Synthesis of ferrocene-functionalized monomers for biodegradable polymer formation†
Brianna M.
Upton
,
Raymond M.
Gipson
,
Selma
Duhović
,
Brian R.
Lydon
,
Nicholas M.
Matsumoto
,
Heather D.
Maynard
and
Paula L.
Diaconescu
*
Department of Chemistry & Biochemistry, University of California, Los Angeles, California 90095, USA. E-mail: pld@chem.ucla.edu
First published on 14th February 2014
Abstract
Cyclic carbonate and δ-valerolactone substrates functionalized with ferrocene were synthesized via alkyne–azide “click” cycloaddition. The cyclic carbonates were polymerized using 1,8-diazabicycloundec-7-ene, 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea, and benzyl alcohol. The resulting polymers were characterized by GPC, NMR spectroscopy, and cyclic voltammetry studies. It was found that polycarbonate molecular weights fall in the range of 4.1–5.2 × 103 g mol−1 with polydispersities as low as 1.26. Electrochemistry studies allowed us to identify the monomer/polymer pair with the most desirable redox potential for biological studies.
Introduction
Biodegradable and biocompatible polymers are finding more and more sophisticated applications in improving human health, ranging from the controlled release of active ingredients in drug delivery systems to tissue engineering and bone repair in surgery.1–7 Although such polymers can be prepared from a variety of (natural) compounds according to specific synthetic routes, their formation by ring-opening polymerization of suitable cyclic monomers such as lactide, lactones, and cyclic carbonates is the preferred method since it offers good control of the resulting polymer structure. The choice of the repetitive unit(s) is important since it allows the modulation of the physico-chemical properties of the polymer, such as crystallinity, glass transition temperature, toughness, stiffness, adhesion, permeability, and degradability.8–10Polymers containing ferrocene have been extensively researched due to potential materials properties,11–14 but far less focus has been placed on the synthesis of biodegradable analogues.15,16 Ferrocene has already been utilized to increase the bioactivity of many pharmaceuticals, such as in the anticancer and antimalarial pharmaceutical drugs ferrocifen17–19 and ferroquine.20 With the abundance of biological activity attributed to the tunable redox couple of ferrocene,21–23 functionalized biodegradable polymers have potential for application in medicinal chemistry by delivering ferrocene units to specific sites in the human body. However, only a few classes of biodegradable ferrocene-containing polymers are known24–27 with only one class of polyesters being reported.26,27
Biodegradable polymers with pendant functionalities have been well researched and used in numerous biological studies.28–30 Previous approaches to ferrocene-functionalized biodegradable polymers utilized the synthesis of polyaspartamide bearing reactive amine functional groups, followed by the coupling of the side chain with a ferrocene carboxylic acid moiety (Chart 1)31 or polycondensation between diol-terminated Schiff bases and 1,1′-ferrocenediyl diacid chlorides. To avoid difficulty with functionalization, we propose the use of the highly developed azide–alkyne “click” reaction to synthesize functionalized monomers for use in ring opening polymerization.32–35 An improved route to functionalized polycarbonates utilized monomers derived from the commercially available 2,2-bis(methoxy)propionic acid (bis-MPA).36,37 In this paper, we employed the Huisgen alkyne–azide click reaction to functionalize δ-valerolactone and cyclic carbonates with ferrocene. We then used the novel monomers in ring opening polymerization and obtained ferrocene-functionalized biodegradable polymers.
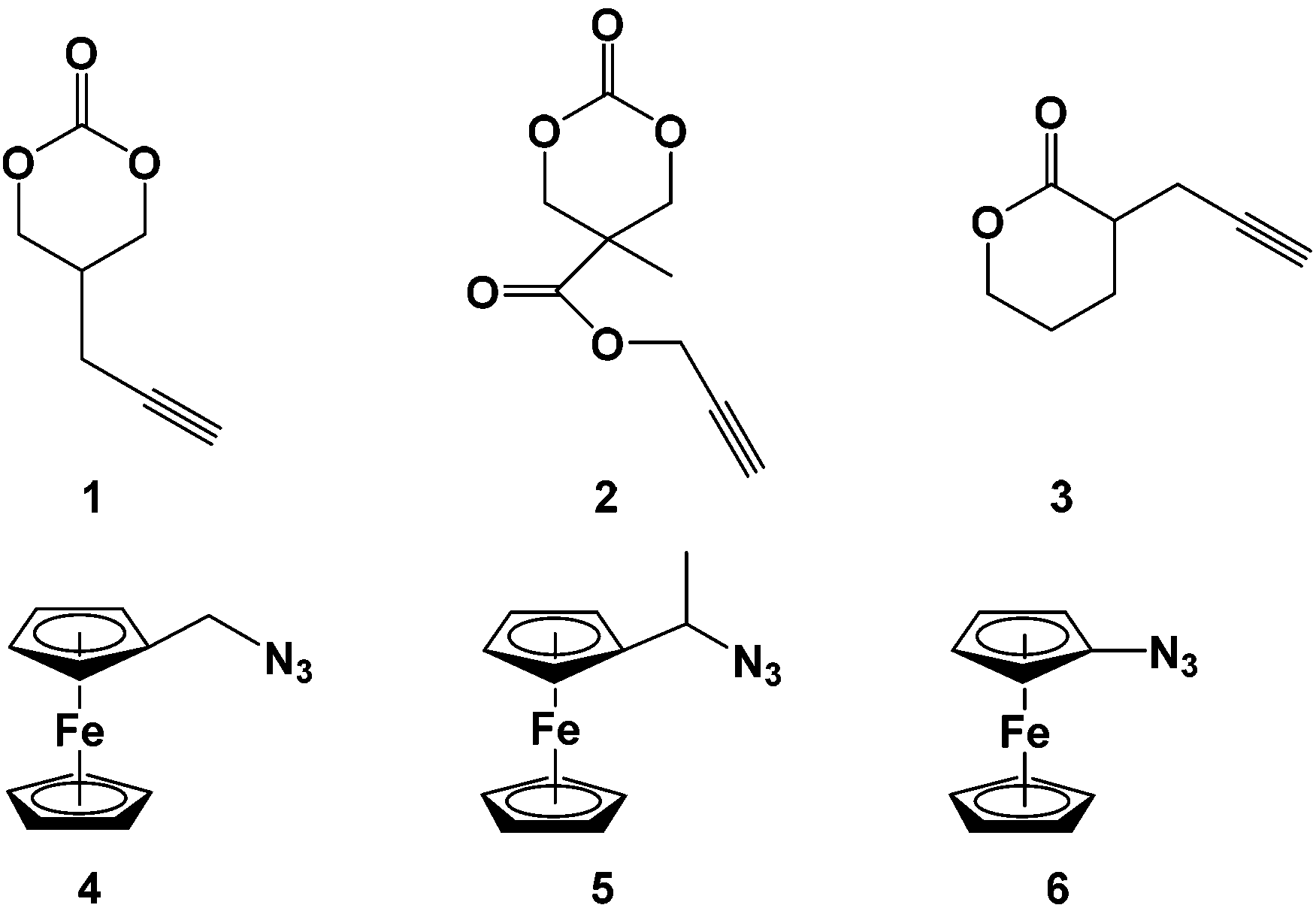 | ||
| Chart 1 Alkyne-functionalized carbonates 1–2 and lactone 3 and azidoferrocene precursors 4–6. | ||
Results and discussion
Synthesis and characterization of monomers
Nine novel ferrocene-functionalized monomers were prepared via click chemistry using 5-(propynyl)-1,3-dioxan-2-one (1), propargyl 5-methyl-2-oxo-1,3-dioxane-5-carboxylate (2), and α-propargyl-δ-valerolactone (3) as alkynes (Chart 1). Cyclic carbonate 1 was prepared in only three steps from dimethyl malonate, while carbonate 2 was prepared by a protecting group strategy in five steps from 2,2-bis(methoxy)propionic acid (bis-MPA). δ-Valerolactone derivative 3 was synthesized in one step from its lactone precursor.33 Compounds 1-methylazidoferrocene (4) and 1-azidoethylferrocene (5) were obtained in four and three steps, respectively, using known precursors,38,39 while 1-azidoferrocene (6) was prepared in two steps from ferrocene.40With the necessary alkyne and azide precursors, click reactions were employed to synthesize nine lactone and cyclic carbonate monomers (Chart 2). A stoichiometric amount of copper sulfate pentahydrate reduced in situ by sodium ascorbate was used as the pre-catalyst and all reactions were performed in a mixed solution of THF and water for five hours. Upon completion, the reactions were quenched with the addition of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 15% NH3 (aq.) and dichloromethane biphasic solution resulting in an intensely blue aqueous layer and a yellow-orange dichloromethane layer. The aqueous layer was separated and the organic layer was dried to give the product as a yellow-orange solid. All click reactions reached high conversion with the same general reaction conditions, the lowest being M6 with a 68.9% yield. The monomer products were readily identified by 1H NMR spectroscopy via three characteristic peaks: the triazole proton between 7.85 and 7.24 ppm, the methylene bridge of the alkyne functionality between 3.17 and 2.68 ppm, and the ferrocene signal between 4.27 and 4.10 ppm. The isolated monomers (Chart 2) were air and thermally stable and could be stored at −30 °C without any decomposition.
:1 15% NH3 (aq.) and dichloromethane biphasic solution resulting in an intensely blue aqueous layer and a yellow-orange dichloromethane layer. The aqueous layer was separated and the organic layer was dried to give the product as a yellow-orange solid. All click reactions reached high conversion with the same general reaction conditions, the lowest being M6 with a 68.9% yield. The monomer products were readily identified by 1H NMR spectroscopy via three characteristic peaks: the triazole proton between 7.85 and 7.24 ppm, the methylene bridge of the alkyne functionality between 3.17 and 2.68 ppm, and the ferrocene signal between 4.27 and 4.10 ppm. The isolated monomers (Chart 2) were air and thermally stable and could be stored at −30 °C without any decomposition.
