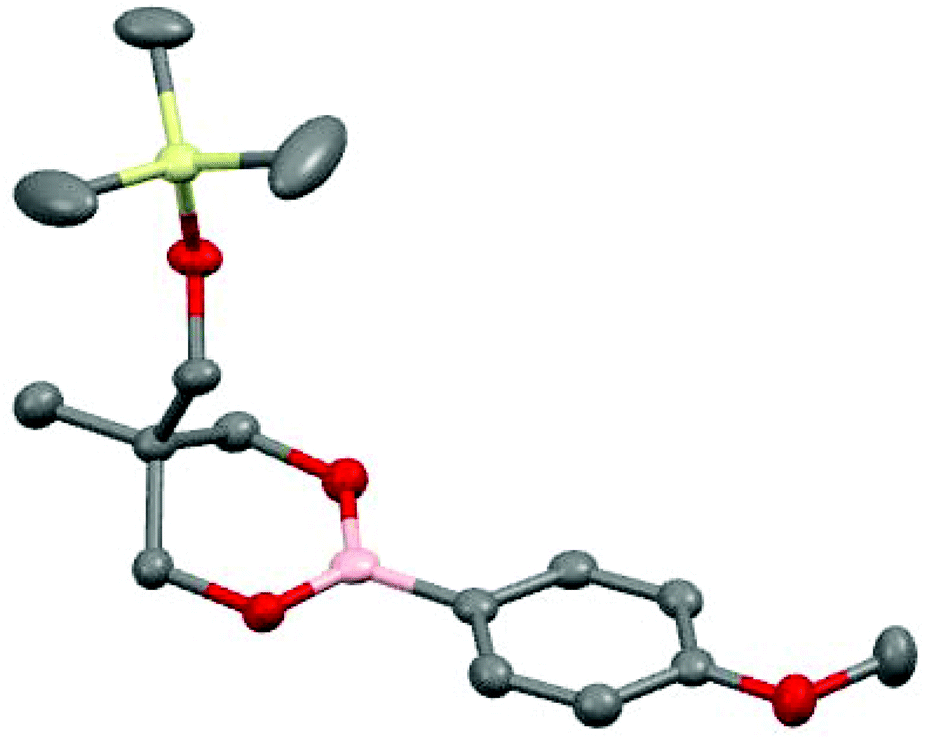 | ||
| Fig. 2 ORTEP drawing of 2k. Hydrogen atoms omitted for clarity. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | Yieldb (%) | Entry | R | Yieldb (%) | ||
| a Reaction conditions: boronic acid (8 mmol) suspended in anhydrous CH2Cl2 (4 mL mmol−1) under N2. Tris(hydroxymethyl)ethane (1 eq., 8 mmol) was then added and stirred until homogeneous (∼0.5 h), then MgSO4 added and stirred for an additional (0.25 h). Filtration and concentration gives crude intermediate. Redissolved in anhydrous THF (2 mL mmol−1) under N2 and TEA (2 eq. 16 mmol) was added. At 0 °C, TMSCl (1.5 eq., 12 mmol) added and left to stir overnight. Purification via silica gel column chromatography (hexane 8–2 EtOAc). b Isolated yields over two steps. | |||||||
| 1 | 2a |
|
96 | 10 | 2j |
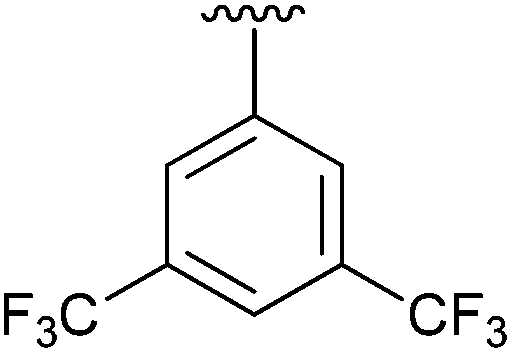
|
63 |
| 2 | 2b |
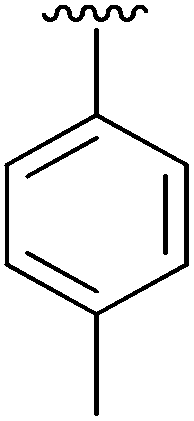
|
95 | 11 | 2k |
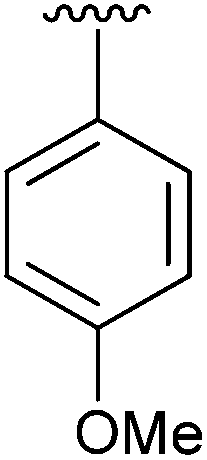
|
94 |
| 3 | 2c |
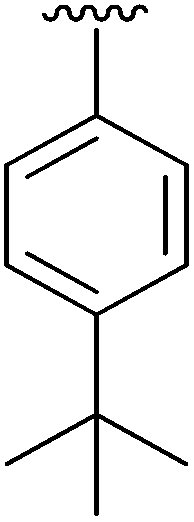
|
49 | 12 | 2l |
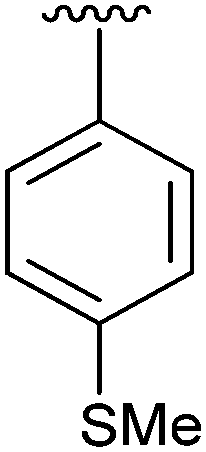
|
37 |
| 4 | 2d |
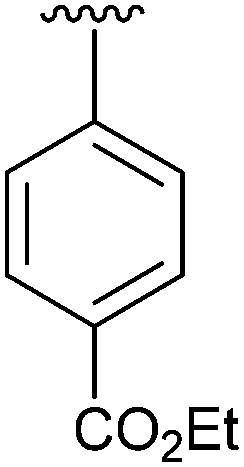
|
56 | 13 | 2m |
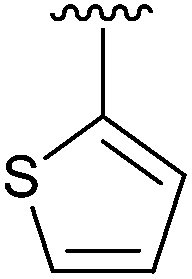
|
17 |
| 5 | 2e |
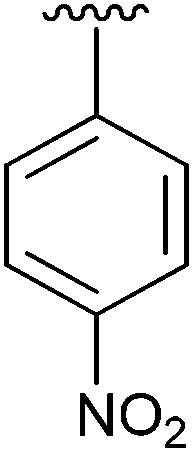
|
70 | 14 | 2n |
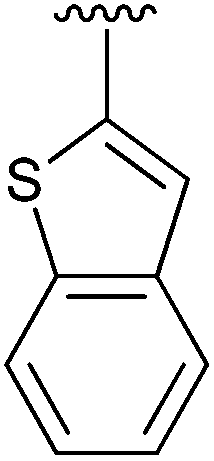
