N-heterocyclic silylene complexes in catalysis: new frontiers in an emerging field
Burgert
Blom
,
Daniel
Gallego
and
Matthias
Driess
*
Department of Chemistry: Metalorganics and Inorganic Materials, Technische Universität Berlin, Strasse des 17. Juni 135, Sekr. C2, D-10623 Berlin, Germany. E-mail: matthias.driess@tu-berlin.de
First published on 13th January 2014
Abstract
The present account is a review of all N-heterocyclic silylene (NHSi) transition metal complexes that have been employed in catalytic transformations, reported up to the present time (2013). NHSi transition metal complexes now enjoy indefatigable attention since their facile isolation was realised by the report of the first isolable NHSis by West and Denk in 1994. Despite considerable research activity since then, in comparison to ubiquitous N-heterocyclic carbene (NHC) complexes, NHSi complexes are still comparatively rare. Accordingly, in comparison to the plethora of reports associated with NHC complexes, implicated in catalytic processes, only scant examples exist for NHSi complexes. Some of these reports include Heck or Suzuki type coupling, alkyne cyclotrimerisation, ketone hydrosilylation, amide reduction or Sonogashira cross-coupling reactions, and are discussed in detail here. These endeavours pave the way for new families of catalysts based on NHSis and highlight the potential future applications of these emerging and rather unexplored complexes in novel catalytic processes.
 Burgert Blom | Burgert Blom obtained a Ph.D. degree from the University of Bonn (Germany) in 2011 in the field of metal–main group multiple bonding. He was awarded the Dr Edmund |
 Daniel Gallego | Daniel Gallego obtained a bachelor's degree with honours in Colombia from Universidad Nacional de Colombia. He then joined the joint Erasmus Mundus Master program ASC (Advanced Spectroscopy in Chemistry) in Lille (France) and Leipzig (Germany) where he obtained his Master's degree in 2011. Currently, he is a doctoral student in the group of Prof. Driess, exploring the use of novel bidentate NHSi and NHGe ligands as new scaffolds in supporting metals for catalysis. |
 Matthias Driess | Matthias Driess obtained his Ph.D. degree in 1988 and completed his habilitation at the University of Heidelberg (Germany) in 1993. Since 2004 he is full professor at the Department of Chemistry of the Technische Universität Berlin (Germany). Since 2007 he serves as spokesperson of the Cluster of Excellence UniCat in the Berlin area. He received several awards, including the Wacker Silicone Award in 2010, and is a member of the German National Academy of Sciences (Leoplodina). He has published more than 240 papers. |
Introduction
Since the seminal work on the first N-heterocyclic silylene (NHSi) complex in 1977 by Welz and Schmid,1 which was a thermolabile iron complex (Scheme 1), many subsequent NHSi complexes across the transition metals have been reported. Despite these impressive efforts, in comparison, the number of reported N-heterocyclic carbene (NHC) stabilised complexes still far out-weighs those based on NHSi ligands. This fact is most likely precipitated by the prevalence of the latter in key catalytic processes and their use in stabilising metal complexes in unusual oxidation states.2 | ||
| Scheme 1 Seminal example of an NHSi transition metal complex in 1977. | ||
We have recently reported a comprehensive review of all existing N-heterocyclic silylene (NHSi) complexes of group IV through XII transition metal elements.3 We showed that these complexes represent an exciting new frontier which lies at the interface of ‘classical’ organometallic and contemporary main group chemistry, but can still be considered an emerging field. These emerging complexes have the ability to activate a range of small molecules in unique ways, highlighting their importance. Moreover, they possess the propensity to perform other interesting stoichiometric transformations, such as alkyne hydrosilylation mediated by a nickel based NHSi ligand, or silane activation, facilitated by a Ru NHSi complex, in addition to various other transformations.
In the present report, we wish to present a focussed and detailed discussion on NHSi complexes so far implicated exclusively in catalytic transformations, reported up to the present time (2013), and we will do so from a chronological perspective. Our group has been intensively involved in NHSi research in the last decade, and indeed most (but not all) of the examples of catalytically active NHSi complexes discussed here have evolved from these efforts. Only very few such examples exist to date, but these nevertheless show a “proof of concept” that NHSi ligands indeed hold promise as a new class of ligands likely to play an increasingly important role in future catalytic developments, and are certainly more than simple isoelectronic replacements for more traditional ligands.