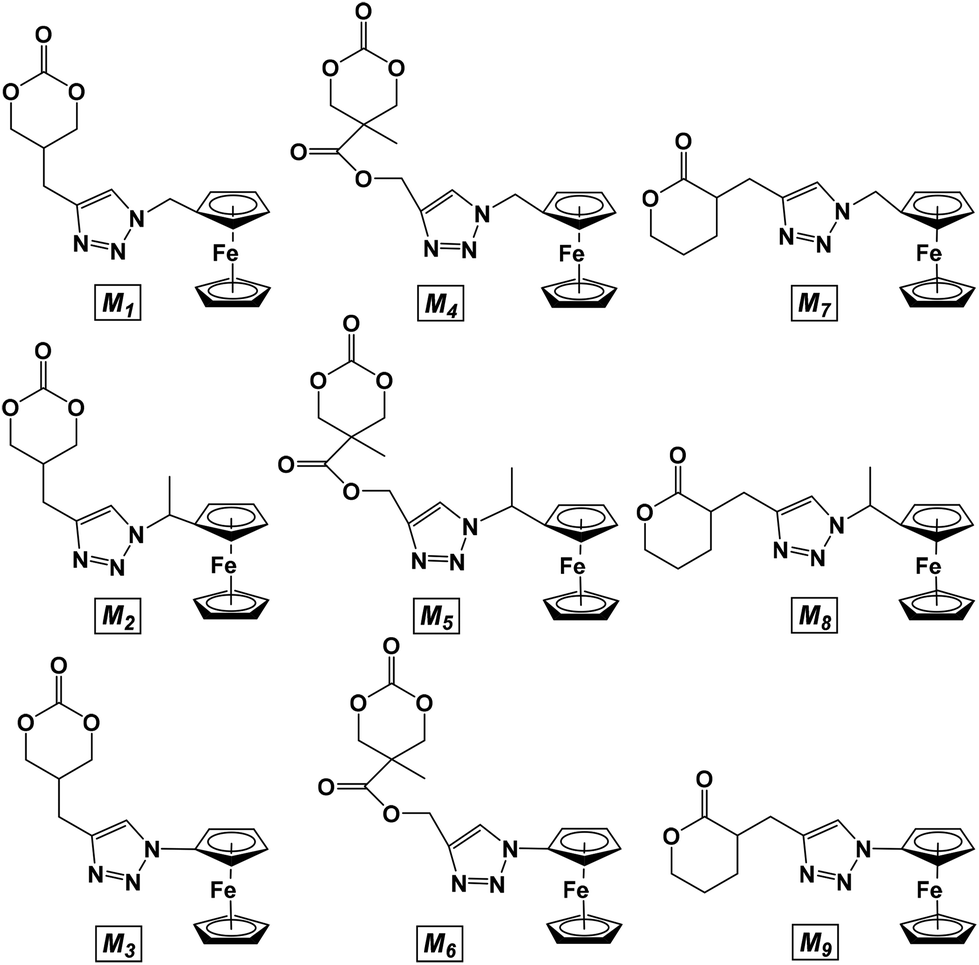 | ||
| Chart 2 Monomers M1 to M9 post azide–alkyne click reaction. | ||
Ring-opening polymerization reactions
Lewis acidic metals are known as excellent catalysts for the ring opening polymerization of cyclic carbonates and lactones.41–44 Based upon prior work, the polymerization of cyclic carbonate M1 was attempted with Sn(oct)2, Ti(Oi-Pr)4, and Al(Oi-Pr)3. However, no polymerization was observed even after heating to high temperatures. The aluminum diamide catalyst (o-[(TMS)Ph-N]2-C6H4)Al(NMe2)(HNMe2) reported by Bergman et al. was also employed without success.45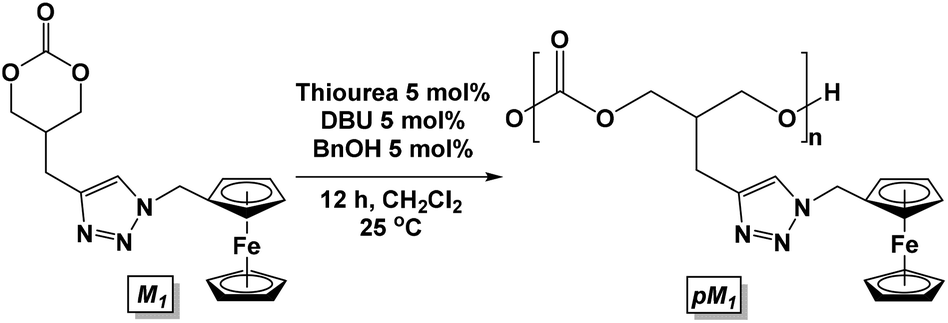 | (1) |
Based upon the successful ring-opening polymerization of other functionalized monomers,46,47 we turned our attention to organocatalysts. Dove et al.48 used the following catalytic conditions for the polymerization of a norbornene-functionalized 5-methyl-2-oxo-1,3-dioxane-5-carboxylic acid monomer: benzyl alcohol (BnOH, 1 eq.), 1,8-diazabicycloundec-7-ene (DBU, 1 mol%), and 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea (thiourea, 5 mol%). Utilizing the same conditions, the polymerization of M1 was attempted. While the polymerization appeared to be complete in less than 10 min, the compound could not be isolated by the addition of methanol and was dried to an orange oil. Analysis by gel permeation chromatography (GPC) showed a small Mn, suggesting the formation of oligomers with a low molecular weight. Catalyst loading was then lowered to 5 mol% for BnOH, DBU, and thiourea (eqn (1)). Monitoring by 1H NMR spectroscopy showed 75% conversion after 12 h. The resulting polycarbonate pM1 was isolated by precipitation from dichloromethane with cold methanol. The GPC trace showed that pM1 had a Mn of 4088 g mol−1 relative to a poly(methyl methacrylate) standard and a PDI of 1.26.
Attempting to increase Mn further, catalyst loading was reduced to 1 mol% for DBU and benzyl alcohol, but those reaction conditions yielded almost no conversion (11%) after 24 h and the product was isolated as an orange oil. The GPC trace once again showed a small Mn. Therefore, we decided to use the above conditions to carry out the polymerization of monomers M1 to M6 and obtained similar conversions and Mn (Table 1). Integration of 1H NMR spectra for polymers pM1–pM6 revealed that the ferrocene appendage was retained during all polymerizations for each repeating unit.
| Monomer | Time | Conversion | M n (10−3 g mol−1) | PDI |
|---|---|---|---|---|
| M1 | 12 h | 75% | 4.0 | 1.26 |
| M2 | 12 h | 73% | 4.3 | 1.81 |
| M3 | 12 h | 71% | 4.0 | 1.33 |
| M4 | 24 h | 62% | 5.2 | 1.69 |
| M5 | 24 h | 70% | 4.9 | 1.61 |
| M6 | 24 h | 76% | 4.7 | 1.36 |
Thermogravimetric analysis was conducted with all six polycarbonates (Fig. S3–S8†). The thermograms of all polymers show broad decomposition temperatures. The thermograms also show three thermal steps ascribed to the vaporization and/or pyrolysis of the polycarbonate functionality, the ester linkage, and ferrocene, respectively.