|
57 |
| 6 | 2f |
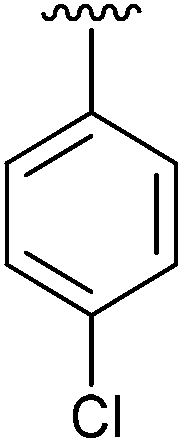
|
98 | 15 | 2o |
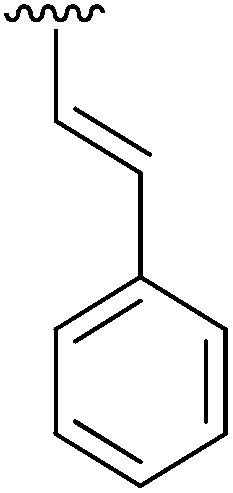
|
38 |
| 7 | 2g |
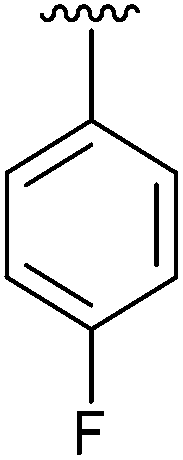
|
97 | 16 | 2p |
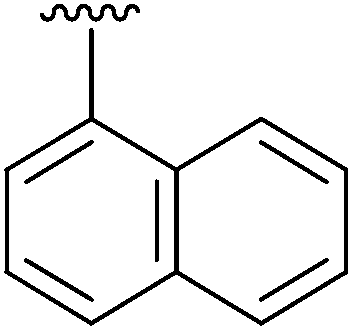
|
59 |
| 8 | 2h |
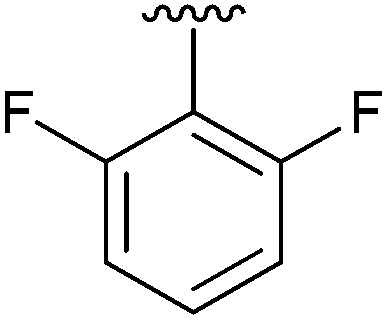
|
15 | 17 | 2q |
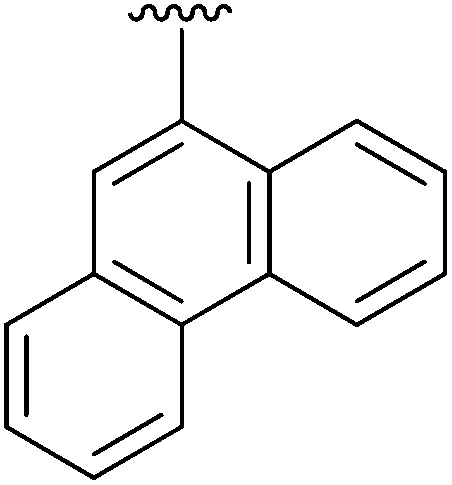
|
28 |
| 9 | 2i |
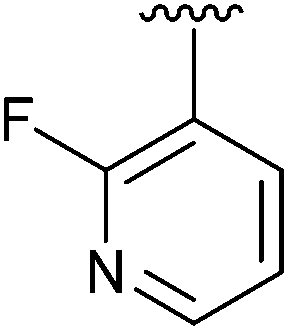
|
37 | 18 | 2r |
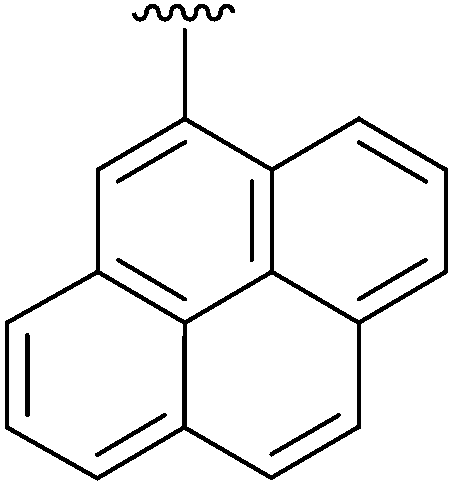
|
27 |
Previously, we have shown that silyl-protected dioxaborinanes provide superior yields as a boron reagent than boronic acids, trifluoroborates and triol borates within rhodium-catalysed conjugate addition reactions to arylidene Meldrum's acids.7 We therefore attempted to extend the application of the silyl-protected dioxaborinanes by utilising them in Suzuki cross-coupling reactions.
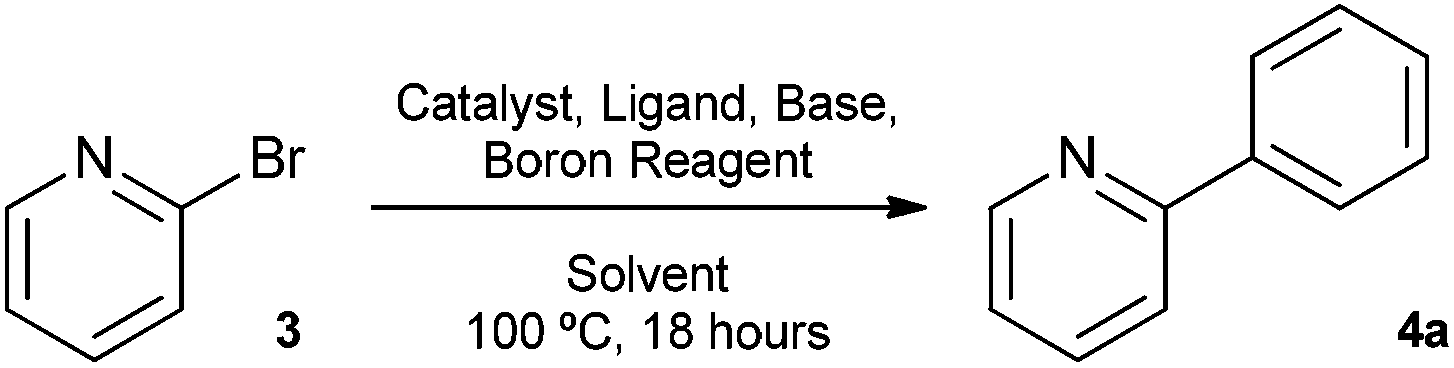
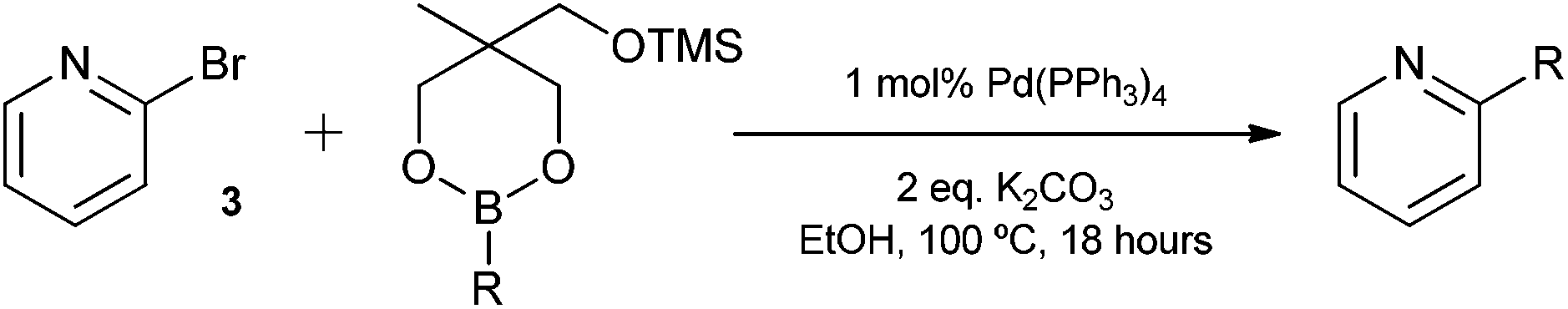
The Suzuki cross-coupling reaction between boronic acids and aryl halides has developed into one of the most important cross-coupling reactions and is a powerful and general method for the formation of carbon–carbon bonds.8 In particular, the construction of carbon–carbon bonds between heteroaromatics is of great interest as a variety of pharmaceutical