Polymer-based biocompatible fluorescent sensor for nano-molar detection of Zn2+ in aqueous medium and biological samples†
Kamalpreet
Kaur
a,
Manjot
Kaur
b,
Amanpreet
Kaur
c,
Jasminder
Singh
b,
Narinder
Singh
a,
Susheel K.
Mittal
*b and
Navneet
Kaur
*c
aDepartment of Chemistry, Indian Institute of Technology Ropar (IIT Ropar), Rupnagar, Punjab 140001, India. E-mail: nsingh@iitrpr.ac.in
bSchool of Chemistry & Biochemistry, Thapar University, Patiala, 147004, India. E-mail: smittal@thapar.edu
cCentre for Nanoscience and Nanotechnology (UIEAST), Punjab University, Chandigarh, 160014, India. E-mail: navneetkaur@pu.ac.in; Tel: +91-1722534464
First published on 6th January 2014
Abstract
Chemosensors are prepared through condensation reactions between polymeric amines and three different aldehydes; the activities of the three products were evaluated in DMF–H2O (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) solvent system. Chemosensor 2c demonstrated selectivity for Zn2+ enhancement of the fluorescence intensity at 420 nm. Upon binding of 2c with Zn2+, an approximately 9 fold enhancement in fluorescence intensity was observed due to the combined effect of ESIPT, C
:3, v/v) solvent system. Chemosensor 2c demonstrated selectivity for Zn2+ enhancement of the fluorescence intensity at 420 nm. Upon binding of 2c with Zn2+, an approximately 9 fold enhancement in fluorescence intensity was observed due to the combined effect of ESIPT, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N isomerisation and the two-channel quenching of PET. The investigation of the photophysical properties of 2c revealed that the sensor at low pH led to a shift in the emission profile because of the presence of two types of nitrogen i.e. sp2 and sp3 nitrogens and the pH dependent fluorescence profile is due to the protonation at sp3 hybridised nitrogen that influences the charge transfer transitions of 2c. However, the salt effect has not much influence on the photophysical properties of 2c; and 2c was found to form organic nano-particles (ONP) in a DMF–H2O (1:99, v/v) solvent system and this material was found to detect a minimum of 40 nM concentration of Zn2+. The receptor 2c was found to be biocompatible through MTT assay and potent to detect Zn2+ in real biological conditions of microbial cells. The compounds 2a, 2b and 2c while immobilized in PVC based membrane, exhibited their selectivity for zinc ions as ion selective electrodes. These electrodes responded linearly in a wide concentration range with a lower detection limit of 10 μM. The electrodes have been successfully used as indicator electrodes in potentiometric titration of zinc ions with EDTA.
N isomerisation and the two-channel quenching of PET. The investigation of the photophysical properties of 2c revealed that the sensor at low pH led to a shift in the emission profile because of the presence of two types of nitrogen i.e. sp2 and sp3 nitrogens and the pH dependent fluorescence profile is due to the protonation at sp3 hybridised nitrogen that influences the charge transfer transitions of 2c. However, the salt effect has not much influence on the photophysical properties of 2c; and 2c was found to form organic nano-particles (ONP) in a DMF–H2O (1:99, v/v) solvent system and this material was found to detect a minimum of 40 nM concentration of Zn2+. The receptor 2c was found to be biocompatible through MTT assay and potent to detect Zn2+ in real biological conditions of microbial cells. The compounds 2a, 2b and 2c while immobilized in PVC based membrane, exhibited their selectivity for zinc ions as ion selective electrodes. These electrodes responded linearly in a wide concentration range with a lower detection limit of 10 μM. The electrodes have been successfully used as indicator electrodes in potentiometric titration of zinc ions with EDTA.
Introduction
The selective recognition of trace levels of transition metal ions is an in demand and developing research area.1 These metal ions play important roles in biology and environment.2 The design, synthesis and development of metal chemosensors with high sensitivity, selectivity, applicability in aqueous or semi-aqueous medium is highly desirable.3 Zn2+ is the second most abundant transition metal ion in the human body and its concentration varies from sub μM to mM.4 For proper physiological functions, it is vital to maintain Zn2+ homeostasis; which is maintained by the import/export between extracellular space and cell organelles.5 Hence, to understand the physiological function; it is crucial to monitor the Zn2+ levels in living cells and body fluids. Numerous scientific endeavours have been focused on the development of chemosensors for in vitro and in vivo detection of Zn2+.6,7 However, for metal ion detection in body fluids and in the cellular environment, the chemosensor must be able to operate in an aqueous/semi-aqueous medium;8 while most of the reported Zn2+ sensors have poor water solubility and are susceptible to the interference of other metal ions.9 This poor solubility is due to the presence of hydrophobic moieties in the design of the chemosensor such as anthracene,9a,b naphthalimide,9c,d pyrene9fetc. Contrary to these types of chemosensors, the binding units constituted by the polyamine10 scaffolds may offer good solubility in aqueous medium and the polyamine derivatives can form stable metal chelates.11 However, the selective binding and sensing of metal cations can be achieved by functionalization of polyamines with fluorescent/chromogenic moieties. For cellular applications, the biocompatibility of the sensor for living cells is another important concern. In a continuation of our interest in sensor development,12 the present manuscript is focused on developing a water soluble, biocompatible, selective, sensitive sensor with direct cellular applications. The polyamine (1) on condensation with different aldehydes gave substituted polyamines 2a–c, which were evaluated as fluorescent chemosensors; secondly these compounds are used in an ion selective electrode (ISE). Generally, ISEs have the problems of receptor leaching, short lifetimes and suffer from interference by species with chemical behaviour similar to primary ions like Cd2+ and Hg2+ interfering with a Zn2+ selective electrode.13 The advantage of receptors made up of polymeric scaffolds may help to stop the leaching process with retained properties of chemical sensing with a longer lifetime.Results and discussion
The present investigation deals with the development of solution phase and matrix bound (ISE) fluorescent chemosensors for analysis of Zn2+ in neutral aqueous/semi-aqueous medium. It is well documented that the imine linkages provide effective binding sites for Zn2+.14 We envisioned that the imine linked receptors appended on the polyamine backbone will improve the (semi)aqueous solubility of the receptor to afford a selective, sensitive fluorescence response to Zn2+. Further, this interaction will help in the determination of Zn2+ concentration using the polymer appended with an ionophore as an electroactive component in the ISE as well.The synthesis of compounds 2a–c is detailed in Scheme 1 and these compounds were synthesized by the condensation reaction between the polymeric polyamine 1 and the corresponding aldehydes. The resultant imine linked product was characterized by spectroscopic methods i.e. a singlet for –CHN– in the 1H NMR spectra at 8.43, 8.96 and 7.84 ppm and a characteristic band for –CHN– in the IR spectra at νmax: 1629, 1664 and 1611 cm−1 corresponding to the structures of 2a, 2b and 2c respectively (Fig. S1–9†). A 10 μM concentration of 2a–c in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solvent system exhibits absorption bands between 310–425 nm (Fig. S10†); these bands are assigned to charge transfer transitions due to imine linkages.15 The fluorescence spectra recorded with 10 μM concentration of 2a–c in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solvent system have unique fluorescence signatures for each compound (Fig. 1). The probe 2a exhibits dual channel emission with weak emission at λmax = 345 nm and moderate emission at λmax = 480 nm. The receptor 2a exists in keto and enol tautomers through an excited state intramolecular proton transfer (ESIPT) mechanism.16 The emission at lower wavelength is due to the enol form; while the keto form emits at a higher wavelength. The fluorescence signature of 2b was recorded under the same conditions as that used for 2a and appears as an intense fluorescence emission band at λmax = 350 nm. The receptor 2b is unable to exhibit the ESIPT mechanism involving keto–enol tautomerism; this may be due to geometrical restrictions. The fluorescence profile of receptor 2c depicted the fluorescence spectra with two equal intensity emission bands at λmax = 416 and 480 nm. The emission bands are explained on the basis of the ESIPT mechanism through keto–enol tautomerism involving the –OH and sp2 nitrogen of the imine linkage. The longer wavelength band found at 480 nm is because of the keto form and the fluorescence emission band observed at 416 nm is attributed to fluorescence emission from the excited enol state. The compounds 2a–c differ only in the substitution at the end group; otherwise every compound is fabricated with –CHN– and –OH groups. To gain more insights about the modulation of the fluorescence signatures of 2a–c due to structural features, DFT calculations were performed using Becke's three parameterized Lee–Yang–Parr (B3LYP) exchange functional with 6-31G* basis sets, on Gaussian-09 programs17 and fragments of each receptor were optimised by taking a small segment of the polymer (Fig. S22†). The fully optimised structures of fragments of receptors 2a–c indicate the energy equivalence for the enol and keto states (Table S1†). The enol form of all the receptors has approximately the same type of symmetry to show H-bonding between –OH groups and the –sp2 hybridised nitrogens. Therefore, both the tautomeric forms should exist in equilibrium and the receptors 2a–c may exhibit the ESIPT phenomenon. DFT calculations predict the keto form of each receptor to be energetically more stable than the enol form. Since we could optimize a part of each of the receptors 2a–c, the practical inability of 2b to offer ESIPT seems to be the consequence of some structural features of the polymer backbone. All the receptors (2a–c) bear the proton acceptor site i.e. sp2 hybridised nitrogen and also bear an –OH, ionisable moiety under basic conditions. Thus the fluorescence properties of these compounds are expected to modulate with the change in the pH of the solution. Compound 2a was dissolved readily in DMF–H2O (7:3, v/v) and fluoresces with λmax at 345 nm and 480 nm under neutral conditions; however the lowering in pH from 8.96 to 2.0 led to an enhancement in the fluorescence intensity by two times (Fig. S12A†). The phenomenon can be explained as: the receptor under neutral conditions may undergo cis–trans isomerism at the –CN– linkage and thus is under the influence of non-radiative decay.18 The protonation of the sp2 nitrogen at lower pH restricts the cis–trans isomerism and also retards the non-radiative decay; thus the fluorescence intensity is increased (Fig. S12A†). The increase in pH to 12.0 leads to switch “ON” the fluorescence intensity of 2a due to deprotonation of –OH; which restricts the non-radiative decay due to vibronic coupling in –OH (Fig. S12B†).19 Under neutral conditions, receptor 2b shows a fluorescence maximum at λmax = 350 nm. The effect of pH on receptor 2b was studied in DMF–H2O (7:3, v/v) solvent system. A two-fold increase in fluorescence intensity is observed with a decrease in pH from 10.68 to 2.0 (Fig. S13A†). However, with a slight increase in pH from 10.68 to 10.96, a decrease in the fluorescence intensity at 350 nm is observed with the simultaneous appearance of a new band at 440 nm (Fig. S13B†). With a further increase in the pH to 12.0; an abrupt increase in fluorescence of the latter band is observed. Interestingly, the lowering in pH of the solution of 2c from 8.65 to 2.0 leads to a shift in the emission profile (Fig. S14A†). In fact, compound 2c bears two types of nitrogens i.e. sp2 and sp3 nitrogens and the pH dependent fluorescence profile seems to be due to the protonation at the sp3 hybridised nitrogen, because a close binding of H+ to the fluorophore may lead to affect the ring current and consequently influence the charge transfer positions.20 A comparison of the effect of base on the fluorescence profile of 2a and 2c leads to conclude that relatively 2c is less prone to be influenced under basic conditions due to the different acidity of the –OH group in the two compounds. The perturbation of high ionic strength was ruled out by comparison of the fluorescence spectra of 2a–c (recorded with 10 μM concentration of sensor in DMF–H2O (7:3, v/v) solvent system) with the respective fluorescence spectrum recorded upon addition of 100 equiv. of tetrabutylammonium nitrate under the same concentration of sensor and solvent system.