Development of NHSi complexes as catalysts
Preliminary considerations
Transition metal silylene complexes can be classified into four distinct classes (Chart 1). Type A are analogues of Fischer or Schrock type carbene/alkylidene complexes (LnM = CR2), for example [(CO)4Os![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si{SpTol}{Ru(η5-C5Me5)}] or [PtH(PCy3)2Si(SEt)2]+BPh4− reported by Tilley and co-workers in the 1990s.4 Type B represents “base stabilised” silylene ligands coordinated to transition metal fragments, exemplified by the seminal work of Zybill and co-workers: [(CO)4Fe{Si(OtBu)2←Donor}] (Donor = THF or HMPA).5 Members of type C represent NHSi complexes where unsaturated functionalities and/or additional R groups may exist in the ligand backbone, while type D represents NHSi halide or hydride complexes (Chart 2). We concentrate our discussion in this review on complexes belonging to classes C and D exclusively that have been shown to effect catalytic transformations.
Si{SpTol}{Ru(η5-C5Me5)}] or [PtH(PCy3)2Si(SEt)2]+BPh4− reported by Tilley and co-workers in the 1990s.4 Type B represents “base stabilised” silylene ligands coordinated to transition metal fragments, exemplified by the seminal work of Zybill and co-workers: [(CO)4Fe{Si(OtBu)2←Donor}] (Donor = THF or HMPA).5 Members of type C represent NHSi complexes where unsaturated functionalities and/or additional R groups may exist in the ligand backbone, while type D represents NHSi halide or hydride complexes (Chart 2). We concentrate our discussion in this review on complexes belonging to classes C and D exclusively that have been shown to effect catalytic transformations.
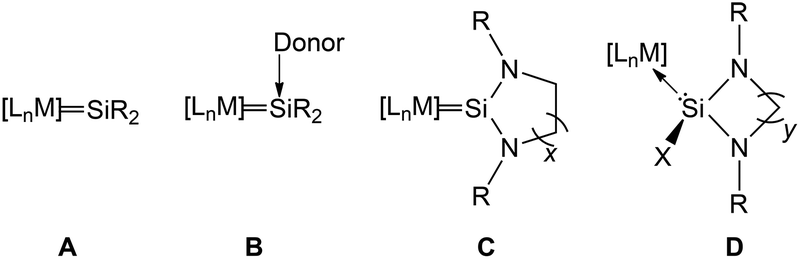 | ||
| Chart 1 Transition metal silylene complexes categorised in four classes: in type C x = 1 or 2; R = bulky aromatic or aliphatic group; in type D y = 1, 2 or 3; X = a halogen or H; R = bulky aromatic or aliphatic group (adapted from ref. 3). | ||
 | ||
| Chart 2 “Free” NHSi metallylenes as a new class of supporting ligands for new catalysts. | ||
Chart 2 provides an overview of the existing NHSi metallylene ligands, some of which have been employed in the synthesis of catalytically active metal complexes, and are used throughout the discussion in this review.6
A chronological account of catalytically active NHSi complexes
The first appearance of an NHSi complex implicated in a catalytic process was that by Fürstner and co-workers in 2001. They reported a novel dinuclear NHSi Pd complex prepared by treatment of 1 with Pd(PPh3)4 affording the complex [Pd2(μ2-1-)2(PPh3)2] (9).7 In this seminal work, two NHSi (1) ligands act as bridging ligands between the palladium centres forming a diamond-like core. Moreover, the dinuclear complex 9 was found to be effective in the Suzuki cross-coupling reaction of aryl boronic acids with bromoarenes (Scheme 2). | ||
| Scheme 2 Seminal example of an NHSi complex (9) effectively enabling Suzuki cross-coupling reactions. | ||
For the first time the ability of an NHSi to act as supporting ligands in a catalytically active complex was, perhaps fortuitously, uncovered.
It would take several more years for the next report to appear in this new area established by Fürstner and co-workers. In fact, only some seven years later did Roesky and co-workers communicate the second example of a catalytically active NHSi complex. They reported a mononuclear Pd-based NHSi complex obtained by reacting the NHSi 1 with the Pd precursor [Pd(η3-C3H5)Cl]2 affording [PdCl(η3-C3H5)(←:1)] (10) in moderate yields (54%) (Scheme 3). Complex 10 was also found to be effective in the Heck coupling of styrene and bromoacetophenone.8 They also report a systematic study showing the effect of temperature on the yield of the coupled product, where at 140 °C, a 99% yield is achieved in 24 h, compared to 45% achieved in the same time at 80 °C.