compounds are structured around heteroaryl motifs, often containing a pyridine moiety.9 With this in mind, we decided to choose 2-bromopyridine as the substrate for Suzuki cross-coupling reactions with our silyl-protected dioxaborinanes to identify any potential benefit the protecting group has over standard boronic acids. Typically, a combination of solvents, bases and ligands are required to obtain high yields in Suzuki cross-coupling reactions, especially with difficult substrates.10 Therefore, a common set of conditions were chosen to identify the optimum conditions for the cross-coupling reaction with our dioxaborinanes (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Base | Boron reagent | Solvent | Yieldj (%) |
| a Reaction conditions: catalyst, ligand and base dissolved in solvent under argon. Boron reagent in solvent then added followed by 2-bromopyridine (1 mmol). Reaction mixture then stirred for 18 hours at 100 °C. Purification via silica gel column chromatography (EtOAc 5–95 hexane). b 2.5 mol% Pd2dba3, 5 mol% PCy3. c 1.7 eq. d 2.5 mol% Pd(OAc)2, 5 mol% SPhos. e 3 eq. f 0.5 mol% Pd(PPh3)4. g 2 eq. h 1 mol% Pd(PPh3)4. i 1.5 eq. j Isolated yields. | |||||
| 1 | Pd2dba3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
K3PO4c |
2a (1 eq.) | Dioxane 2–1 H2O | 0 |
| 2 | Pd(OAc)2d |