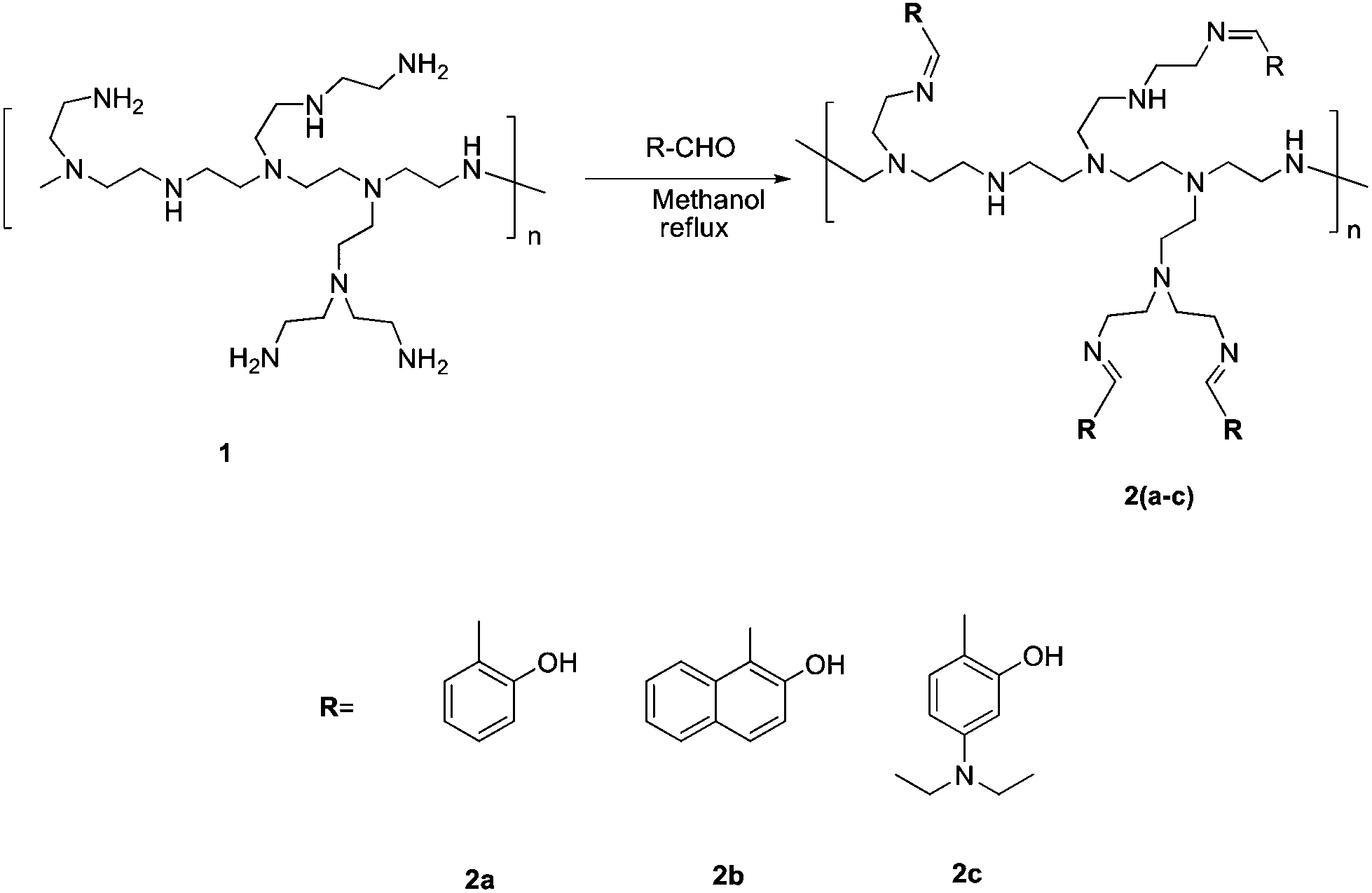 | ||
| Scheme 1 | ||
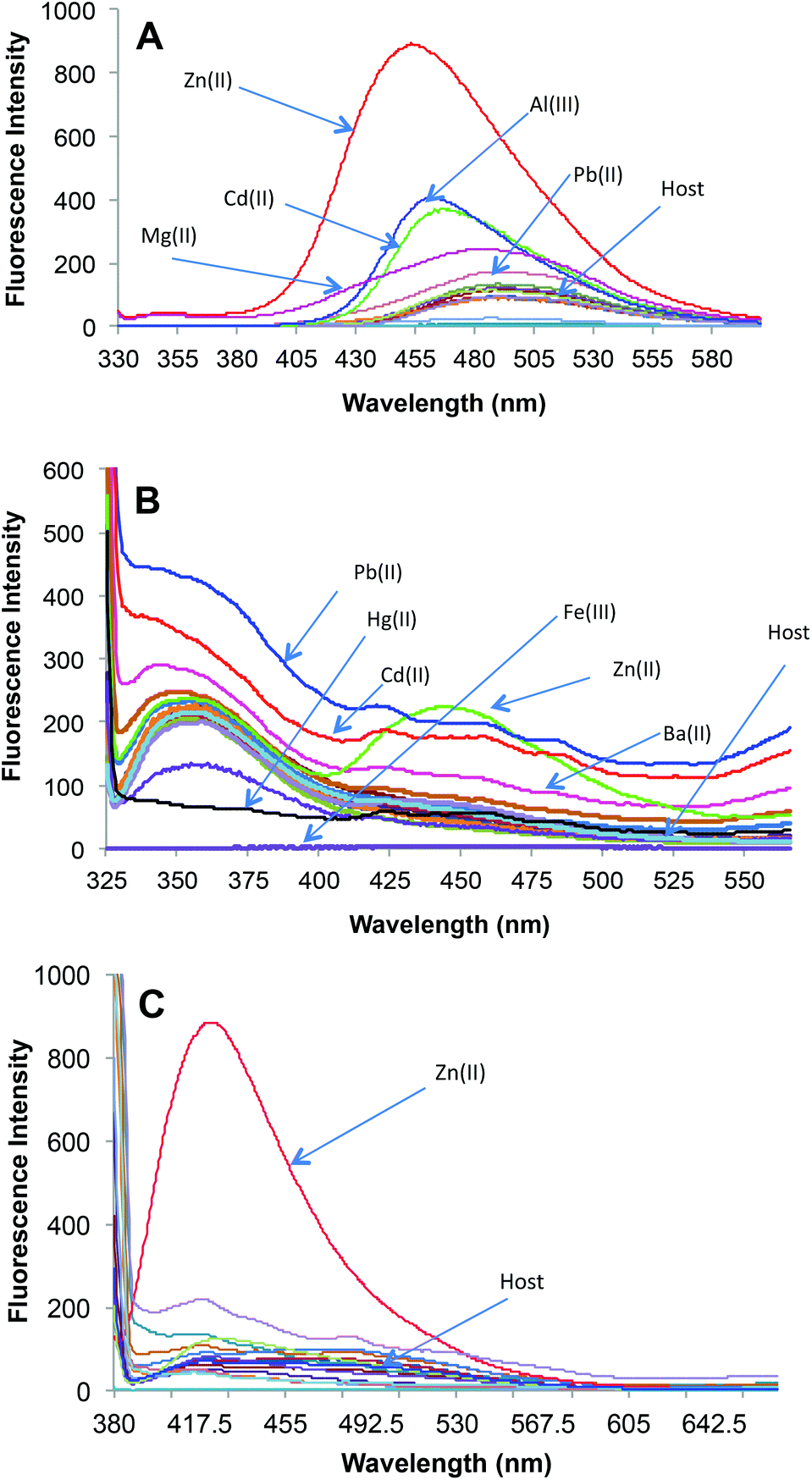 | ||
| Fig. 1 (A) Changes in fluorescence intensity of receptor 2a (10 μM) upon addition of 50 μM of a particular metal nitrate salt in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) with λex = 310 nm. (B) Changes in fluorescence intensity of receptor 2b (10 μM) upon addition of 50 μM of a particular metal nitrate salt in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) with λex = 309 nm. (C) Changes in fluorescence intensity of receptor 2c (10 μM) upon addition of 50 μM of a particular metal nitrate salt in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) with λex = 370 nm. | ||
It was found that the fluorescence profiles of 2a–c are influenced by the change in pH of the solution; thus all the further studies were conducted under HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solvent system. To evaluate the extent to which Zn2+ will fit in the proposed design concept of 2a; 50 μM of different metal nitrate salts such as (Na+, K+, Mg2+, Ca2+, Ba2+, Sr2+, Al3+, Cr3+, Mn2+, Fe3+, Co2+, Ni2+, Cu2+, Zn2+, Ag+, Pb2+, Cd2+ and Hg2+) were added to HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solution containing 10 μM of 2a (Fig. 1A). A significant increase of fluorescence is observed for complex (2a·Zn2+) formed upon addition of Zn2+ and relatively smaller enhancements were observed upon addition of Cd2+, Al3+ and Mg2+. The binding of Cd2+ and Al3+ with 2a may not bring much interference in bio-analytical methods due to the scarcity of both these metal ions in biological systems. However, Mg2+ seems to be a major interfering cation for zinc estimation in biological systems. The [Zn2+] dependent fluorescence response of 2a was observed in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solution and, as per expectations, the enhancement in fluorescence intensity of 2a is directly proportional to [Zn2+] within the concentration range of 0–50 μM (Fig. 2). An enhancement factor of 325% is observed when the mole ratio (2a/Zn2+) is 1:1 and higher [Zn2+]total results in further evident fluorescent change, suggesting the formation of multicavities for the encapsulation of Zn2+. The encapsulation of Zn2+ by the sensor 2a has engaged the lone pairs of –OH and the sp2 nitrogen; thus restricting the electron donating ability of the groups, which results in a decrease in PET efficiency. Besides, Zn2+ could be detected up to 10 μM; a concentration found in many biological systems. The association constant (Ka) of Zn2+ with 2a is determined by the equation: log[Imax − I]/[I − Imin] = log[cation] − log β and is found to be 2.6 × 104.21 The sensor 2a seems to be sensitive for Zn2+ along a fairly good concentration range of Zn2+ with a detection limit of 60 nM making it a well suited method for bioanalytical applications. Since, for a sensor, selectivity is an important criteria for practical applicability, so competitive metal binding experiments were performed to estimate Zn2+ (10 μM) in the presence of any of Na+, K+, Mg2+, Ca2+, Ba2+, Sr2+, Al3+, Cr3+, Mn2+, Fe3+, Co2+, Ni2+, Cu2+, Ag+, Pb2+, Cd2+ and Hg2+ (50 μM) to check 2a as a selective fluorescent sensor for Zn2+ (Fig. S23†). The sensor activity of 2a for Zn2+ is influenced by the presence of Ca2+, Mg2+, Cu2+, Al3+ and Cd2+; this interference will limit the application of 2a as a sensor for Zn2+. However, when structural features of 2a are compared with those of 2b and 2c, it was envisaged that they may show improved photophysical properties. This can be due to: (a) insertion of bulkier group, i.e., naphthyl (design of 2b) may provide rigidity to the receptor and hence rule out the possibility of a flexible coordination sphere for many metal ions; (b) recently, Prof. A. P. de Silva has reported the “path selective PET” where the molecule needs to possess a minimum two paths for PET.22 These paths are arranged by building a “receptor1–spacer1–fluorophore–spacer2–receptor2” system and this system is realized as a high generation version of the basic “fluorophore–spacer–receptor” format.23 The advantage of this approach lies in the fact that only strongly binding metal ions switch “OFF” the PET process from two channels and binding of metal ions to one donor group may not be able to block two channels so they are not potent to switch “ON” the fluorescence