After the ring-opening polymerization of cyclic carbonates M1–M6 was successfully carried out, the polymerization of δ-valerolactone derivatives M7–M9 was targeted. Poor monomer solubility limited polymerization studies to the use of dichloromethane as solvent. Attempts to polymerize lactone M7 with the same organocatalytic system as described above yielded no reaction, even after heating to 100 °C for 24 h. Polymerization was also attempted with Bergman's aluminum catalyst; once again, heating to 100 °C for 24 h did not result in polymerization. Furthermore, no polymerization was observed when using the highly active organocatalyst 1,5,7-triaza/bicycle[4.4.0]dec-5-ene (TBD).49
DFT calculations
While our results agree with previous reports on the difficulty to polymerize functionalized valerolactones,50,51 we decided to investigate the thermodynamic favorability of the ring opening reaction by density functional theory (DFT) calculations. Based upon known methods,52 we calculated the ring strain energy for all monomers (Table S1†).53 The three lactone monomers show relatively large, positive free energies, with the largest value obtained for monomer M9 (ΔG = 6.89 kcal mol−1). By comparison, the ring strain of monomers M1–M6 was found to be 0.03–0.70 kcal mol−1. Although these values are positive, they are about an order of magnitude lower than those obtained for the three lactone monomers.Cyclic voltammetry studies
In order to transport the ferrocene moiety to the target cells under optimized pharmacological conditions, it is necessary to favor the stable lower (ferrocene) over the labile higher (ferrocenium) oxidation state. It was proposed that, once in the lysosomal compartment at a pH of ∼5, the ferrocenium cation state, expected to be the biologically active component, would be enzymatically generated in concentrations dependent on the metallocene's electrochemical reduction potential, the lowest positive potential versus Fc0/+ reflecting the highest cation stability.54Electrochemical properties of polymeric and monomeric species were investigated using cyclic voltammetry in THF with tetraisopropylammonium tetrakis(3,5-trifluoromethyl)phenylborate (TPABArF) as the supporting electrolyte. The anodic and cathodic peak currents for both cyclic carbonates increased linearly with the square root of the scan rate, indicating that both monomers show electrochemically reversible redox events (Fig. S1†). However, the anodic and cathodic peak potentials (Fig. S2†) of both monomers and polymers become slightly more separated with increasing scan rates, consistent with slow electron transfer when compared to an ideal Nernstian system.
Lactones M7 and M9 have lower redox potentials than their cyclic carbonate counterparts (0.04 V for M7 compared to 0.37 V for M4, Fig. 1). The introduction of a methylene spacer between the pendant ferrocene and the triazole ring lowered the redox potentials of both lactone (−0.01 V for M7 compared to 0.04 V for M9) and cyclic carbonate (0.02 V for M4 compared to 0.37 V for M6) substrates (Table 2). A similar change was also observed for the cyclic carbonate polymers (0.37 V for pM6 compared to 0.10 V for pM4). This effect can be attributed to electron donation from the methylene spacer shifting the redox potential in the cathodic direction.55 Interestingly, when comparing the redox potentials of the cyclic carbonate monomers and their respective polycarbonates (Fig. 2 for M4 and pM4), the redox potential changes more substantially and in the opposite direction when a methylene group is present (+80 mV difference between M4 and pM4) than when it is absent (−5 mV difference between M6 and pM6). Overall, the electrochemical properties indicate that M4 and pM4 show appropriate redox potentials E1/2 of 0.02 and 0.10 V versus Fc0/+, respectively, for biological studies.
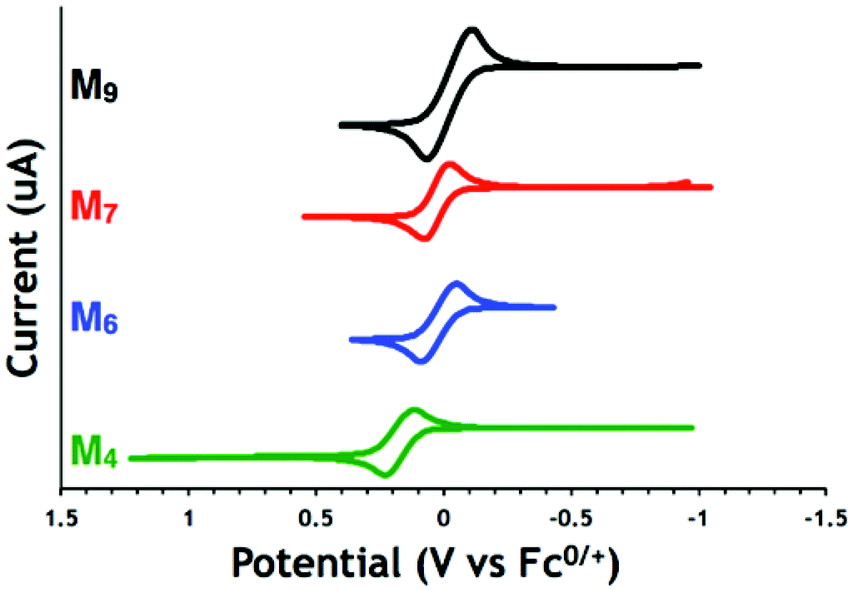 | ||
| Fig. 1 Cyclic voltammograms of lactone (M7 and M9) and cyclic carbonate (M4 and M6) monomers in THF with TPABArF as the supporting electrolyte at a 2 mm Pt disk electrode, 10 mV s−1 scan rate (referenced to the ferrocene/ferrocenium couple). | ||
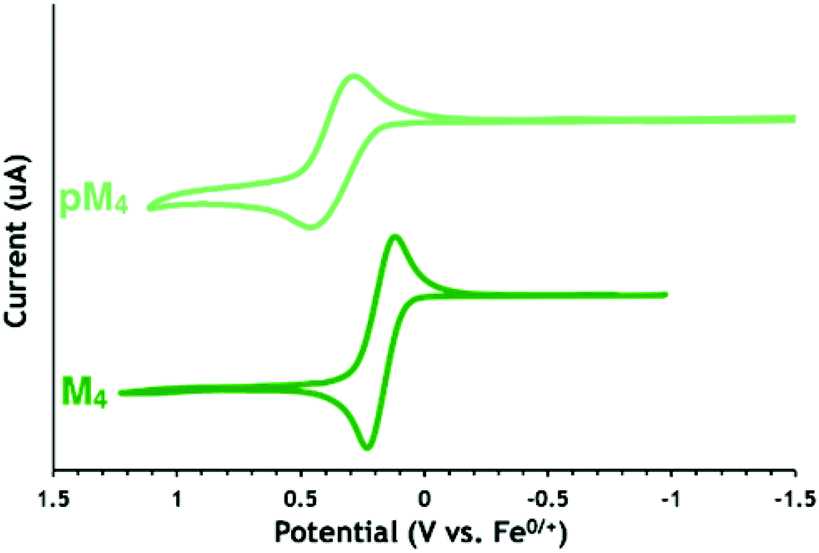 | ||
| Fig. 2 Cyclic voltammograms of polycarbonate pM4 (top) and monomer M4 (bottom) in THF with TPABArF as the supporting electrolyte at a 2 mm Pt disk electrode, 10 mV s−1 scan rate (referenced to the ferrocene/ferrocenium couple). | ||
| Compound | E 1/2 (V vs. Fc0/+) | |
|---|---|---|
| Carbonate monomers | M4 | 0.02 |
| M6 | 0.37 | |
| Lactone monomers | M7 | −0.01 |
| M9 | 0.04 | |
| Carbonate polymers | pM4 | 0.10 |
| pM6 | 0.37 |
Conclusions