K3PO4e |
2a (0.8 eq.) | DMF 9–1 H2O | 77 |
| 3 | Pd(PPh3)4f |
K2CO3g |
2a (1.5 eq.) | Ethanol | 70 |
| 4 | Pd(PPh3)4h |
K2CO3g |
2a (1.5 eq.) | Ethanol | 98 |
| 5 | Pd(PPh3)4h |
K2CO3g |
Ph-B(OH)2i |
Ethanol | 74 |
| 6 | Pd(PPh3)4h |
K2CO3g |
2a (1.3 eq.) | Ethanol | 73 |
| 7 | Pd(PPh3)4h |
K2CO3g |
2a (1.1 eq.) | Ethanol | 70 |
Surprisingly, when using conditions pioneered by Fu et al., that can deliver excellent yields using aryl chlorides as substrates at room temperature, the Suzuki cross-coupling failed to give any of the desired product (Table 2, entry 1).11 Conditions typified by Buchwald et al., afforded 2-phenylpyridine 4a only in a modest yield (Table 2, entry 2).12 However, when using standard Suzuki cross-coupling conditions of 1 mol% Pd(PPh3)4 in ethanol, a near quantitative yield was obtained (Table 2, entry 4). Lowering the catalytic loading and dropping the equivalents of dioxaborinane only lead to a drop-off in yield. Comparatively, when using the same conditions but using phenylboronic acid as the coupling partner, a lower yield was obtained (Table 2, entry 5). This interesting result prompted a further study to compare the advantage silyl-protected dioxaborinanes may have over boronic acids and other boronic acid ester derivatives using a more challenging substrate.