intensity. The design of 2c is fabricated from the basic design of 2a; however with another PET channel from the sp3 nitrogen. The cation binding properties of 2b (10 μM) are evaluated from the change in its fluorescence intensity upon addition of a particular metal nitrate salt (50 μM) in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solvent system, when excited at 309 nm (Fig. 1B). However, the fluorescence profile of 2b is perturbed by the addition of many of the tested metal ions. Thus receptor 2b is found to be non-selective towards any of the tested metal ions. With the envisaged two paths PET phenomenon in 2c, the metal binding studies were conducted to further validate the concept. The probe 2c (10 μM) was tested against a series of metal ions Na+, K+, Mg2+, Ca2+, Ba2+, Sr2+, Al3+, Cr3+, Mn2+, Fe3+, Co2+, Ni2+, Cu2+, Ag+, Pb2+, Cd2+ and Hg2+ (50 μM) in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solvent system, when excited at 370 nm (Fig. 1C). The primary metal binding studies suggested improvement in binding selectivity towards Zn2+ by probe 2c as compared to 2a. None of the other metal ions i.e. Na+, K+, Mg2+, Ca2+, Ba2+, Sr2+, Al3+, Cr3+, Mn2+, Fe3+, Co2+, Ni2+, Cu2+, Ag+, Pb2+, Cd2+ and Hg2+ could show a high enhancement of fluorescence intensity w.r.t. Zn2+. This may be due to the strongly fitting capacity of Zn2+ in the coordination sphere of the binding pocket laced with –CN– and –OH groups of pendant arms in the polymeric chain and extra stability being provided by the sp3 nitrogen present on the aromatic ring. The quantum yields for 2c and 2c·Zn2+ were found to be 0.21 ± 0.01 and 0.66 ± 0.01 respectively depicting high quantum efficiency for the complex (Table S4†). In order to identify the working range of this polymeric sensor (2c), fluorescence titrations were performed between 2c and Zn2+ (Fig. 3). The fluorescence changes in the probe 2c were recorded by adding aliquots of Zn2+ and interpreted to be linear in the range 0–60 μM. It has been found that the addition of Zn2+ from nM to μM led to a tremendous increase in the fluorescence intensity of the probe i.e. 9 fold with binding constant of 2.7 × 104. The minimum concentration at which polymeric sensor 2c can detect the presence of Zn2+ in the given media has been found to be 40 nM. The addition of EDTA quenches the fluorescence of the receptor 2c·Zn2+ complex (Fig. S15†), making receptor 2c a reversible chemosensor. The kinetics of the quenching of the fluorescence intensity due to the 2c·Zn2+ complex
(20 μM) upon the addition of EDTA (40 μM) was determined by the initial rate method by monitoring the quenching of the band at 418 nm due to the 2c·Zn2+ complex as a function of time. A plot of log[A∞/A∞ − At] versus time was plotted over a time interval of 0–20 min and the value of the rate constant was determined to be 3.90 × 10−2 min−1 (Fig. S16 and 17†). The competitive metal ion binding studies were conducted to find whether 2c can detect Zn2+ selectively even in the presence of other metal ions. To the solution of 2c·Zn2+ (10 μM) fixed amounts of different metal ions (50 μM) were added and their fluorescence profiles were studied in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solvent system (Fig. S24†). The data revealed high selectivity of 2c for Zn2+ as none of the added metal ions could displace Zn2+ from the binding pocket of the polymeric sensor. To investigate the binding behaviour of Zn2+ with 2c, the UV-Vis spectrum was recorded. The UV-Vis absorption profile exhibited λmax at 370 nm, which can be ascribed to intramolecular charge transfer transitions with a clear isosbestic point at 341 nm (Fig. S10C†). In order to understand the role of the polymer backbone, a linear receptor (3) was synthesized by a literature method24 (Scheme S1†) and this material was found to be non-selective for any metal ion (Fig. S25†). Although, 2c and 3 bear similar types of binding sites, this experiment highlights the importance of the polymer backbone to induce selectivity in the sensor system. The effect of pH on the binding affinity of 2c for Zn2+ was also recognised (Fig. S26†). A decrease in pH enhances the fluorescence intensity, which may be due to cancellation of PET channels; however, with an increase in pH we observed the formation of some white precipitates and the analysis of these precipitates revealed the formation of ZnO nanoparticles.
