Transporting platinum drugs from a copper chaperone to ATPase: the mechanistic implication of drug efflux mediated cisplatin resistance†
Dechen
Xu
,
Zhaoyong
Xi
,
Linhong
Zhao
and
Yangzhong
Liu
*
CAS Key Laboratory of Soft Matter Chemistry, CAS High Magnetic Field Laboratory, Department of Chemistry & Collaborative Innovation Center of Suzhou Nano Science and Technology, University of Science and Technology of China, 230026 Hefei, Anhui, China. E-mail: liuyz@ustc.edu.cn; Tel: (+86) 551-63600874
First published on 13th January 2014
Abstract
Cisplatin transfers from Atox1 to ATPase, which could be associated with drug efflux from the cell.
Introduction
Cisplatin is one of the most effective anticancer drugs in the treatment of a wide array of human malignancies.1 However, the clinical effectiveness of cisplatin is limited by the intrinsic and acquired resistance.2 The cellular transportation of cisplatin, including the influx and efflux, is one of the crucial processes associated with the drug resistance.3–5 Several copper proteins, which regulate the cellular copper homeostasis, have been proved to be also involved in the cellular transportation of cisplatin.6 Ctr1 has been proved to facilitate the drug uptake of cisplatin, and the low expression level of Ctr1 has been found in the drug resistant cells.7 P-type ATPase, including ATP7A (also known as Menkes protein, MNK) and ATP7B (also known as Wilson protein, WLN), has been proved to efflux platinum drugs out of cells, and the elevated ATPase level was observed in the drug resistant cells.8–11 It has been demonstrated that the acquisition of cisplatin resistance is accompanied by changes in the cellular pharmacology of copper.12 The cross-resistance of copper and cisplatin further supports the interaction of cisplatin with copper proteins.13Recent investigations reveal that the intracellular copper delivery system is also associated with the drug resistance of cisplatin. Overexpression of Atox1 resulted in higher resistance to cisplatin,14 and the loss of Atox1 reduced the accumulation and cytotoxicity of cisplatin in mouse fibroblasts.15 The crystallographic data show that cisplatin binds to the copper coordinating cysteines (Cys12 and Cys15) of Atox1.16 The platination of Atox1 leads to the unfolding and aggregation of the protein.14,17 The in cell NMR study reveals that the platination of Atox1 also occurs in cells; however, the protein aggregation progress slowly according to NMR spectra.17
The cellular copper homeostasis is maintained by the copper-induced interaction between Atox1 and P-type ATPase, and the relocation of ATPase from the trans-Golgi network to the plasma membrane results in the copper efflux from the cell.18 As both Atox1 and ATPase are involved in the cisplatin resistance, and the metal binding domains (MBD) of ATPase are very similar to Atox1,19 it can be estimated that cisplatin binds to the MBD of ATPase in a similar way to that in the Atox1. Also because the copper induced interaction between Atox1 and ATPase is the basis of the copper transportation in cells, we speculated that the platinum transfer occurs from Atox1 to ATPase, which could be associated with the drug efflux from cells.
To verify this hypothesis, we studied the reaction of cisplatin to the fourth MBD of MNK (MNK4). Results show that cisplatin can directly coordinate to MNK4. The interaction with the cisplatin/Atox1 complex reveals that MNK4 can acquire platinum from Atox1. This result indicates that platinum is able to transfer from Atox1 to ATPase, which could be associated with the drug efflux from the cell.