The highly developed and well tolerated alkyne–azide click reaction provides a facile method for the functionalization of cyclic monomers with ferrocene. Alkyne functionalized cyclic monomers were combined with azidoferrocene derivatives using this reaction to yield nine new monomeric species. Six polycarbonates, which incorporated ferrocene moieties, were synthesized using organocatalytic ring-opening polymerization and characterized by GPC, NMR spectroscopy, and cyclic voltammetry studies. The electrochemistry studies indicate that M4 and pM4 show desirable redox potentials for biological studies.Experimental section
General considerations
All experiments were performed under a dry nitrogen atmosphere using standard Schlenk techniques or in an MBraun inert-gas glovebox. Solvents were purified using a two-column solid state purification system by the method of Grubbs56 and transferred to the glove box without exposure to air. All other materials were used as received unless either wise stated. Benzyl alcohol and 1,8-diazabicycloundec-7-ene (DBU) were dried over calcium hydride, distilled, and stored under inert atmosphere. 1-Hydroxymethylferrocene,57 1-hydroxyethylferrocene,58 α-propargyl-δ-valerolactone,59 and 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea60 were synthesized according to published procedures. Thin-layer chromatography (TLC) was conducted with silica gel plates and visualized using potassium permanganate staining. EMD silica gel 60 (particle size 40–63 μm) was used for flash column chromatography. NMR solvents were obtained from Cambridge Isotope Laboratories, degassed, and stored over activated molecular sieves prior to use. 1H NMR spectra were recorded on Bruker300 or Bruker500 spectrometers at room temperature in CDCl3. Chemical shifts are reported with respect to internal solvent, 7.26 ppm (CDCl3) for 1H NMR spectra. 13C NMR spectra were recorded on a Bruker500 spectrometer. Electrochemical measurements were carried out at room temperature under an inert atmosphere in anhydrous THF with TPABArF as the supporting electrolyte using a CHI630D potentiostat. The electrochemical cell consisted of a platinum disk (2 mm) as the working electrode, glassy carbon disk (3 mm) as the counter electrode, and silver wire (0.25 mm) as the pseudo-reference electrode. Steady-state experiments were conducted with a platinum disk microelectrode (0.25 μm). All potentials are reported versus a ferrocene internal standard, which was added to the electrochemical cell at the end of each measurement. Gel permeation chromatography (GPC) for polycarbonates pM1–pM6 was conducted on a Shimadzu HPLC system equipped with a refractive index detector RID-10A, one Polymer Laboratories PLgel guard column, and two Polymer Laboratories PLgel 5 μm mixed D columns. LiBr (0.1 M) in DMF at 40 °C was used as an eluent (flow rate: 0.60 mL min−1). Calibration was performed using near-monodisperse poly(methyl methacrylate) standards from Polymer Laboratories. Chromatograms were analyzed using EZStart 7.2 chromatography software. Thermogravimetric analysis was conducted on a Perkin Elmer Pyris Diamond TG/DTA Thermogravimetric/Differential Thermal Analyzer. The TGA instrument operated under an argon atmosphere and between 23 °C and 500 °C. Experiments used a heating rate of 10 °C min−1 and samples were heated in a platinum crucible.Synthesis of 5-(prop-2-yn-1-yl)-1,3-dioxan-2-one (1)
2-(Prop-2-yn-1-yl)propane-1,3-diol (1.000 g, 8.8 mmol) and pyridine (4.2 mL, 52.1 mmol) were combined in 40 mL dichloromethane (DCM) and cooled to −78 °C. A solution of triphosgene (1.301 g, 4.4 mmol) in 20 mL DCM was added dropwise over 30 minutes. After the addition, the reaction was allowed to warm to room temperature and stir for one hour. After one hour, the reaction was quenched with saturated NH4Cl solution. The organic layer was separated and washed with 1 M HCl solution (3 × 100 mL), saturated NaHCO3 (1 × 100 mL), and water (1 × 100 mL). The organic layer was dried over MgSO4, filtered and concentrated yielding a white solid. The white solid was recrystallized in ethyl acetate layered with cyclohexane yielding colorless, needle-like crystals. Yield: 0.883 g, 71.9% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 4.48 (dd, 2H, OCH2CH), 4.28 (dd, 2H, OCH2CH), 2.41 (br m, 1H, OCH2CH), 2.37 (m, 2H, HCCCH2) 2.08 (t, 1H, HCCCH2). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 148.10 (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 78.72 (OCH2CH), 71.69 (HCCCH2), 70.84 (HCCCH2), 30.19 (HCCCH2), 17.31 (OCH2CH). Anal. Calcd for C7H8O3: C, 59.99, H, 5.75. Found: C, 59.94; H, 5.79.
C), 78.72 (OCH2CH), 71.69 (HCCCH2), 70.84 (HCCCH2), 30.19 (HCCCH2), 17.31 (OCH2CH). Anal. Calcd for C7H8O3: C, 59.99, H, 5.75. Found: C, 59.94; H, 5.79.