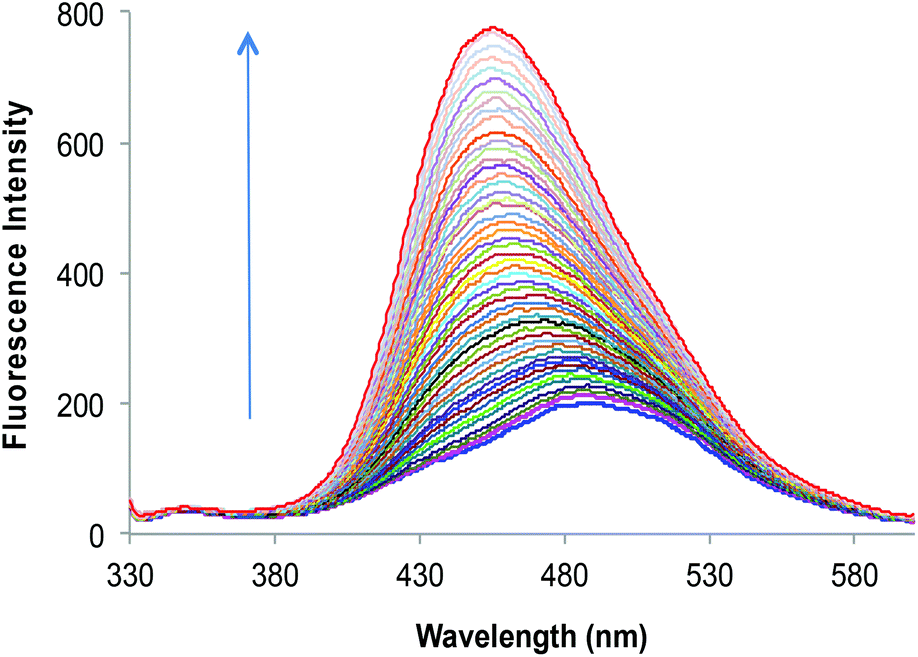 | ||
| Fig. 2 Fluorescence emission spectra of 2a (10 μM) upon successive addition of Zn2+ (0–50 μM) in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v). | ||
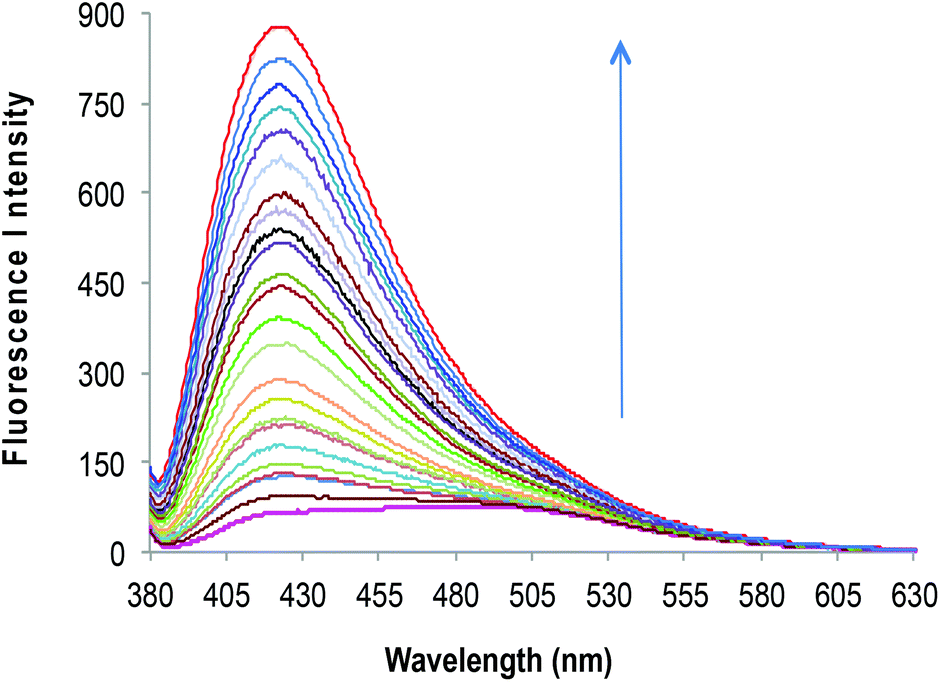 | ||
| Fig. 3 Fluorescence emission spectra of 2c (10 μM) upon successive addition of Zn2+ (0–50 μM) in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v). | ||
To evaluate the maximum water content up to which the sensor can exhibit its activities, the photophysical properties were evaluated in different solvent combinations of DMF–H2O and it was found that an excess of water fraction (i.e. 99% of H2O in DMF–H2O combination) led to the formation of organic nano-particles (ONP) of 2c (Fig. 4A). Thus it was decided to evaluate Zn2+ recognition studies with a solvent system of DMF–H2O (1:99, v/v). The complexation of 2c(ONP) with Zn2+ triggered fluorescence with an emission band at 418 nm for 2c(ONP)·Zn2+. Complete addition of 6 equiv. of Zn2+ to the 10 μM solution of 2c(ONP) leads to a significant enhancement of fluorescent intensity of 2c(ONP) (13 times).
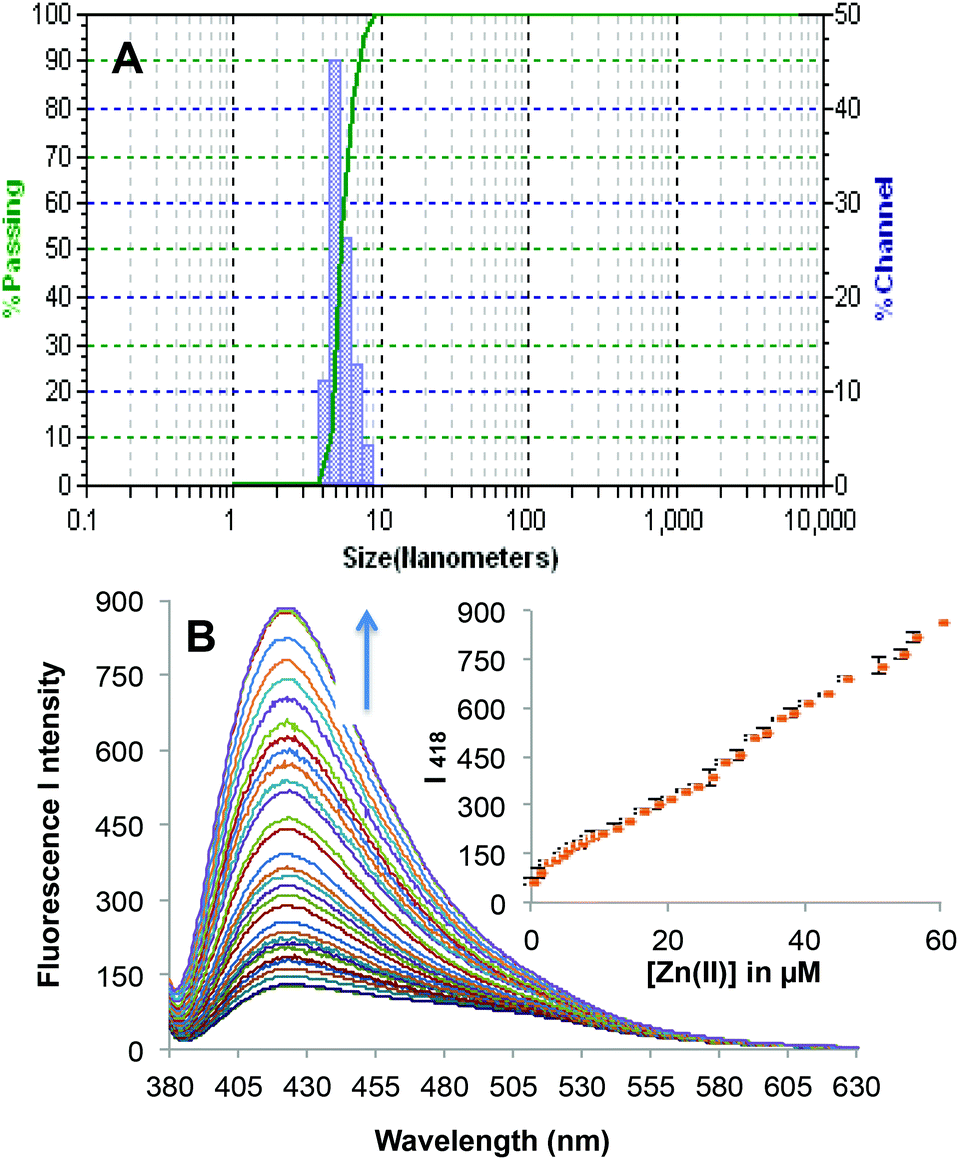 | ||
| Fig. 4 (A) Particle size analysis of organic nano-particles (ONP) of 2c as determined with DLS. (B) Fluorescence emission spectra of 2c (10 μM) upon successive additions of Zn2+ (0–60 μM) in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (1:99, v/v). | ||
The cytotoxicity of 2c was determined using the MTT assay with HeLa cells cultured in nutrient mixture. The cells were incubated at 37 °C with 5% CO2 environment for 24 h. Only marginal cell death (∼95% cell survival) was caused by 2c when the experiments were performed at probe concentration, which is used for recognition studies (10 μM). To investigate the potential cytotoxic effects of 2c at higher concentrations, the dose dependent culture studies were performed. This is important because sometimes higher concentrations of probe may be needed for certain biological applications. The results indicate that 2c is not very toxic to HeLa cells if probe concentrations remain below 18 μM, i.e. with ∼85% cell survival over 24 h for 18 μM concentration of 2c. At much higher concentrations of 2c, the probe becomes more toxic. For example, the IC50 value was found to be 43 μM over 24 h (Fig. S17†). To explore the real application of 2c for Zn2+ recognition in a real biological system such as microbial cells (Saccharomyces cerevisiae), the microbe cells were cultured separately in normal medium, and also microbes were cultured in a medium enriched with Zn2+. The culture content of both cases was divided into two parts and one part of each was treated with probe 2c (incubated for 3 h at 37 °C). The content of all the four cultures was centrifuged and then washed with a DMF–H2O (7:3, v/v) solvent mixture. All the four slides were investigated under a fluorescence microscope and the results are presented in Fig. 5. Thus, in the presence of probe 2c in Zn2+ enriched microbe cells, the cells were illuminated under the fluorescence microscope. Furthermore, the level of Zn2+ in spiked tap water was estimated and compared with AAS. The analysis results obtained by using 2c and by AAS are summarized in Table S5.†
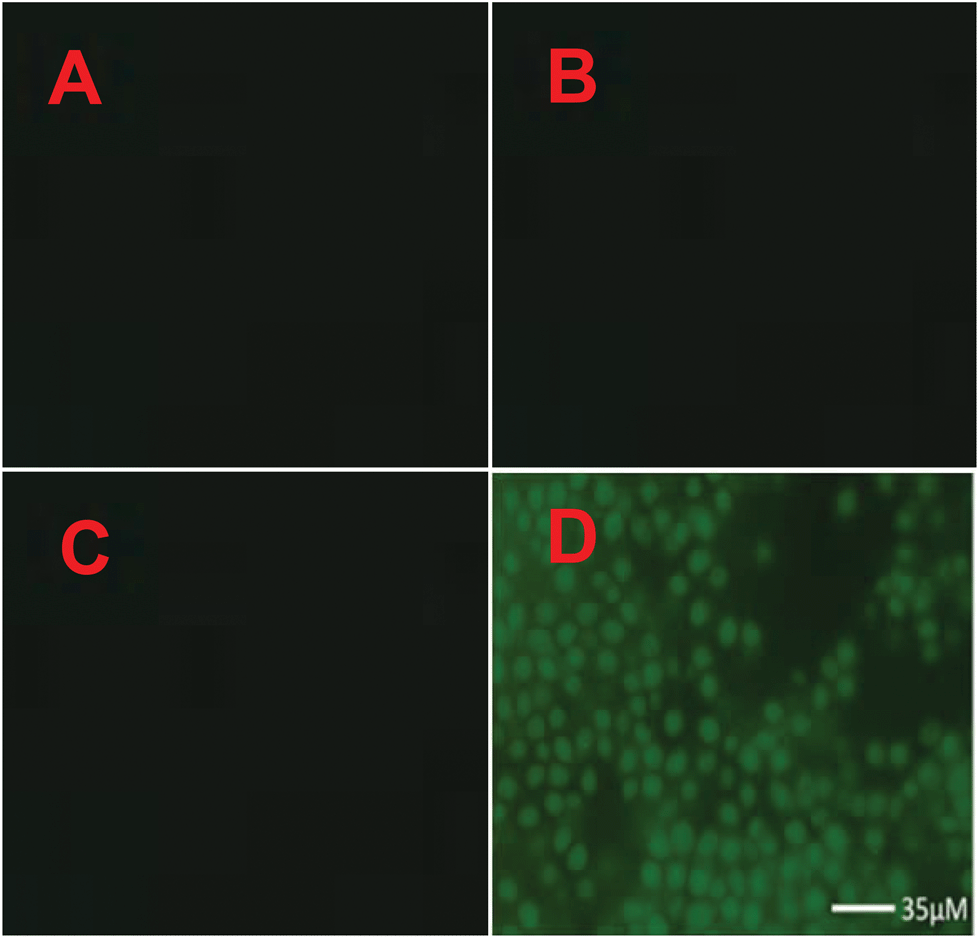 | ||
| Fig. 5 Microscopic images of: (A) microbe cells cultured in normal medium; (B) microbe cells cultured in medium enriched with Zn2+; (C) microbe cells cultured in normal medium and treated with 2c; (D) microbe cells cultured in medium enriched with Zn2+ and treated with 2c. Before performing microscopy, the microbe cells were washed with a DMF–H2O (7:3, v/v) solvent mixture. | ||