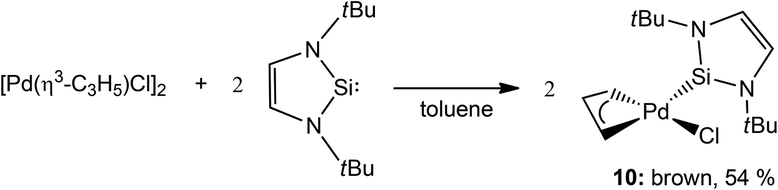 | ||
| Scheme 3 Synthesis of the second example of an NHSi complex (10) as an active Heck coupling pre-catalyst. | ||
The effect of electron withdrawing/donating substituents on the catalyst rates was also systematically investigated by performing Heck coupling of various aromatic compounds with styrene. It was found that electron donating substituents on the aromatic ring generally afford lower yields of the coupled product, while electron withdrawing substituents result in higher yields.
Besides these two important examples, the remainder of the reports in this area stems from our group, given our prolonged interest in NHSis, and these reports have appeared recently and somewhat simultaneously.
Accordingly, we first reported NHSi cobalt complexes, useful in catalysis, using our bidentate NHSi ligand, 8, as a supporting chelating or even mono-dentate ligand.6g The synthesis is straightforward and involves the reaction of Na[C5H5] with anhydrous CoBr2, in the presence of a reducing agent (KC8), along with 8 (Scheme 4). This procedure affords the complex [(η5-C5H5)Co(η2-8)] (11), which features a resonance signal in the 29Si NMR spectrum at δ = 82.0 ppm, and is downfield shifted from that of the free ligand (δ = 43.3 ppm) 8, as expected for coordination. In close analogy, the Ge analogue of 11 can also be accessed synthetically (12).
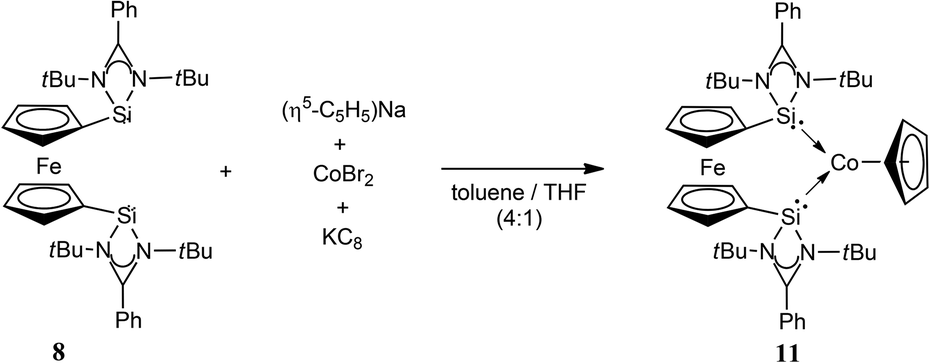 | ||
| Scheme 4 Synthesis of the novel cobalt-based bis-NHSi complex 11. The Ge analogue (12) can also be accessed in an analogous way. | ||
Complex 11 is catalytically active in the [2 + 2 + 2] cyclotrimerisation reaction of phenylacetylene, yielding two isomeric forms of triphenylbenzene (Scheme 5), while surprisingly 12 is inactive catalytically. A plausible reason was proposed for the inactivity of the latter: stronger coordination of the Ge atoms to Co, thereby hampering alkyne coordination to the Co centre, an obvious key step in the catalytic process. This seemed reasonable given the lack of detailed mechanistic studies on this system in this report. However, it was shown in a subsequent study (vide infra) that Si is more strongly coordinating, so most likely the catalytic inactivity of 12 is a consequence of instability under the catalytic conditions, rather than an effect of coordination strength.
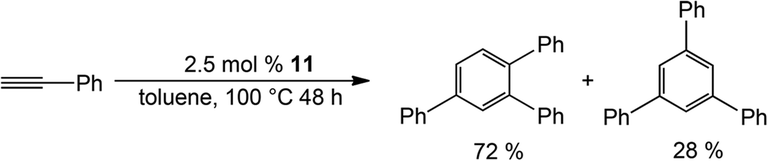 | ||
| Scheme 5 Catalytic cyclotrimerisation using bis-silylene Co complex 11 as a pre-catalyst. | ||
Remarkably, repeating the cyclotrimerisation catalysis of phenylacetylene in CH3CN as a solvent, mediated by 11 as a catalyst, affords substituted pyridines in moderate yields (Scheme 6).