General procedure for click reactions
The ferrocene-based azide (1 eq.) and alkyne functionalized carbonate (1 eq.) were combined in 20 mL of THF. An aqueous solution of copper sulfate pentahydrate (1 eq.) was added. An aqueous solution of sodium ascorbate (0.57 eq.) was added dropwise and the reaction was allowed to stir for 5 hours at room temperature. The reaction was then quenched via the addition of a 1:1 15% NH3 solution and DCM solution. The reaction mixture was allowed to stir for an additional 10 minutes. The organic layer was then separated, dried over MgSO4, and concentrated to an oily yellow solid. The crude product was recrystallized in ethyl acetate layered with pentane three times to give the pure product as a yellow-orange powder.
Details for M1
Yield: 0.619 g, 87.2% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 7.25 (s, 1H, CCHN), 5.28 (s, 2H, NCH2Cp), 4.47 (dd, 2H, OCH2CH), 4.28–4.17 (m, 11H, OCH2CH and fc), 2.80 (m, 2H, CHCH2C), 2.68 (br m, 1H, CHCH2C). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 148.28 (OC), 142.93 (CH2CCH), 121.07 (CH2CCH), 80.43 (OCH2CH), 71.41 (fc), 69.25 (fc), 68.95 (Cp), 50.18 (NCH2Cp), 31.13 (CHCH2C), 23.82 (OCH2CH). Anal. Calcd for C18H19FeN3O3: C, 56.71; H, 5.02; N, 11.02. Found C, 56.62; H, 5.16; N, 10.86.
Details for M2
Yield: 0.978 g, 74.9% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 7.25 (s, 1H, CCHN), 5.63 (m, 1H, NCHCH3Cp), 4.39 (dd, 2H, OCH2CH), 4.27–4.12 (m, 11H, OCH2CH and fc), 2.68 (m, 2H, CHCH2C), 2.56 (br s, 1H, CHCH2C), 1.84 (d, 3H, NCHCH3). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 148.41 (OC), 142.62 (CH2CCH), 119.80 (CH2CCH), 87.16 (OCH2CH), 71.51 (fc), 69.06 (fc), 68.98 (fc), 56.61 (NCHCH3Cp), 31.11 (CHCH2C), 23.72 (OCH2CH), 21.31 (NCHCH3Cp). Anal. Calcd for C19H21FeN3O3: C, 57.74; H, 5.36; N, 10.63. Found: C, 58.69; H, 5.70; N, 10.39.
Details for M3
Yield: 0.579 g, 76.8% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 7.56 (s, 1H, CCHN), 5.29 (s, 1H, CH2CHC), 4.84 (s, 2H, OCH2CH), 4.39–4.21 (m, 11H, OCH2CH and fc), 2.86 (m, 2H, CHCH2C). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 148.31 (OCO), 143.18 (CH2CCH), 121.66 (CH2CCH), 95.12 (OCH2CH), 71.41 (fc), 70.20 (fc), 68.27 (fc), 63.61 (fc), 31.07 (CHCH2C), 23.76 (OCH2CH). Anal. Calcd for C17H17FeN3O3: C, 55.61; H, 4.67; N, 11.44. Found: C, 55.19; H, 4.77; N, 11.08.
Details for M4
Yield: 0.743 g, 83.1% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 7.52 (s, 1H, CCHN), 5.26 (br s, 4H, OCH2CCHNCH2), 4.64 (d, 2H, OCH2CCH3), 4.27–4.14 (m, 11H, OCH2CH and fc), 1.25 (s, 3H, OCH2CCH3). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 171.04 (COCOCH2), 147.44 (OC), 80.64 (OCH2CCH3), 72.93 (CH2CCH), 69.19 (fc), 69.00 (fc), 68.94 (fc), 59.20 (OCH2CCHN), 50.20 (NCH2Cp), 40.29 (CHCH2C), 17.32 (OCH2CCH3). Anal. Calcd for C20H21FeN3O5: C, 54.69, H, 4.82; N, 9.57. Found: C, 54.54; H, 4.78; N, 9.33.
Details for M5
Yield: 0.459 g, 79.4% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 7.44 (s, 1H, CCHN), 5.66 (q, 1H, NCHCH3Cp), 5.24 (s, 2H, OCH2CCHNCH2), 4.62 (m, 2H, OCH2CCHNCH2), 4.27–4.12 (m, 11H, OCH2CH and fc), 1.87 (d, 3H, NCHCH3Cp), 1.23 (s, 3H, OCH2CCH3). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 171.03 (COCOCH2), 147.44 (OC), 141.22 (CH2CCH), 121.74 (CH2CCH), 87.12 (OCH2CH), 68.99 (fc), 68.39 (fc), 67.86 (fc), 66.18 (fc), 59.23 (OCH2CCHN), 56.86 (NCHCH3Cp), 40.27 (CHCH2C), 21.34 (NCHCH3Cp), 17.27 (OCH2CCH3). Anal. Calcd for C21H23FeN3O5: C, 55.65; H, 5.11; N, 9.27. Found: C, 55.60; H, 5.16; N, 9.39.
Details for M6
Yield: 0.512 g, 68.9% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 7.85 (s, 1H, CCHN), 5.40 (s, 2H, OCH2CCHNCH2), 4.86 (m, 2H, OCH2CCHNCH2), 4.73–4.69 (m, 2H, OCH2CH), 4.28–4.19 (m, 9H, fc), 1.33 (s, 3H, OCH2CCH3). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 171.20 (COCOCH2), 147.53 (OC), 141.83 (CH2CCH), 123.40 (CH2CCH), 93.40 (OCH2CH), 70.26 (fc), 67.93 (fc), 66.88 (fc), 62.20 (NCp), 59.19 (OCH2CCHN), 40.44 (CHCH2C), 17.18 (OCH2CCH3). Anal. Calcd for C19H19FeN3O5: C, 53.67; H, 4.50; N, 9.88. Found: C, 53.52; H, 4.57; N, 9.90.