Results and discussion
To investigate the platination of MNK, we first studied the reaction of the protein with cisplatin. The binding of cisplatin to MNK4 was characterized using ESI-MS (Fig. 1). The reactions were performed on apo-MNK4 with 1.3 molar equivalents of cisplatin. The ESI-MS spectra were recorded directly at different reaction times. The +6 charged peak at 1318.33 corresponds to the apo-MNK4 with molecular weight 7903.98 (calcd 7903.92). Two major peaks from the platinated MNK4 increased with the reaction time. The mass increases indicate the binding of [Pt(NH3)2Cl]+ or [Pt(NH3)2]2+ to the protein (Table S1†). This result is similar to the binding of cisplatin to Atox1,14,17 while the ammine ligands are more stable in the reaction of MNK4. Also similar to the reaction of Atox1,14,20 the platination of MNK4 led to the formation of multimers of this protein as shown in the gel electrophoresis (Fig. S1†).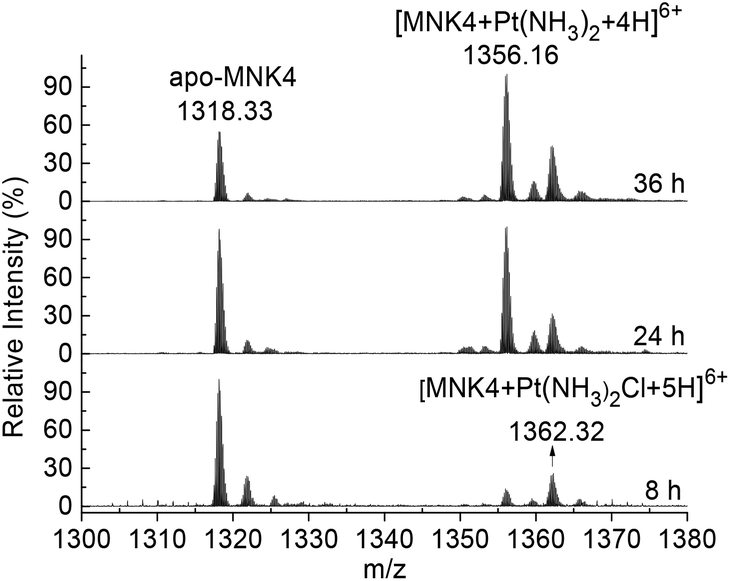 | ||
| Fig. 1 Time dependent ESI-MS spectra of MNK4 in the reaction with cisplatin. The reaction was carried out on apo-MNK4 with 1.3 fold molar equivalents of cisplatin at 25 °C in 20 mM potassium phosphate buffer, pH 7.4, in the presence of 4-fold dithiothreitol (DTT) as a reducing agent. The reaction time is labeled in each spectrum. The +6 charged peaks are shown in the spectra. | ||
The platinum transfer from Atox1 to MNK4 was verified by anion exchange chromatography. Due to the lower isoelectric point (pI) value of MNK4 (4.36) than that of Atox1 (6.68), two proteins can be well separated on the anion exchange column (Fig. S2†). The platination of MNK4 (Pt-MNK4) lowers the retention of the protein on the anion exchange column; therefore the platination level of MNK4 can be detected on the chromatography (Fig. 2).
 | ||
| Fig. 2 Anion exchange chromatography analyzes the platinum transfer to MNK4 from the Pt-Atox1 complex. (A) WT-MNK4; (B) NEM-MNK. The ratios of Pt-Atox1 to MNK4 are labeled in the figure. | ||
The platinated Atox1 (Pt-Atox1) was obtained by incubation of apo-Atox1 with 1.3 fold molar equivalents of cisplatin at 25 °C for 3 h. The excess cisplatin was removed using a Hitrap desalting column. By the incubation of Pt-Atox1 with apo-MNK4 for 10 h at 25 °C, a broad new peak emerged on the anion exchange chromatography, indicating the formation of platinated MNK4 (Fig. 2A). More platinated adducts were generated with increasing the ratio of Pt-Atox1 to MNK4. This result indicates that platinum can transfer from Atox1 to MNK4.
As the copper binding residues are the most potent platination sites, we tested whether the chemical modification of cysteine residues of MNK4 can influence the platinum transfer. The modification of cysteine residues was achieved by alkylation of the thiol group using N-ethylmaleimide (NEM).21 After the incubation of alkylated MNK4 (NEM-MNK4) with Pt-Atox1, the sample was analysed by anion exchange chromatography. In comparison to the reaction of wild type MNK4 (WT-MNK4), the result showed that no new products were generated in the reaction of NEM-MNK4 (Fig. 2B). This result indicated that the alkylation of cysteine in MNK4 blocks platinum transfer from Atox1 to MNK4, which confirmed the reaction of WT-MNK4 and the platination site of cysteine residues.
To prove the platination of MNK4 in the reaction of Pt-Atox1, MNK4 and Pt-MNK4 were collected from the chromatography (Fig. S3†). The ICP-MS analysis clearly indicated the binding of platinum to MNK4, and the amount of platinum bound to MNK4 increases with the ratio of Pt-Atox1 to MNK4 (Fig. 3). This result provides direct evidence of platination of MNK4 in the reaction with Pt-Atox1.