The ion-selective electrodes (ISE) were prepared with the development of membranes with different amounts of ionophore and were tested for better response towards Zn2+ ions. The membrane with the composition: ionophore:PVC:plasticiser:lipophilic additive in the ratio of 10:29:56:5 gave the best results. The presence of lipophilic additives generally improves the performance characteristics of various cation selective electrodes with the minimisation of ohmic resistance. Experiments were conducted to study the electrode response with different ionophores 2a–c. Potentiometric readings were recorded with the electrode immersed in different concentrations of Zn2+ ions in the concentration range of 10−8 to 10−1 M. The representative plots of electrode potential (E) for the ISE of ionophore 2a against the logarithm of primary ion concentration are shown in Fig. S18.† Slopes of the calibration curves for Zn2+ ion using electrodes with ionophores 2a–c are shown in Table S2.† It can be seen (Fig. S18†) that the electrode exhibited a near Nernstian slope in the working range 10−5 to 10−1 M with the detection limit as 10−5 M. The effect of interfering ions on the potentiometric response of the electrode was measured by a fixed interference method. Keeping the concentration of the interfering ion fixed at 10−3 M, the concentration of the primary ion (Zn2+) was varied from 10−8 M to 10−1 M. Selectivity coefficient values of the Zn2+ selective electrodes prepared with ionophores 2a–c for different interfering ions is shown in Table S3.† It can be seen from the values of selectivity coefficients that the electrode has limited interference from a large number of metal ions. Electrode response in aqueous environments with different pH in the range 2–11 was also studied. Adjustment of the desired pH was done by the dropwise addition of HNO3 (0.1 M) or NaOH (0.1 M) solution. Plots of electrode potentials (E) against pH of the electrode with ionophore 2a are shown in Fig. S19.† Mostly, for a given solution of the Zn2+ ion, the electrode response remains stable in the pH range 4–8. At lower pH, electrodes start responding to H+ ions and at higher pH, the Zn2+ ions are precipitated due to the formation of Zn(OH)2. The electrodes were used as indicator electrodes for the titration of Zn2+ solution against EDTA. The pH of the solution was adjusted to 5–6 with buffer. The representative plots between electrode potentials of electrodes against volume of EDTA used for the titration are shown in (Fig. S20†) which show that with all the ionophores 2a–c sharp end points were obtained corresponding to a 1:1 stoichiometry of the Zn2+–EDTA complex.
Experimental section
All the starting materials including polymer 1 were purchased from Sigma Aldrich and were used without any further purification. 1H NMR and 13C NMR spectra were recorded in DMSO-d6 on a Bruker Avance II 400 spectrometer (which operated at 400 MHz for 1H NMR and at 100 MHz for 13C NMR with TMS as internal standard; chemical shifts are expressed in ppm). IR spectra were recorded on a Bruker Tensor 27 spectrometer for the neat samples. The absorption spectra were recorded on a Specord 250 Plus Analytikjena spectrometer spectrophotometer. The fluorescence measurements were performed on a Perkin Elmer L55 Fluorescence spectrophotometer. ISE measurements were carried out using Equiptronics potentiometer (EQ-602) with accuracy ±0.1 mV.Synthesis of 2a
Compound 2a was prepared by dissolving 1 (650 mg) and salicylaldehyde (488 mg) in dry methanol separately. The two solutions were mixed and the reaction mixture was refluxed with stirring for 6 h. Upon completion of reaction, the semi solid material was separated out. The material was washed with CH3OH (50 mL) and dried under vacuum. Yield = 80%; IR (νmax, cm−1): 1629 (–CHN–); 1H NMR (400 MHz, DMSO-d6) δ: 8.43 (brs, CHN), 7.13–7.35 (m, ArH), 6.69–6.84 (m, ArH), 3.57 (s, CH2), 3.19–3.24 (m, CH2), 2.48–2.73 (m, CH2); 13C NMR (100 MHz, DMSO-d6) δ: 166.1, 160.8, 131.7, 129.2, 118.4, 116.2, 58.5, 56.7, 54.4, 52.6, 49.7, 48.7, 47.2, 41.3.