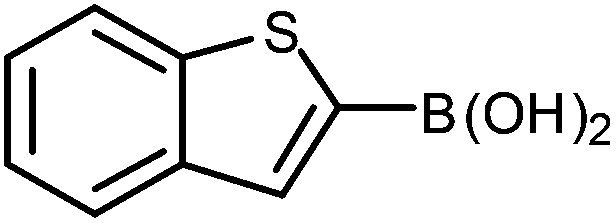
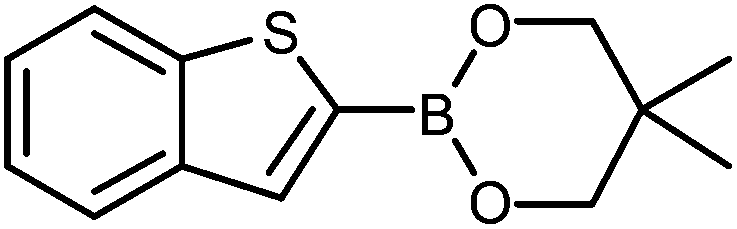
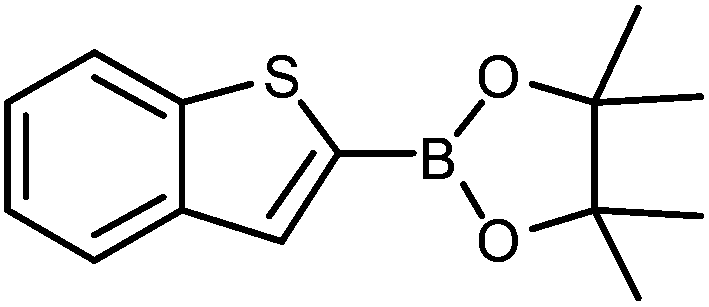
The construction of carbon–carbon bonds between two heteroaryl reagents can be problematic for palladium-catalysed coupling reactions due to hetero-atom lone pairs being able to coordinate to the metal centre and subsequently poison the catalyst.13 Benzo[b]thiophen-2-ylboronic acid, its neopentyl glycol and pinacol derivatives, along with silyl-protected dioxaborinane derivative 2n, were therefore chosen as boron reagents in the Suzuki cross-coupling with 2-phenylpyridine under our optimised conditions (Table 3).
|
|
||
|---|---|---|
| Entry | Boron reagent | Yieldb (%) |
| a Reaction conditions: Pd(PPh3)4 (0.01 mmol, 1 mol%), K2CO3 (2 mmol, 2 eq.) dissolved in ethanol (2 mL) under argon. Boron reagent (1.5 mmol, 1.5 eq.) in ethanol (2 mL) was added followed by 2-bromopyridine (1 mmol, 1 eq.). Reaction mixture stirred for 18 hours at 100 °C. Purification via silica gel column chromatography (hexane 95–5 EtOAc). b Isolated yields. | ||
| 1 |
|
50 |
| 2 |
|
67 |
| 3 |
|
91 |
| 4 | 2n | 92 |
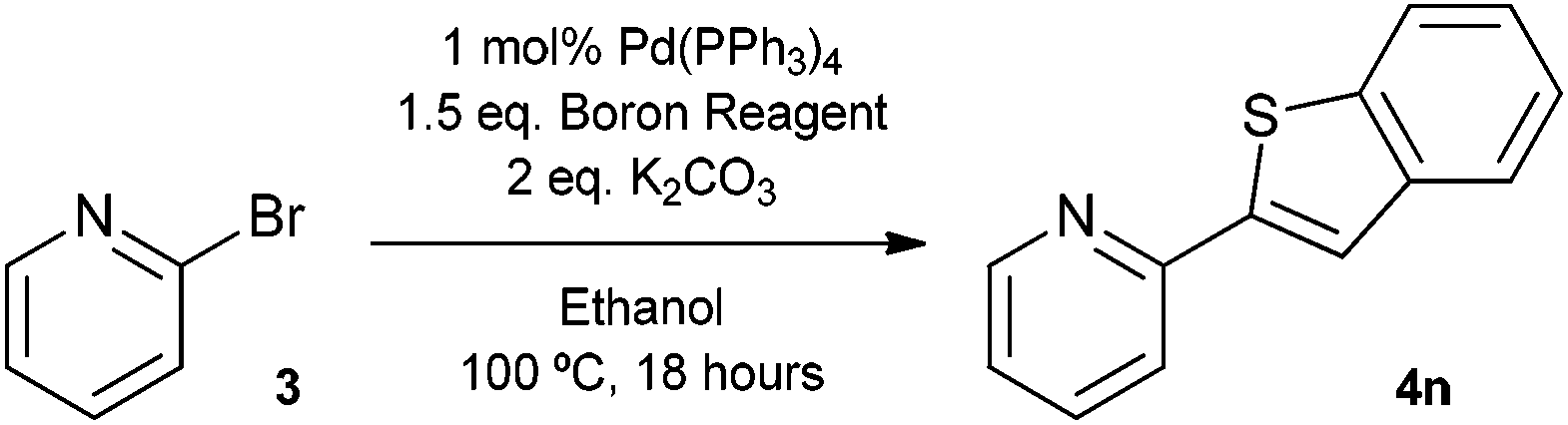
The unprotected boronic acid delivered only a moderate yield of product 4n, whilst the neopentyl glycol protected species provided just a slight improvement. Surprisingly, the structurally similar protecting group of the silyl-protected dioxaborinane provided a far superior yield in comparison. This leads us to believe that the –OTMS group is playing a role within the reaction mechanism. Recent investigations into the rate of hydrolysis of boronic acid protecting groups have revealed that a ‘slow-release’ mechanism from the protected species to the free boronic acid suppresses the formation of competing side-products.14 The silyl-protected dioxaborinanes could be operating in a similar fashion and slowly releasing the boronic acid under mild conditions for Suzuki cross-coupling reactions without the need for additional water to be added. However, it is also plausible that under basic conditions, the trimethylsilyl group is removed leading to the formation of a triol borate, which is a known active species formed prior to oxidative insertion. Further investigations are underway to determine the precise mechanism of the silyl-protected dioxaborinanes with coupling reactions. The pinacol-protected benzothiophene derivative also furnished a high yield of 4n showing that the silyl-protected dioxaborinane competes favourably with existing protecting groups, and offers an alternative option for the preparation of a stable boron protecting group that has high reactivity in carbon–carbon bond forming reactions.