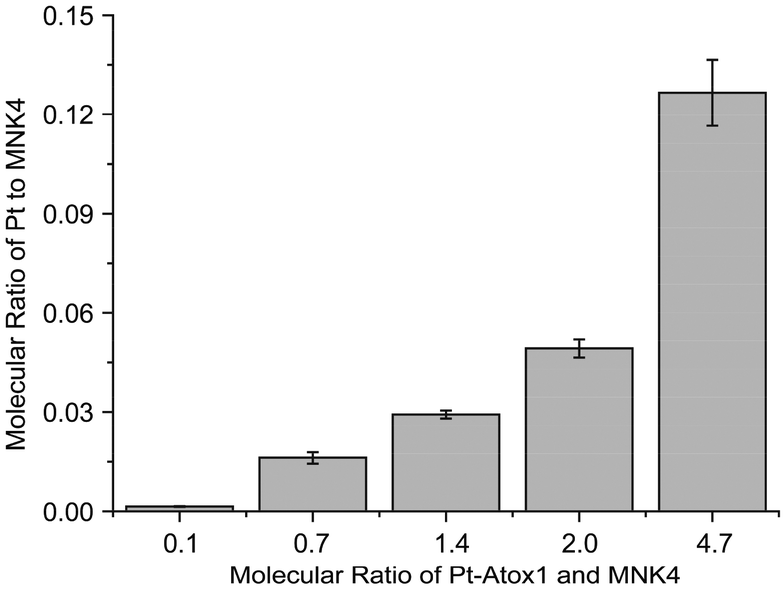 | ||
| Fig. 3 The platinum content in MNK4 after the reaction with Pt-Atox1 in different ratios. The reaction was performed at 25 °C for 10 h, then MNK4 was separated by anion exchange chromatography and the Pt content was determined by ICP-MS. | ||
To characterize the platinated adduct of MNK4 in the reaction with Pt-Atox1, the products were analyzed using ESI-MS. The reaction was performed on apo-MNK4 with Pt-Atox1 in potassium phosphate buffer (20 mM, pH 7.4) for 10 h at 25 °C. After desalting with a Hitrap desalting column, the sample was directly analysed by ESI-MS. The mass spectrum showed a set of multiple charged peaks of the platinated MNK4 (Fig. S4†). The binding of the phosphate ligand to the platinated adducts [MNK4 + Pt(NH3)(PO4)] was a major product as the reaction was performed in phosphate buffer (Fig. 4). The calculated isotope pattern confirmed the assignment (Fig. S5†). This result further proved the platinum transfer from Atox1 to MNK4. Different from the direct platination of MNK4 with cisplatin, the release of one ammine ligand was detected in the reaction of Pt-Atox1. The loss of ammine on platinum often happens when cisplatin coordinates to a sulfur ligand. Apo-Atox1 and apo-MNK4 are still the major peaks on the ESI-MS spectra because the platinated protein has much less ionization efficiency,22 or the release of platinum may occur during the ionization process23,24 (Fig. S4†). The observation of Pt-MNK4 reveals that platinum could be transferred from Atox1 to MNK4.
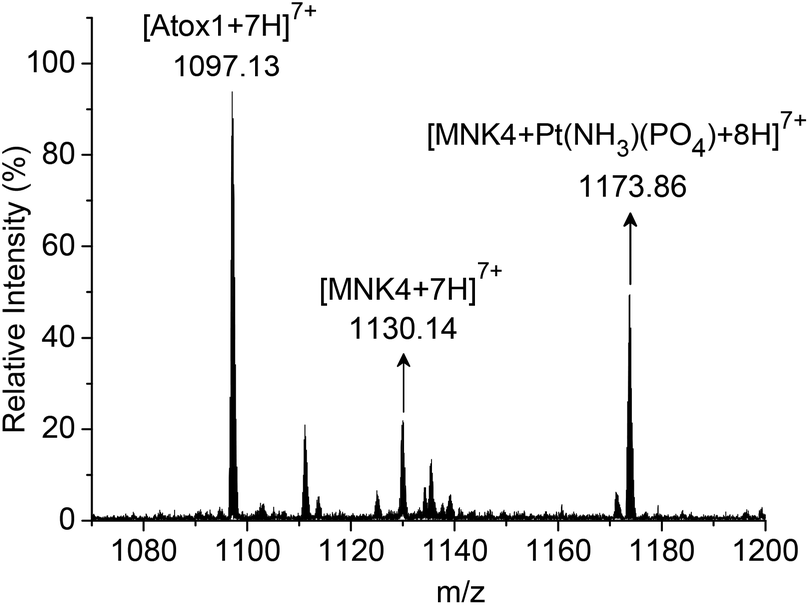 | ||
| Fig. 4 ESI-MS analysis of products from the reaction of MNK4 with the Pt-Atox1 complex. The reaction was performed in 20 mM potassium phosphate buffer at 25 °C for 10 h. The m/z region with +7 charged peaks is shown in the spectra. | ||
The platinum transfer from Atox1 to MNK4 was also identified on chitin columns (Fig. 5). The chitin beads can specifically capture the proteins with a chitin binding domain (CBD) tag. The fusion protein with a CBD tag at the N-terminus of Atox1 (CBD-Atox1) was expressed to study the interaction of Pt-Atox1 and MNK4. The fusion protein can be conveniently separated from the cell lysates on the chitin column. The platinated Atox1 was generated by 3 h reaction of cisplatin with the CBD-Atox1 protein bound to the chitin beads. After removing the excess cisplatin from the column, MNK4 was added for the further reactions. After a 10 h rotation of the column at 25 °C, the supernatant containing MNK4 was separated from the column and the content of Pt in the solution was determined by ICP-MS (Fig. 5). The binding of platinum to MNK confirms the platinum transfer from Atox1 to MNK4. To prove the platination at the copper coordination sites (Cys12 and Cys15) of Atox1, we also performed experiments on the fusion protein with the Cys-to-Ser mutation on these two residues (CBD-Atox1S). The result shows that only about half the amount of platinum was detected in the MNK protein, which confirms the importance of Cys12 and Cys15 in the platinum binding (Fig. 5B). The platinum bound to the CBD-Atox1S fusion protein suggests the non-specific binding of platinum to the protein, either to the Atox1S or to the CBD domain.