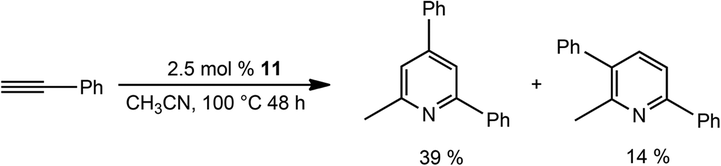 | ||
| Scheme 6 Catalytic formation of pyridines using bis-silylene Co complex 11 as a pre-catalyst in CH3CN. | ||
The reaction of silylene 8 with two molar equivalents of [(η5-C5H5)Co(CO)2] in hexane accordingly affords the related dinuclear Co complex 13, with concomitant loss of two equivalents of CO (Fig. 1). However, in the case of complex 13, no catalytic studies were undertaken, but it principally also represents a potentially active catalyst precursor, and could facilitate mechanistic investigations into this catalyst process.
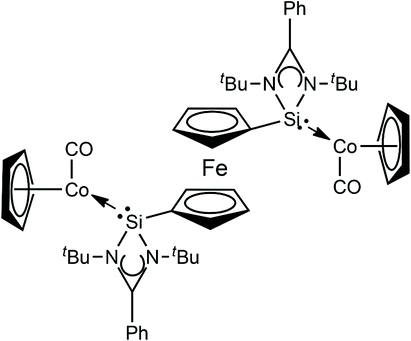 | ||
| Fig. 1 Structure of dinuclear Co complex 13 based on NHSi 8. | ||
The X-ray structure of complex 11 was in accord with the spectroscopic findings and is shown in Fig. 2. The rather short Co–Si bond lengths (2.1252(14) and 2.1200(14) Å respectively) hint at a rather strong σ-donor ability of ligand 8.
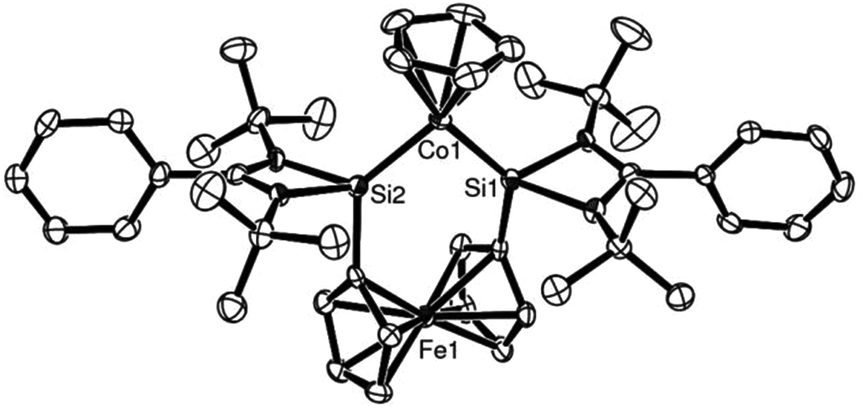 | ||
| Fig. 2 ORTEP representation of complex 11: an active alkyne cyclotrimerisation catalyst precursor. (Thermal ellipsoids at 50% probability, H atoms omitted for clarity.) | ||
On continued exploration of the versatility of the NHSi 4, the first pincer bis-NHSi was obtained in our group by its salt metathesis reaction. We explored the coordination ability of 7 to an iridium centre, obtaining the octahedral complex [SiCSi]IrHCl(coe) (14) by oxidative addition of an aromatic C–H bond in 7 with 0.5 molar equivalent of [IrCl(coe)2]2 (Scheme 7). In this work, we found that complex 14 served as a pre-catalyst for the C–H borylation of arenes using pinacolborane (HBPin).9 For comparative purposes, we also prepared a series of iridium pincer complexes with the Ge analogue (15) and two phosphane-based ligands: tBu2P and the more electronically related (iPrNCH2)2P (17 and 18 respectively, Scheme 8). The catalytic evaluation of this series showed an interesting contrast between the metallylenes versus the phosphane based pincer ligand systems. We found that both 14 and 15 are substantially more effective in this catalytic transformation compared to the isoelectronic P(III) analogues 17 and 18 (Table 1), highlighting the fact that the use of Si or Ge as donor ligands plays a substantial role in the catalytic activity, and are not simple isoelectronic substitutes. The latter (Si and Ge) are much stronger σ-donor ligands, which tune the catalytic activity in this key catalytic process.