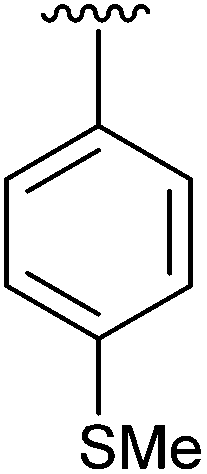
Having found the optimum conditions for the Suzuki cross-coupling using the silyl-protected dioxaborinane, the scope of the reaction was then explored (Table 4). Pleasingly, the desired product was obtained with all the dioxaborinanes that were screened against 2-bromopyridine. Electron-donating substituents on the aryl ring of the dioxaborinane gave near quantitative yields of the bis-aryl compound (Table 4, entries 2, 11, 12). This is as expected as increased nucleophilicity of the aryl ring accelerates transfer to the palladium catalyst during the transmetallation step of the catalytic cycle.15 The alkenyl dioxaborinane 2o also gave a near quantitative yield of 2-styrylpyridine, a result which is in accordance with Suzuki and Miyaura's findings in their seminal publication.16 Generally, aryl halides were tolerated giving good yields in some cases (Table 4, entries 6, 7), but electron-withdrawing substituents on the aryl ring caused a decrease in yields (Table 4, entries 4, 5, 8, 9). Heteroaromatics were also tolerated giving good yields (Table 4, entries 13, 14) but bulkier groups and larger aromatics gave decreased yields due to sterics. The use of silyl-protected dioxaborinanes with 2-bromopyridine in Suzuki cross-coupling reactions allows for the efficient formation of 2-substituted pyridines; products which can be further functionalised via C–H activation methodology due to the directing group nature of the pyridine moiety.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | Yieldb (%) | Entry | R | Yieldb (%) | ||
| a Reaction conditions: Pd(PPh3)4 (0.01 mmol, 1 mol%), K2CO3 (2 mmol, 2 eq.) dissolved in ethanol (2 mL) under argon. Silyl-protected dioxaborinane (1.5 mmol, 1.5 eq.) in ethanol (2 mL) was added followed by 2-bromopyridine (1 mmol, 1 eq.). Reaction mixture stirred for 18 hours at 100 °C. Purification via silica gel column chromatography (hexane 95–5 EtOAc). b Isolated yields. | |||||||