Synthesis of 2b
To synthesize compound 2b, polymer 1 (650 mg) and 2-hydroxynaphthaldehyde (688 mg) were dissolved separately in dry methanol. The two solutions were then mixed and the reaction mixture was refluxed with stirring for 8 h. Upon completion of reaction, the semi-solid material was separated out. The material was washed with CH3OH (50 mL) and dried under vacuum. Yield = 86%; IR (νmax, cm−1): 1664 (–CHN–); 1H NMR (400 MHz, DMSO-d6) δ: 8.95 (brs, CHN), 7.99 (d, ArH), 7.54–7.72 (m, ArH), 7.26 (d, ArH), 7.23 (d, ArH), 7.09 (t, ArH), 6.70 (s, ArH), 3.21–3.62 (m, CH2), 2.82 (s, CH2), 2.52 (s, CH2); 13C NMR (100 MHz, DMSO-d6) δ: 160.7, 139.8, 134.5, 132.8, 132.7, 131.3, 131.1, 127.8, 123.9, 113.8, 62.4, 62.0, 57.8, 56.4, 54.1, 53.8, 52.3.
Synthesis of 2c
Compound 2c was prepared by dissolving polymer 1 (650 mg) and 4-(diethylamino)salicylaldehyde (772 mg) in dry methanol separately. The two solutions were mixed and the reaction mixture was refluxed with stirring for 8 h. The semi solid material was separated out upon completion of reaction. The material was washed with CH3OH (50 mL) and dried under vacuum. Yield = 76%; IR (νmax, cm−1): 1611(–CHN–); 1H NMR (400 MHz, DMSO-d6) δ: 7.84 (brs, CHN), 6.62–6.75 (m, ArH), 5.81–5.84 (m, ArH), 5.63 (s, ArH), 3.00–3.22 (m, CH2), 2.76–2.98 (m, CH2), 2.23–2.45 (m, CH2); 13C NMR (100 MHz, DMSO-d6) δ: 168.9, 163.6, 156.7, 153.9, 138.6, 134.8, 60.0, 59.0, 57.9, 55.0, 53.7, 52.2, 48.9, 48.8.
Metal recognition studies
The recognition studies were performed at 25 ± 1 °C, and the solutions were shaken for a sufficient time before recording the spectrum to ensure uniformity of solutions. The cation binding ability of receptors 2a–c (10 μM) in a HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solvent system was determined by preparing standard solutions of receptors 2a–c (10 μM) along with fixed amounts of a metal nitrate (50 μM). For titrations, volumetric flasks each containing a standard solution of receptor 2a and 2c (10 μM each) along with varied amounts of a zinc nitrate (0–50 μM) for receptor 2a and 2c under HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solvent system were used. However under the DMF–H2O (1:99, v/v) solvent system for 2c the Zn2+ concentrations were varied up to 60 μM. To evaluate any possible interference due to different cations for the estimation of Zn2+, solutions were prepared containing 2a and 2c (10 μM each) and Zn2+ (10 μM) along with both with and without other interfering metal ions (50 μM) in DMF–H2O (7:3, v/v) HEPES buffered solution (10 mM, pH = 7.0).
Kinetics studies
The rate constant for the quenching of the fluorescence intensity of the complex was determined by treating a 20 μM solution of the zinc complex of receptor 2c with 40 μM of EDTA at 25 ± 1 °C in HEPES buffered (10 mM, pH = 7.0) DMF–H2O (7:3, v/v) solvent system. The fluorescence spectra of solutions were recorded directly after the addition and successively after regular intervals of 2 min over a period of 20 minutes. The fluorescence intensity at 418 nm was measured as a function of time.
Cytotoxicity of 2c
The cytotoxicity of 2c was determined through an MTT assay, using HeLa cells seeded in a 96-well flat-bottomed microplate in growth medium (100 μL) and incubated at 37 °C under a 5% CO2 atmosphere for 24 h. The 2c as test compound and blank analysis were performed and 10 μL of MTT in PBS was added to each well. The microplate was incubated at 37 °C under a 5% CO2 atmosphere for another 3 h. The medium was then removed, and DMSO (100 μL) was added to each well. The absorbance spectrum of each solution was measured at 570 nm. The dose dependent cytotoxicity of 2c was determined using different concentrations of 2c; however, using the same conditions as mentioned above.Recognition of Zn2+ in microbe cells
To investigate the Zn2+, the culture medium was taken in culture tubes guarded with plug. For inoculation, the dry microbes (Saccharomyces cerevisiae) were dissolved in water and one drop of this suspension was added to each tube of medium. The cultures were a clear solution (to the naked eye) at the beginning and with the growth of microbes, cloudiness appeared. The culture tubes were marked as A, B, C, D; where tubes A and C contains normal culture medium; while tubes B and D were enriched with Zn2+ (0.01 M). All the four tubes were placed in an incubator at 27 °C. After incubation, the content of each tube was centrifuged and washed three times with double distilled water. The content of tubes C and D was treated with sensor 2c and again content was centrifuged and washed with DMF–H2O (7:3, v/v) solvent combination. Finally, the microbes of all the tubes were submitted for fluorescence microscopy.
ISE fabrication with 2a–c
Membranes with each of 2a–c were prepared by mixing poly vinyl chloride (PVC), ortho-nitrophenyl octyl ether (o-NPOE), and sodium tetraphenyl borate (TPB) in appropriate amounts in tetrahydrofuran (THF) as a solvent. The solvent was evaporated slowly and the viscous solution was poured in glass rings placed on glass plates. Solutions were further evaporated slowly at room temperature overnight. The membranes so prepared were removed from the glass plate and fixed to one end of the glass tubes 5 cm in length and 1.8 cm in diameter, flattened on one side by using an epoxy resin as adhesive. Each membrane was equilibrated with Zn2+ solution (0.1 M) for about 5 to 6 days by putting Zn2+ solution (0.1 M) on both sides of the membrane. After equilibration, the electrodes were washed thoroughly with distilled water to remove excess salt and kept immersed in distilled water when not in use. Membrane electrodes were filled one-fourth with zinc nitrate solution and immersed in a beaker containing the test solution of varying concentrations of zinc. The potentiometric measurements were made with a digital potentiometer having an accuracy of ±0.1 mV. All the measurements were done through using the following cell assembly:| SCE|Zn2+ (0.1 M)||Membrane||Zn2+ (test solution)|SCE |
Buffer solutions were not used to avoid the presence of any unwanted ions that might affect the potential. The effect of interfering ions on the potentiometric response of the electrode was measured in terms of selectivity coefficients (KA,B) determined by the fixed interference method (FIM). EMF values were measured by changing the activity of the primary ions but the activity of interfering ions remains fixed throughout the experiment. Intersection of the extrapolated linear portions of the curves drawn between EMF and the primary ion concentration was taken as the value of aA, and is applied in the Nikolsky–Eisenmann equation to find the selectivity coefficient values.
| KA,B = aA/(aB)zA/zB |
Here, aA is the activity of the primary ion A and aB is the activity of interfering ion B. zA and zB are charges on the ions A and B, respectively. The concentration of the primary ion was varied from 10−8 M to 10−1 M while that of the interfering ion was fixed at 10−3 M for all the membranes.
Conclusion
In summary, we have developed three polymer based Schiff base fluorescent sensors 2a–c. Receptor 2c is acting as an “on–off” fluorescent chemosensor for the biocompatible, operative in real microbe cell, selective, and sensitive detection of Zn2+. On binding with Zn2+, a 9 fold enhancement in fluorescence intensity is observed due to the combined effect of ESIPT, CN isomerisation and the two-channel quenching of PET. The polymeric sensor is able to detect zinc in (1:99) DMF–H2O to a minimum concentration of 40 nM. Receptors 2a, 2b and 2c while immobilized in a PVC based membrane, exhibited their selectivity for Zn2+ ions as ion selective electrodes. These electrodes responded linearly in a wide concentration range with a lower detection limit of 10 μM. The electrodes have been successfully used as indicator electrodes in potentiometric titration of Zn2+ ions with EDTA.
Acknowledgements
This work was supported with research grant (SR/FT/CS-97/2010(G) from Department of Science and Technology (DST), Government of India.Notes and references
- (a) J. Liu, K. Wu, S. Li, T. Song, Y. Han and X. Li, Dalton Trans., 2013, 42, 3854 RSC; (b) Y. Fu, L. Mu, X. Zeng, J.-L. Zhao, C. Redshaw, X.-L. Ni and T. Yamato, Dalton Trans., 2013, 42, 3552 RSC; (c) G. Sivaraman, T. Anand and D. Chellappa, Analyst, 2012, 137, 5881 RSC; (d) Q.-b. Mei, Y.-h. Guo, B.-h. Tong, J.-N. Weng, B. Zhang and W. Huang, Analyst, 2012, 137, 5398 RSC; (e) G. Sivaraman, T. Anand and D. Chellappa, RSC Adv., 2012, 2, 10605 RSC; (f) J. Hatai, S. Pal, G. P. Jose, T. Sengupta and S. Bandyopadhyay, RSC Adv., 2012, 2, 7033 RSC; (g) R. Y. Tsien, in Fluorescent and Photochemical Probes of Dynamic Biochemical Signals inside Living Cells, ed. A. W. Czarnik, American Chemical Society, Washington, DC, 1993, p. 130 Search PubMed; (h) D. P. Murale, H. Liew, Y.-H. Suh and D. G. Churchill, Anal. Methods, 2013, 5, 2650 RSC; (i) X.-C. Fu, J. Wu, C.-G. Xie, Y. Zhong and J.-H. Liu, Anal. Methods, 2013, 5, 2615 RSC.