 | ||
| Fig. 5 (A) Scheme for the identification of platinum traffic on chitin columns, and (B) the Pt content in the MNK4 solutions after incubation with platinated CBD-Atox1 or CBD-Atox1S captured on chitin columns. | ||
Copper is an essential element and is utilized as a cofactor in a large variety of enzymes. The amount of copper is strictly regulated by the copper homeostasis system as the unsuitable level of copper could lead to diseases.25 The copper transfer from Atox1 to MNK is accomplished through the copper induced protein hetero-dimer, in which the copper ion coordinates to the CXXC motifs of two proteins.26 It has been proved that copper transport proteins, including Atox1 and MNK, are also involved in the subcellular distribution of platinum drugs.6 Due to the similar coordination preference of Pt(II) and Cu(I), platinum could also be transferred through the interaction of Atox1 and MNK. The cisplatin bridged homo-dimer of Atox1 in the crystalline highly supports this hypothesis, as the MBDs of MNK are similar to Atox1.16 ESI-MS data and gel electrophoresis results showed no distinct difference between MNK4 and Atox1 in the reaction with cisplatin. Thus, MNK4 can bind to the cisplatin/Atox1 complex and remove Pt from Atox1. Although it has been postulated that the platinum induced aggregation of Atox1 could be associated with the drug resistance, the protein aggregation occurs rather slowly,17 implying that the platinum transfer ATPase can be more efficient to reduce the drug efficacy. As the DNA is the target of platinum drugs, the binding of cisplatin to ATPase certainly decreases the platination of DNA; in addition, the platinum efflux through ATPase further reduces the cellular accumulation of cisplatin. Recent results showed that Pt-Atox1 also interacts with the second MBD of WLN protein and transfers Pt to the down-stream protein.27,28 This result further supports the hypothesis that the platinum transfer between Atox1 and ATPase could be associated with the platinum efflux from cells, and is correlated to the cisplatin resistance.
Conclusions
In conclusion, the metal binding domain of ATP7A (MNK4) is highly reactive to cisplatin and the conserved copper coordination residues are the favorable binding sites of platinum. The MNK4 also reacts with the platinated Atox1, resulting in the platinum transfer from Atox1 to MNK4. This result indicates that the copper homeostasis system could be also involved in the efflux of platinum drugs and associated with drug resistance of the cell.This work was supported by the National Science Foundation of China (U1332210, 21171156), the National Basic Research Program of China (973 Program 2012CB932502) and USTC-NSRL Association Funding (KY2060190005).
Notes and references
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- M. A. Fuertes, C. Alonso and J. M. Perez, Chem. Rev., 2003, 103, 645–662 CrossRef CAS PubMed.
- M. T. Kuo, H. H. W. Chen, I. S. Song, N. Savaraj and T. Ishikawa, Cancer Metast. Rev., 2007, 26, 71–83 CrossRef CAS PubMed.
- L. Galluzzi, L. Senovilla, I. Vitale, J. Michels, I. Martins, O. Kepp, M. Castedo and G. Kroemer, Oncogene, 2012, 31, 1869–1883 CrossRef CAS PubMed.
- O. Y. Dmitriev, Biochem. Cell Biol., 2011, 89, 138–147 CrossRef CAS PubMed.
- R. Safaei, Cancer Lett., 2006, 234, 34–39 CrossRef CAS PubMed.
- A. K. Holzer, K. Katano, L. W. J. Klomp and S. B. Howell, Clin. Cancer Res., 2004, 10, 6744–6749 CrossRef CAS PubMed.