Details for M7
Yield: 1.507 g, 88.3% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 7.32 (s, 1H, CCHN), 5.19 (d, 2H, NCH2Cp), 4.15 (m, 11H, fc and OCH2CH2), 3.11 (m, 1H, OCCH), 2.90 (m, 2H, OCCHCH2), 2.06 (m, 1H, OCH2CH2CH2), 1.81 (m, 2H, OCH2CH2CH2), 1.58 (m, 1H, OCH2CH2CH2). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 174.17 (OC), 144.77 (CCHN), 121.92 (CCHN), 81.18 (OCOCH2), 68.95 (fc), 68.86 (fc), 68.29 (fc), 49.86 (CHNCH2Cp), 39.86 (OCCHCH2), 26.99 (OCCHCH2), 24.15 (OCH2CH2CH2), 21.95 (OCH2CH2CH2). Anal. Calcd for C19H21FeN3O2: C, 60.17; H, 5.58; N, 11.08. Found: C, 60.78; H, 5.87; N, 11.42.
Details for M8
Yield: 1.498 g, 85.7% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 7.24 (s, 1H, CCHN), 5.58 (m, 1H, NCHCH3Cp), 4.11 (m, 11H, fc and OCH2CH2), 3.06 (m, 1H, OCCH), 2.81 (m, 2H, OCCHCH2), 2.01 (m, 1H, OCH2CH2CH2), 1.76 (m, 5H, OCH2CH2CH2 and NCHCH3Cp), 1.52 (m, 1H, OCH2CH2CH2). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 174.12 (OCCH), 144.40 (CH2CCH), 120.22 (CH2CCH), 87.72 (OCOCH2CH2CH2), 68.94 (fc), 68.21 (fc), 67.73 (fc), 56.34 (NCHCH3Cp), 39.76 (OCCHCH2), 27.06 (CHCH2CCH), 24.15 (OCOCH2CH2CH2), 21.92 (OCOCH2CH2CH2), 21.50 (NCHCH3Cp). Anal. Calcd for C20H23FeN3O2: C, 61.08; H, 5.89; N, 10.69. Found: C, 61.46; H, 6.03; N, 10.84.
Details for M9
Yield: 0.736 g, 70.4% 1H NMR (300 MHz, 25 °C, CDCl3), δ (ppm): 7.60 (s, 1H, CCHN), 4.82 (d, 4H, OCH2CH2CH2), 4.30 (br m, 11H, fc and OCH2CH2), 3.17 (m, 2H, CHCH2C), 2.95 (m, 1H, OCCHCH2). 13C NMR (125 MHz, 25 °C, CDCl3), δ (ppm): 174.28 (OCCH), 122.18 (CH2CCH), 95.06 (OCOCH2CH2CH2), 70.13 (fc), 68.37 (fc), 68.32 (fc), 63.60 (fc), 39.76 (OCCHCH2), 26.98 (CHCH2CCH), 24.35 (OCOCH2CH2CH2), 22.01 (OCOCH2CH2CH2). Anal. Calcd for C18H19FeN3O2: C, 59.20; H, 5.24; N, 11.51. Found: C, 59.27; H, 5.10; N, 11.30.
General procedure for polymerizations
The corresponding monomer was weighed, added to a scintillation vial and dissolved in 0.3 mL dichloromethane (DCM). 1-(3,5-Bis(trifluoromethyl)phenyl)-3-cyclohexylthiourea (5 mol%) was weighed and added to a separate vial along with 1,8-diazabicycloundec-7-ene (5 mol%), benzyl alcohol (5 mol%), and 0.3 mL DCM. This solution was added to the monomer solution and allowed to stir for 12 or 24 h (depending on the monomer). After completion, the reaction was quenched with benzoic acid and cold methanol was added to precipitate the polymer as an orange solid.Acknowledgements
This work was supported by NSF (CAREER grant 0847735 to PLD) and the Sloan Foundation. NMM acknowledges support for an NSF Graduate Research Fellowship. The NMR spectroscopy studies are based upon work supported by the National Science Foundation under Equipment grant no. CHE-1048804 and CHE-9974928.Notes and references
- L. S. Nair and C. T. Laurencin, in Advances in Biochemical Engineering/Biotechnology, Springer, Berlin/Heidelberg, 2006, vol. 102, pp. 47–90 Search PubMed.
- T. Hayashi, Prog. Polym. Sci., 1994, 19, 663 CrossRef CAS.
- H. T. Yoshito Ikada, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef.
- R. Langer, Acc. Chem. Res., 2000, 33, 94–101 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- S. Inkinen, M. Hakkarainen, A.-C. Albertsson and A. Södergård, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS PubMed.
- A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- C. Jérôme and P. Lecomte, Adv. Drug Delivery Rev., 2008, 60, 1056–1076 CrossRef PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- N. Masato, P. W. Cyr, L. Kun, E. H. Sargent and I. Manners, Adv. Funct. Mater., 2008, 18, 470–477 CrossRef.
- P. W. Cyr, E. J. D. Klem, E. H. Sargent and I. Manners, Chem. Mater., 2005, 17, 5770–5773 CrossRef CAS.
- P. W. Cyr, M. Tzolov, M. A. Hines, I. Manners, E. H. Sargent and G. D. Scholes, J. Mater. Sci., 2003, 13, 2213–2219 CAS.
- K. Kulbaba and I. Manners, Macromol. Rapid Commun., 2001, 22, 711–724 CrossRef CAS.
- R. D. A. Hudson, J. Organomet. Chem., 2001, 637, 47–69 CrossRef.
- R. Horikoshi and T. Mochida, Eur. J. Inorg. Chem., 2010, 5355–5371 CrossRef CAS.
- M. J. Meegan and D. G. Lloyd, Curr. Med. Chem., 2003, 10, 181 CrossRef CAS.