- (a) L. Xu, Y. Xu, W. Zhu, X. Sun, Z. Xu and X. Qian, RSC Adv., 2012, 2, 6323 RSC; (b) S. Sen, T. Mukherjee, B. Chattopadhyay, A. Moirangthem, A. Basu, J. Marek and P. Chattopadhyay, Analyst, 2012, 137, 3975 RSC; (c) S. Sen, S. Sarkar, B. Chattopadhyay, A. Moirangthem, A. Basu, K. Dhara and P. Chattopadhyay, Analyst, 2012, 137, 3335 RSC; (d) R. Nishiyabu, H. Kobayashi and Y. Kubo, RSC Adv., 2012, 2, 6555 RSC; (e) L. Wang, B. Zheng, Y. Zhao, J. Du and D. Xiao, Anal. Methods, 2012, 4, 2369 RSC; (f) A. P. de Silva, H. Q. Nimal Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed.
- (a) L. Huang, F. Hou, J. Cheng, P. Xi, F. Chen, D. Bai and Z. Zeng, Org. Biomol. Chem., 2012, 10, 9634 RSC; (b) R. Han, X. Yang, D. Zhang, M. Fan, Y. Ye and Y. Zhao, New J. Chem., 2012, 36, 1961 RSC; (c) H. Zheng, M. Yan, X.-X. Fan, D. Sun, S.-Y. Yang, L.-J. Yang, J.-D. Li and Y.-B. Jiang, Chem. Commun., 2012, 48, 2243 RSC; (d) L. Liu, X. Dong, Y. Xiao, W. Lian and Z. Liu, Analyst, 2011, 136, 2139 RSC; (e) S. Jang, P. Thirupathi, L. N. Neupane, J. Seong, H. Lee, W. I. Lee and K.-H. Lee, Org. Lett., 2012, 14, 4746 CrossRef CAS PubMed.
- (a) J. G. Park, Y. Qin, D. F. Galati and A. E. Palmer, ACS Chem. Biol., 2012, 7, 1636 CrossRef CAS PubMed; (b) H. W. Mbatia and S. C. Burdette, Biochemistry, 2012, 51, 7212 CrossRef CAS PubMed; (c) G. Meloni, T. Polanski, O. Braun and M. Vašák, Biochemistry, 2009, 48, 5700 CrossRef CAS PubMed; (d) A. R. Henriques, A. Chalfun-Junior and M. Aarts, Braz. J. Plant Physiol., 2012, 24, 3 CrossRef CAS; (e) S. O. Dahms, I. Könnig, D. Roeser, K.-H. Gührs, M. C. Mayer, D. Kaden, G. Multhaup and M. E. Than, J. Mol. Biol., 2012, 416, 438 CrossRef CAS PubMed; (f) R. A. Colvin, W. R. Holmes, C. P. Fontaine and W. Maret, Metallomics, 2010, 2, 306 RSC.
- (a) C. J. Frederickson, J.-Y. Koh and A. I. Bush, Nat. Rev. Neurosci., 2005, 6, 449 CrossRef CAS PubMed; (b) A. I. Bush, W. H. Pettingell, G. Multhaup, M. Paradis, J.-P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, Science, 1994, 265, 1464 CAS.
- (a) N. Y. Baek, C. H. Heo, C. S. Lim, G. Masanta, B. R. Cho and H. M. Kim, Chem. Commun., 2012, 48, 4546 RSC; (b) H. Sasaki, K. Hanaoka, Y. Urano, T. Terai and T. Nagano, Bioorg. Med. Chem., 2011, 19, 1072 CrossRef CAS PubMed; (c) Y. Jeong, G.-w. Jin, S. K. Kim, J. Yoon and J.-S. Park, Bull. Korean Chem. Soc., 2011, 32, 37 CrossRef CAS; (d) G. Masanta, C. S. Lim, H. J. Kim, J. H. Han, H. M. Kim and B. R. Cho, J. Am. Chem. Soc., 2011, 133, 5698 CrossRef CAS PubMed; (e) D. Buccella, J. A. Horowitz and S. J. Lippard, J. Am. Chem. Soc., 2011, 133, 4101 CrossRef CAS PubMed; (f) P. Du and S. J. Lippard, Inorg. Chem., 2010, 49, 10753 CrossRef CAS PubMed; (g) Z. Xu, J. Yoon and D. R. Spring, Chem. Soc. Rev., 2010, 39, 1996 RSC; (h) Z. Xu, K.-H. Baek, H. N. Kim, J. Cui, X. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132, 601 CrossRef CAS PubMed; (i) Y. Zhou, H. N. Kim and J. Yoon, Bioorg. Med. Chem. Lett., 2010, 20, 125 CrossRef CAS PubMed; (j) K. M. K. Swamy, M. J. Kim, H. N. Kim, H. R. Jeon, J. Y. Jung and J. Yoon, Bull. Korean Chem. Soc., 2010, 31, 3611 CrossRef CAS; (k) L. E. McQuade and S. J. Lippard, Inorg. Chem., 2010, 49, 9535 CrossRef CAS PubMed; (l) K. Hanaoka, Y. Muramatsu, Y. Urano, T. Terai and T. Nagano, Chem.–Eur. J., 2010, 16, 568 CrossRef CAS PubMed; (m) B. A. Wong, S. Friedle and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 7142 CrossRef CAS PubMed; (n) A. C. Esqueda, J. A. Lopez, G. Andreu-de-Riquer, J. C. Alvarado-Monzon, J. Ratnakar, A. J. M. Lubag, A. D. Sherry and L. M. De Leon-Rodriguez, J. Am. Chem. Soc., 2009, 131, 11387 CrossRef CAS PubMed; (o) Z. Xu, G.-H. Kim, S. J. Han, M. J. Jou, C. Lee, I. Shin and J. Yoon, Tetrahedron, 2009, 65, 2307 CrossRef CAS PubMed; (p) H. M. Kim and B. R. Cho, Acc. Chem. Res., 2009, 42, 863 CrossRef CAS PubMed; (q) H. M. Kim, M. S. Seo, M. J. An, J. H. Hong, Y. S. Tian, J. H. Choi, O. Kwon, K. J. Lee and B. R. Cho, Angew. Chem., Int. Ed., 2008, 47, 5167 CrossRef CAS PubMed; (r) X. A. Zhang, K. S. Lovejoy, A. Jasanoff and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10780 CrossRef CAS PubMed; (s) K. Komatsu, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13447 CrossRef CAS PubMed; (t) B. P. Joshi, W.-M. Cho, J. S. Kim, J. Yoon and K.-H. Lee, Bioorg. Med. Chem. Lett., 2007, 17, 6425 CrossRef CAS PubMed; (u) G. Crivat, K. Kikuchi, T. Nagano, T. Priel, M. Hershfinkel, I. Sekler, N. Rosenzweig and Z. Rosenzweig, Anal. Chem., 2006, 78, 5799 CrossRef CAS PubMed; (v) K. Kiyose, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2006, 128, 6548 CrossRef CAS PubMed.
- (a) X. Liu, N. Zhang, J. Zhou, T. Chang, C. Fang and D. Shangguan, Analyst, 2013, 138, 901 RSC; (b) X. Zhou, P. Li, Z. Shi, X. Tang, C. Chen and W. Liu, Inorg. Chem., 2012, 51, 9226 CrossRef CAS PubMed; (c) J. E. Kwon, S. Lee, Y. You, K.-H. Baek, K. Ohkubo, J. Cho, S. Fukuzumi, I. Shin, S. Y. Park and W. Nam, Inorg. Chem., 2012, 51, 8760 CrossRef CAS PubMed; (d) H.-Y. Lin, P.-Y. Cheng, C.-F. Wan and A.-T. Wu, Analyst, 2012, 137, 4415 RSC; (e) H. Woo, Y. You, T. Kim, G.-J. Jhon and W. Nam, J. Mater. Chem., 2012, 22, 17100 RSC; (f) J. P. Sumner, J. W. Aylott, E. Monson and R. Kopelman, Analyst, 2002, 127, 11 RSC.
- (a) J. H. Han, S. K. Park, C. S. Lim, M. K. Park, H. M. Kim and B. R. Cho, Chem.–Eur. J., 2012, 18, 15246 CrossRef CAS PubMed; (b) H. N. Kim, S.-W. Nam, K. M. K. Swamy, Y. Jin, X. Chen, Y. Kim, S.-J. Kim, S. Park and J. Yoon, Analyst, 2011, 136, 1339 RSC; (c) L. Xue, C. Liu and H. Jiang, Org. Lett., 2009, 11, 1655 CrossRef CAS PubMed; (d) Y. Zhang, X. Guo, W. Si, L. Jia and X. Qian, Org. Lett., 2008, 10, 473 CrossRef CAS PubMed.