- M. Komatsu, T. Sumizawa, M. Mutoh, Z. S. Chen, K. Terada, T. Furukawa, X. L. Yang, H. Gao, N. Miura, T. Sugiyama and S. Akiyama, Cancer Res., 2000, 60, 1312–1316 CAS.
- K. Nakayama, A. Kanzaki, K. Terada, M. Mutoh, K. Ogawa, T. Sugiyama, S. Takenoshita, K. Itoh, N. Yaegashi, K. Miyazaki, N. Neamati and Y. Takebayashi, Clin. Cancer Res., 2004, 10, 2804–2811 CrossRef CAS.
- G. Samimi, N. M. Varki, S. Wilczynski, R. Safaei, D. S. Alberts and S. B. Howell, Clin. Cancer Res., 2003, 9, 5853–5859 CAS.
- G. Samimi, R. Safaei, K. Katano, A. K. Holzer, M. Rochdi, M. Tomioka, M. Goodman and S. B. Howell, Clin. Cancer Res., 2004, 10, 4661–4669 CrossRef CAS PubMed.
- K. Katano, A. Kondo, R. Safaei, A. Holzer, G. Samimi, M. Mishima, Y. M. Kuo, M. Rochdi and S. B. Howell, Cancer Res., 2002, 62, 6559–6565 CAS.
- R. Safaei, K. Katano, G. Samimi, W. Naerdemann, J. L. Stevenson, M. Rochdi and S. B. Howell, Cancer Chemother. Pharmacol., 2004, 53, 239–246 CrossRef CAS PubMed.
- M. E. Palm, C. F. Weise, C. Lundin, G. Wingsle, Y. Nygren, E. Bjorn, P. Naredi, M. Wolf-Watz and P. Wittung-Stafshede, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6951–6956 CrossRef CAS PubMed.
- R. Safaei, M. H. Maktabi, B. G. Blair, C. A. Larson and S. B. Howell, J. Inorg. Biochem., 2009, 103, 333–341 CrossRef CAS PubMed.
- A. K. Boal and A. C. Rosenzweig, J. Am. Chem. Soc., 2009, 131, 14196–14197 CrossRef CAS PubMed.
- F. Arnesano, L. Banci, I. Bertini, I. C. Felli, M. Losacco and G. Natile, J. Am. Chem. Soc., 2011, 133, 18361–18369 CrossRef CAS PubMed.
- M. A. Cater, S. La Fontaine, K. Shield, Y. Deal and J. F. B. Mercer, Gastroenterology, 2006, 130, 493–506 CrossRef CAS PubMed.
- I. Anastassopoulou, L. Banci, I. Bertini, F. Cantini, E. Katsari and A. Rosato, Biochemistry, 2004, 43, 13046–13053 CrossRef CAS PubMed.
- R. Safaei, P. L. Adams, M. H. Maktabi, R. A. Mathews and S. B. Howell, J. Inorg. Biochem., 2012, 110, 8–17 CrossRef CAS PubMed.
- M. W. Crankshaw and G. A. Grant, in Current Protocols in Protein Science, John Wiley & Sons, Inc., 2001 Search PubMed.
- C. Li, R. Huang, Y. Ding, E. Sletten, F. Arnesano, M. Losacco, G. Natile and Y. Liu, Inorg. Chem., 2011, 50, 8168–8176 CrossRef CAS PubMed.
- T. G. Schaaff, Y. Qu, N. Farrell and V. H. Wysocki, J. Mass Spectrom., 1998, 33, 436–443 CrossRef CAS.
- L. Sleno and D. A. Volmer, J. Mass Spectrom., 2004, 39, 1091–1112 CrossRef CAS PubMed.
- J. R. Prohaska, Am. J. Clin. Nutr., 2008, 88, 826s–829s CAS.
- L. Banci, I. Bertini, F. Cantini, C. T. Chasapis, N. Hadjiliadis and A. Rosato, J. Biol. Chem., 2005, 280, 38259–38263 CrossRef CAS PubMed.
- N. V. Dolgova, S. Nokhrin, C. H. Yu, G. N. George and O. Y. Dmitriev, Biochem. J., 2013, 454, 147–156 CrossRef CAS PubMed.
- M. E. Palm-Espling, C. D. Andersson, E. Bjorn, A. Linusson and P. Wittung-Stafshede, PLoS One, 2013, 8, e70473 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, ESI-MS, gel electrophoresis and anion exchange chromatography. See DOI: 10.1039/c3qi00068k |
| This journal is © the Partner Organisations 2014 |