| 1 | 4a |
|
98 | 10 | 4j |
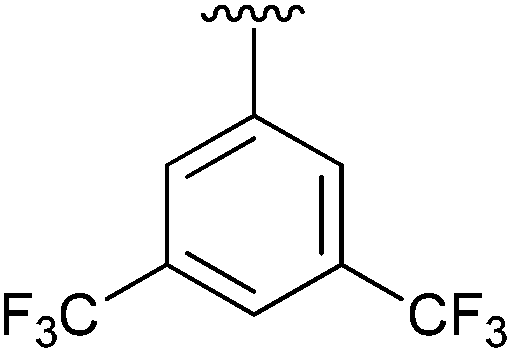
|
88 |
| 2 | 4b |
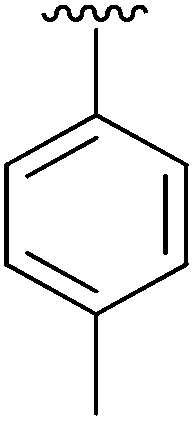
|
96 | 11 | 4k |
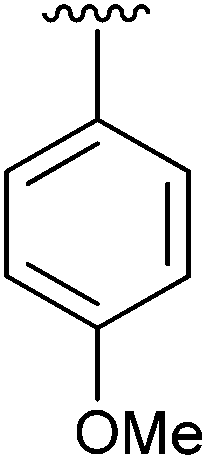
|
97 |
| 3 | 4c |
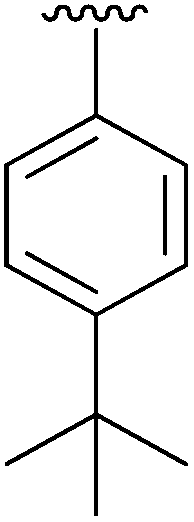
|
80 | 12 | 4l |
|
98 |
| 4 | 4d |
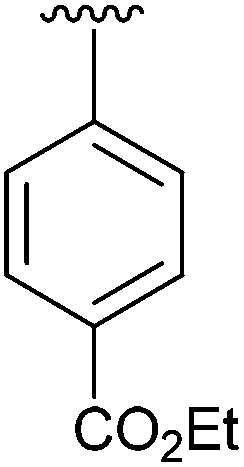
|
48 | 13 | 4m |
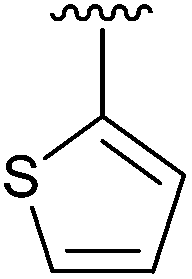
|
79 |
| 5 | 4e |
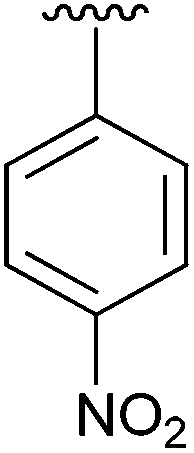
|
67 | 14 | 4n |
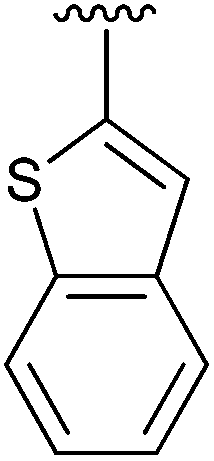
|
92 |
| 6 | 4f |
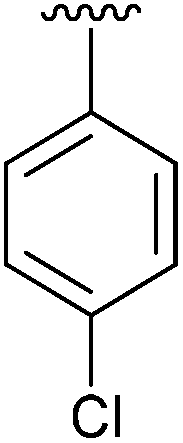
|
87 | 15 | 4o |
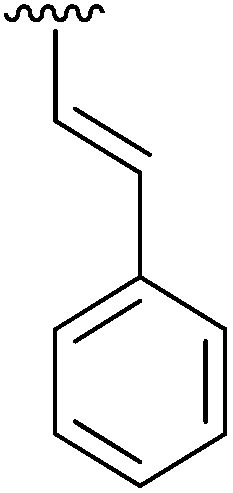
|
99 |
| 7 | 4g |
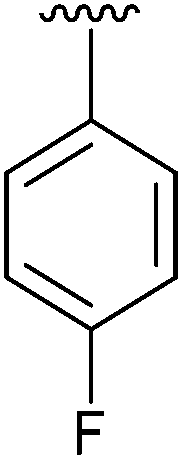
|
98 | 16 | 4p |
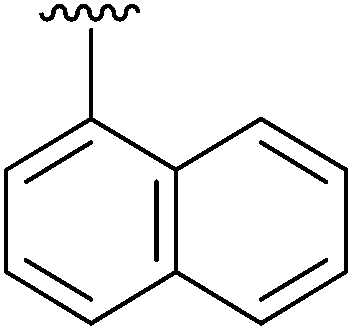
|
77 |
| 8 | 4h |
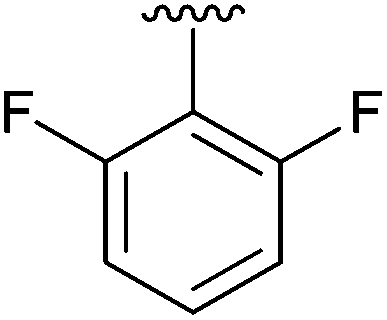
|
42 | 17 | 4q |
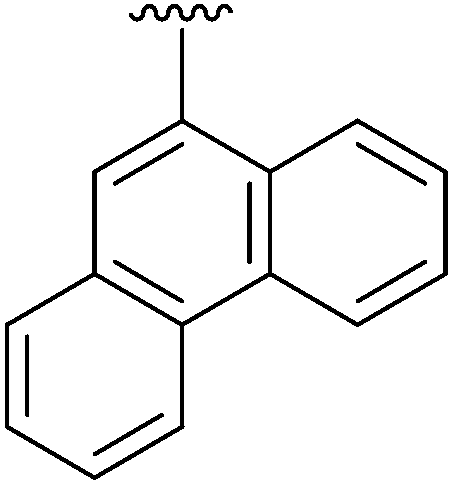
|
47 |
| 9 | 4i |
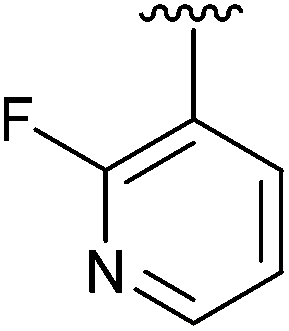
|
9 | 18 | 4r |
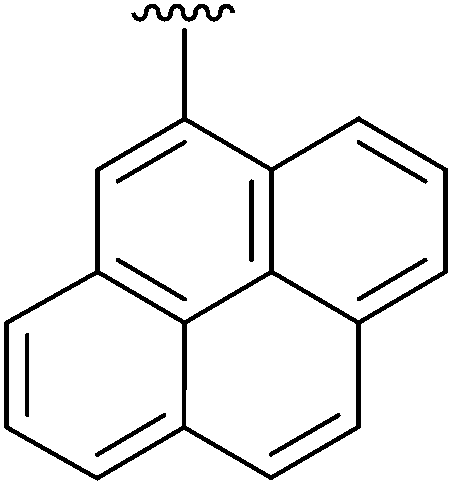
|
20 |
Conclusions
In conclusion, we have shown that a wide range of silyl-protected dioxaborinanes can be prepared on a gram-scale in a mild and simple manner from their parent boronic acid. They are both column- and bench-stable and we have also shown that these dioxaborinanes perform exceptionally well in palladium-catalysed cross-coupling reactions. In the scenario presented here, this allowed for the efficient construction of highly functional bis(hetero)aryl compounds in very good yields from basic starting materials. Mechanistic studies are underway to understand the unique properties of silyl-protected dioxaborinanes within coupling reactions and their application to other reactions are also in progress.Acknowledgements
We are grateful to the University of Bath for funding. We acknowledge the valuable assistance of Dr Anneke Lubben (Mass Spectrometry) and Dr John Lowe (NMR Spectroscopy).Notes and references