- (a) K. Ghosh and I. Saha, Tetrahedron Lett., 2010, 51, 4995 CrossRef CAS PubMed; (b) J.-J. Wang, T.-L. Hu and X.-H. Bu, CrystEngComm, 2011, 13, 5152 RSC; (c) M. Kumar, N. Kumar and V. Bhalla, Chem. Commun., 2013, 49, 877 RSC; (d) A. Misra, M. Shahid and P. Srivastava, Sens. Actuators, B, 2012, 169, 327 CrossRef CAS PubMed; (e) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936 RSC; (f) Y. Zhou, H. N. Kim and J. Yoon, Bioorg. Med. Chem. Lett., 2010, 20, 125 CrossRef CAS PubMed; (g) C. Gao, X. Jin, X. Yan, P. An, Y. Zhang, L. Liu, H. Tian, W. Liu, X. Yao and Y. Tang, Sens. Actuators, B, 2013, 176, 775 CrossRef CAS PubMed; (h) P. A. Kumar, V. T. Ramakrishnan and P. Ramamurthy, J. Phys. Chem. A, 2011, 115, 14292 CrossRef PubMed; (i) X. Zhou, P. Li, Z. Shi, X. Tang, C. Chen and W. Liu, Inorg. Chem., 2012, 51, 9226 CrossRef CAS PubMed; (j) V. Bhalla, Roopa and M. Kumar, Dalton Trans., 2013, 42, 975 RSC; (k) Z. Xu, X. Liu, J. Pan and D. R. Spring, Chem. Commun., 2012, 48, 4764 RSC; (l) E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517 CrossRef CAS PubMed; (m) P. Jiang and J. Guo, Coord. Chem. Rev., 2004, 248, 205 CrossRef CAS PubMed; (n) P. Carol, S. Sreejith and A. Ajayaghosh, Chem.–Asian J., 2007, 2, 338 CrossRef CAS PubMed; (o) E. Kimura and S. Aoki, BioMetals, 2001, 14, 191 CrossRef CAS.
- (a) K. D. Demadis and A. Stathoulopoulou, Ind. Eng. Chem. Res., 2006, 45, 4436 CrossRef CAS; (b) H. Qu, H. Ma, A. Riviere, W. Zhou and C. J. O'Connor, J. Mater. Chem., 2012, 22, 3311 RSC.
- L. J. Teixeira, M. Seabra, E. Reis, M. T. G. da Cruz, M. C. P. de Lima, E. Pereira, M. A. Miranda and M. P. M. Marques, J. Med. Chem., 2004, 47, 2917 CrossRef CAS PubMed.
- (a) A. Kaur, H. Sharma, S. Kaur, N. Singh and N. Kaur, RSC Adv., 2013, 3, 6160 RSC; (b) V. K. Bhardwaj, P. Saluja, G. Hundal, M. S. Hundal, N. Singh and D. O. Jang, Tetrahedron, 2013, 69, 1606 CrossRef CAS PubMed; (c) P. Saluja, N. Kaur, N. Singh and D. O. Jang, Tetrahedron, 2012, 68, 8551 CrossRef CAS PubMed; (d) M. J. Kim, K. Kaur, N. Singh and D. O. Jang, Tetrahedron, 2012, 68, 5429 CrossRef CAS PubMed; (e) P. Saluja, N. Kaur, N. Singh and D. O. Jang, Tetrahedron Lett., 2012, 53, 3292 CrossRef CAS PubMed; (f) H. Sharma, N. Kaur, T. Pandiyan and N. Singh, Sens. Actuators, B, 2012, 166–167, 467 CrossRef CAS PubMed; (g) P. Saluja, H. Sharma, N. Kaur, N. Singh and D. O. Jang, Tetrahedron, 2012, 68, 2289 CrossRef CAS PubMed; (h) P. Saluja, N. Kaur, N. Singh and D. O. Jang, Tetrahedron Lett., 2011, 52, 6705 CrossRef CAS PubMed; (i) N. Singh and D. O. Jang, Tetrahedron Lett., 2011, 52, 5094 CrossRef CAS PubMed; (j) D. Y. Lee, N. Singh and D. O. Jang, Tetrahedron Lett., 2011, 52, 3886 CrossRef CAS PubMed; (k) N. Singh and D. O. Jang, Tetrahedron Lett., 2011, 52, 2608 CrossRef CAS PubMed; (l) D. Y. Lee, N. Singh, M. J. Kim and D. O. Jang, Org. Lett., 2011, 13, 3024 CrossRef CAS PubMed; (m) R. C. Mulrooney, N. Singh, N. Kaur and J. F. Callan, Chem. Commun., 2009, 686 RSC; (n) N. Kaur, N. Singh, D. Cairns and J. F. Callan, Org. Lett., 2009, 11, 2229 CrossRef CAS PubMed; (o) N. Singh, R. C. Mulrooney, N. Kaur and J. F. Callan, Chem. Commun., 2008, 4900 RSC.
- (a) D. W. Kimmel, G. LeBlanc, M. E. Meschievitz and D. E. Cliffel, Anal. Chem., 2012, 84, 685 CrossRef CAS PubMed; (b) X. Xie, M. Pawlak, M.-L. Tercier-Waeber and E. Bakker, Anal. Chem., 2012, 84, 3163 CrossRef CAS PubMed; (c) K. D. Wael, D. Daems, G. V. Camp and L. J. Nagels, Anal. Chem., 2012, 84, 4921 CrossRef PubMed; (d) J. H. Han, G. Cui, S. J. Kim, S. H. Han, G. S. Cha and H. Nam, Analyst, 2001, 126, 2040 RSC; (e) V. S. Vassilev, S. H. Hadjinikolova and S. V. Boycheva, Sens. Actuators, B, 2005, 106, 401 CrossRef CAS PubMed; (f) A. González-Bellavista, J. Macanás, M. Muñoz and E. Fàbregas, Sens. Actuators, B, 2006, 115, 691 CrossRef PubMed.
- (a) S. D. Bella, I. P. Oliveri, A. Colombo, C. Dragonetti, S. Righetto and D. Roberto, Dalton Trans., 2012, 41, 7013 RSC; (b) G. Consiglio, S. Failla, P. Finocchiaro, I. P. Oliveri and S. D. Bella, Dalton Trans., 2012, 41, 387 RSC; (c) G. Consiglio, S. Failla, I. P. Oliveri, R. Purrello and S. D. Bella, Dalton Trans., 2009, 10426 RSC.
- N. Singh, M. S. Hundal, G. Hundal and M. Martinez-Ripoll, Tetrahedron, 2005, 61, 7796 CrossRef CAS PubMed.
- N. Singh, N. Kaur, R. C. Mulrooney and J. F. Callan, Tetrahedron Lett., 2008, 49, 6690 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- D. Ray and P. K. Bharadwaj, Inorg. Chem., 2008, 47, 2252 CrossRef CAS PubMed.
- (a) J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551 CrossRef CAS PubMed; (b) J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483 RSC.
- A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, A. J. Patty and G. L. Spence, J. Chem. Soc., Perkin Trans. 2, 1993, 1611 RSC.
- N. Singh, R. C. Mulrooney, N. Kaur and J. F. Callan, J. Fluoresc., 2009, 19, 777 CrossRef CAS PubMed.
- J. Liu and A. P. de Silva, Inorg. Chim. Acta, 2012, 381, 243 CrossRef CAS PubMed.
- (a) A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385 RSC; (b) S. Uchiyama, K. Iwai and A. P. de Silva, Angew. Chem., Int. Ed., 2008, 47, 4667 CrossRef CAS PubMed; (c) A. P. de Silva, T. P. Vance, M. E. S. West and G. D. Wright, Org. Biomol. Chem., 2008, 6, 2468 RSC; (d) K. Kiyose, H. Kojima, Y. Urano and T. Nagano, J. Am. Chem. Soc., 2006, 128, 6548 CrossRef CAS PubMed; (e) J. Wongkongkatep, Y. Miyahara, A. Ojida and I. Hamachi, Angew. Chem., Int. Ed., 2006, 45, 665 CrossRef CAS PubMed; (f) Y. J. Jang, E. J. Jun, Y. J. Lee, Y. S. Kim, J. S. Kimand and J. Yoon, J. Org. Chem., 2005, 70, 9603 CrossRef CAS PubMed; (g) T. Hirano, K. Kikuchi, Y. Urano, T. Higuchi and T. Nagano, Angew. Chem., Int. Ed., 2000, 39, 1052 CrossRef CAS; (h) S. Uchiyama, T. Santa and K. Imai, Analyst, 2000, 125, 1839 RSC; (i) R. A. Bissell, A. P. de Silva, H. Q. N. Gunaratne, P. L. M. Lynch, G. E. M. Maguire, C. P. McCoy and K. R. A. S. Sandanayake, Top. Curr. Chem., 1993, 168, 223 CrossRef CAS; (j) A. P. de Silva and K. R. A. S. Sandanayake, Angew. Chem., Int. Ed. Engl., 1990, 29, 1173 CrossRef.
- R. Kia, H. Kargar, M. N. Tahir and F. Kianoosh, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o2296 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR spectra, UV-vis absorption, fluorescence profiles and graphs for the fabrication and operation of ISE based upon 2a–c. See DOI: 10.1039/c3qi00031a |
| This journal is © the Partner Organisations 2014 |