- M. A. Musa, M. O. F. Khan and J. S. Cooperwood, Curr. Med. Chem., 2007, 14, 1249–1261 CrossRef CAS.
- G. Leclercq, M. Lacroix, I. Laïos and G. Laurent, Curr. Cancer Drug Targets, 2006, 6, 39–64 CrossRef CAS.
- M. A. L. Blackie, P. Beagley, S. L. Croft, H. Kendrick, J. R. Moss and K. Chibale, Bioorg. Med. Chem., 2007, 15, 6510–6516 CrossRef CAS PubMed.
- S. Golan, B. S. Aytar, J. P. E. Muller, Y. Kondo, D. M. Lynn, N. L. Abbott and Y. Talmont, Langmuir, 2011, 27, 6615–6621 CrossRef CAS PubMed.
- M. F. R. Fouda, M. M. Abd-Elzaher, R. A. Abdelsamaia and A. A. Labib, Appl. Organomet. Chem., 2007, 21, 613–625 CrossRef CAS.
- G. Caldwell, M. G. Meirim, E. W. Neuse and C. E. J. van Rensburg, Appl. Organomet. Chem., 1998, 12, 793–799 CrossRef CAS.
- E. W. Neuse, J. Inorg. Organomet. Polym., 2005, 15, 3–31 CrossRef CAS PubMed.
- E. W. Neuse, Macromol. Symp., 2001, 172, 127–138 CrossRef CAS.
- A. Gul, Z. Akhter, A. Bhatti, M. Siddiq, A. Khan, H. M. Siddiqe, N. K. Janjua, A. Shaheen, S. Sarfraz and B. Mirza, J. Organomet. Chem., 2012, 719, 41–53 CrossRef CAS PubMed.
- A. Gul, Z. Akhter, M. Siddiq, S. Sarfraz and B. Mirza, Macromolecules, 2013, 46, 2800–2807 CrossRef CAS.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2010, 132, 15068–15071 CrossRef CAS PubMed.
- J. E. Gestwiki, C. W. Cairo, L. E. Strong, K. A. Oetjen and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 14922–14933 CrossRef PubMed.
- C. W. Cairo, J. E. Gestwicki, M. Kanai and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 1615–1619 CrossRef CAS PubMed.
- G. Caldwell, M. C. Meirim, E. W. Neuse, K. Beloussow and W.-C. Shen, J. Inorg. Organomet. Polym., 2000, 10, 93–101 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404–7410 CrossRef CAS PubMed.
- C. F. Hansell, P. Espeel, M. M. Stamenovic, I. A. Barker, A. P. Dove, F. E. D. Du Prez and R. K. O'Reilly, J. Am. Chem. Soc., 2011, 133, 13828–13831 CrossRef CAS PubMed.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- R. C. Pratt, F. Nederberg, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2008, 114–116 RSC.
- D. P. Sanders, K. Fukushima, D. J. Coady, A. Nelson, M. Fujiwara, M. Yasumoto and J. L. Hedrick, J. Am. Chem. Soc., 2010, 132, 14724–14726 CrossRef CAS PubMed.
- T. Tang, X. F. Liu, L. Y. Zhang, X. L. Xu and P. R. Zhan, Synth. Commun., 2000, 30, 1657–1660 CrossRef.
- P. J. Graham, R. V. Lindsey, G. W. Parshall, M. L. Peterson and G. M. Whitman, J. Am. Chem. Soc., 1957, 79, 3416–3420 CrossRef CAS.
- S. Ciampi, P. K. Eggers, L. S. Guillaume, M. James, J. B. Harper and J. J. Gooding, Langmuir, 2009, 25, 2530–2539 CrossRef CAS PubMed.
- A. C. Albertsson and M. J. Sjoeling, J. Macromol. Sci., Pure Appl. Chem., 1992, 43 CrossRef.
- H. R. Kricheldorf, I. Kreiser-Saunders and C. Boettcher, Polymer, 1995, 1253 CrossRef CAS.
- K. J. Zhu, R. W. Hendren, K. Jensen and C. G. Pitt, Macromolecules, 1991, 24, 1736 CrossRef CAS.
- G. Rokick, Prog. Polym. Sci., 2000, 259–342 CrossRef.
- J. r. Koller and R. G. Bergman, Organometallics, 2011, 30, 3217–3224 CrossRef CAS.
- C. Thomas, F. Peruch and B. Bibal, RSC Adv., 2012, 2, 12851–12856 RSC.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- R. J. Williams, I. A. Barker, R. K. O'Reilly and A. P. Dove, ACS Macro Lett., 2012, 1, 1285–1290 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- A. Duda and S. Penczek, in ACS Symposium Series, ed. C. Scholz and R. A. Gross, Washington, DC, 2000, vol. 764, p. 160 Search PubMed.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- M. J. Frisch, et al.Gaussian, Inc., Wallingford, CT, 2010.
- K. N. Houk, A. Jabbari, H. K. Hall and C. Aleman, J. Org. Chem., 2008, 73, 2674–2678 CrossRef CAS PubMed.
- G. Caldwell, M. G. Meirim, E. W. Neuse, K. Beloussow and W.-C. Shen, J. Inorg. Organomet. Polym., 2000, 10, 93–101 CrossRef CAS.
- V. S. Sudhit, C. Venkateswarlu, O. T. Muhammet-Musthafa, S. Sampath and S. Chandrasekaran, Eur. J. Org. Chem., 2009, 2120–2129 CrossRef.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- S. Ciampi, P. K. Eggers, G. Le Saux, M. James, J. B. Harper and J. J. Gooding, Langmuir, 2009, 25, 2530–2539 CrossRef CAS PubMed.
- B. Yucel, B. Sanli, H. Soylemez and H. Akbulut, J. Organomet. Chem., 2012, 704, 49–64 CrossRef CAS PubMed.
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404–7410 CrossRef CAS PubMed.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectroscopy, CV and TGA data. See DOI: 10.1039/c3qi00041a |
| This journal is © the Partner Organisations 2014 |