- D. G. Hall, in Boronic Acids, ed. D. G. Hall, Wiley-VCH, Weinheim, Germany, 2005, pp. 1–99 Search PubMed.
- A. N. Cammidge, V. H. M. Goddard, H. Gopee, N. L. Harrison, D. L. Hughes, C. J. Schubert, B. M. Sutton, G. L. Watts and A. Whitehead, Org. Lett., 2006, 8, 4071–4074 CrossRef CAS PubMed.
- Y. Takaya, M. Ogasawara and T. Hayashi, Tetrahedron Lett., 1999, 40, 6957–6961 CrossRef CAS.
- (a) R. A. Batey, A. N. Thadani and D. V. Smil, Org. Lett., 1999, 1, 1683–1686 CrossRef CAS; (b) R. A. Batey and T. D. Quach, Tetrahedron Lett., 2001, 42, 9099–9103 CrossRef CAS; (c) M. Pucheault, S. Darses and J.-P. Genêt, Eur. J. Org. Chem., 2002, 3552–3557 CrossRef CAS.
- For a review of MIDA boronates, see: E. P. Gillis and M. D. Burke, Aldrichimica Acta, 2009, 42, 17–27 CAS.
- Y. Yamamoto, M. Takizawa, X.-Q. Yu and N. Miyaura, Angew. Chem., Int. Ed., 2008, 47, 928–931 CrossRef CAS PubMed.
- J. C. Allen, G. Kociok-Köhn and C. G. Frost, Org. Biomol. Chem., 2012, 10, 32–35 CAS.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS.
- J. Dietrich, C. Hulme and L. H. Hurley, Bioorg. Med. Chem., 2010, 18, 5738–5748 CrossRef CAS PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS.
- J. P. Wolfe, R. A. Singer, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9550–9561 CrossRef CAS.
- I. Kondolff, H. Doucet and M. Santelli, J. Mol. Catal. A: Chem., 2007, 269, 110–118 CrossRef CAS PubMed.
- (a) A. J. J. Lennox and G. C. Lloyd-Jones, Isr. J. Chem., 2010, 50, 664–674 CrossRef CAS; (b) M. Butters, J. N. Harvey, J. Jover, A. J. J. Lennox, G. C. Lloyd-Jones and P. M. Murray, Angew. Chem., Int. Ed., 2010, 122, 5282–5286 CrossRef; (c) A. J. J. Lennox and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2012, 134, 7431–7441 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, copies of NMR spectra for compounds synthesised. CCDC 944020. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob42099j |
| This journal is © The Royal Society of Chemistry 2014 |