 | ||
| Scheme 7 Synthesis of the iridium NHSi complex 14, a pre-catalyst active in the C–H borylation of arenes using HBPin. | ||
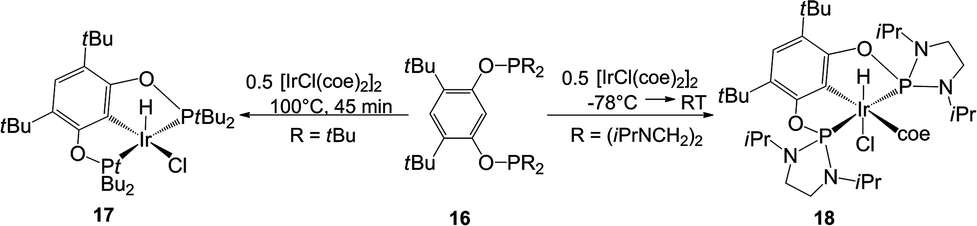 | ||
| Scheme 8 Synthesis of the iridium P(III) analogues of complex 14, for comparison in the catalytic C–H borylation of arenes. | ||
|
|
||||
|---|---|---|---|---|
| Arene | 14 | 15 | 17 | 18 |

|
ArBPin: 90 | ArBPin: 80 | ArBPin: 64 | ArBPin: 21 |
| coaBPin: 3 | coaBPin: 9 | coaBPin: 11 | coaBPin: 35 | |
| coeBPin: 1 | coeBPin: 8 | coeBPin: 7 | coeBPin: — | |
| ArBPin: 53 | ArBPin: 46 | ArBPin: 60 | ArBPin: 55 | |
|
ArBPin: 91 | ArBPin: 39 | ArBPin: 40 | ArBPin: 9 |
(m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :p = 1.6:1) :p = 1.6:1) |
(m:p = 1.5:1) |
(m:p = 1.4:1) |
(m:p = 1.4:1) |
|
| coaBPin: 3 | coaBPin: 8 | coaBPin: 49 | coaBPin: 34 | |
| coeBPin: 3 | coeBPin: 7 | coeBPin: 10 | coeBPin: — | |
| ArBPin: 16 | ArBPin: 12 | ArBPin: 9 | ArBPin: 20 | |
|
ArBPin: 15 | ArBPin: 4 | ArBPin: 10 | ArBPin: 11 |
| coaBPin: 19 | coaBPin & | coaBPin & | coaBPin: 25 | |
| coeBPin: 26 | coeBPin: <5 | coeBPin: <5 | coeBPin: — | |
| ArBPin: 11 | ArBPin: 5 | ArBPin: 8 | ArBPin: 23 | |

|
ArBPin: 11 | ArBPin: 6 | ArBPin: 17 | ArBPin: 5 |
| coaBPin: 30 | coaBPin & | coaBPin & | coaBPin: 20 | |
| coeBPin: 50 | coeBpin: <5 | coeBpin: <5 | coeBpin: — | |
| ArBPin: 10 | ArBPin: 3 | ArBPin: 4 | ArBPin: 19 | |

|
ArBPin: 0 | ArBPin: 0 | — | — |
| — | — | — | — | |

A positive effect on the turnover frequency (TOF) and number (TON) was obtained while an additional equivalent of the co-ligand/hydrogen acceptor COE was added to the catalytic system using 14 and 15 as pre-catalysts. For instance, when benzene was used as the substrate the TON increased ca. twice its value (TON: 9–10 without COE and 16–18 with COE). On the other hand, this effect was higher on the TOF after 3 h of reaction, since almost all of the substrate was consumed in the presence of COE (TOF3h: 1.3 h−1 without COE and 5.3 h−1 with COE). This result suggested that COE plays a crucial role in kinetic stabilization of the active species. In contrast, addition of COE to the C–H borylation reactions using 17 or 18 as pre-catalysts either had little (17) or a detrimental effect on the overall catalytic performance, affecting the selectivity of the reaction towards borylation of the olefin (18) (Table 1).
A general landscape for the mechanism is depicted in Scheme 9. The activation of the pre-catalyst occurs in the presence of HBPin to produce the dihydride species which undergoes concomitant hydrogenation of COE to produce the Ir(I) active species. An oxidative addition of HBPin to Ir(I) occurs subsequently to activate the aromatic C–H bond, forming ArBPin and {Ir(III)(H)2} either by oxidative addition/reductive elimination or a σ-bond metathesis reaction. Finally, the Ir(I) active species can be obtained either by (a) release of H2 or (b) COE coordination to the free site of this species and its further hydrogenation. A competitive reaction is present where a borylation of the olefin occurs instead. The difference in activity of 14 and 15 comes from the fact that in using bis-NHSi or bis-NHGe, the Ir centre is more electron-rich than with the phosphine ligands, increasing the activation barrier for the reductive elimination to produce H2. Thus, addition of COE produces a beneficial effect on the catalytic performance.
 | ||
| Scheme 9 Proposed mechanism for the C–H borylation of arenes by [ECE] Ir-pincer complexes (E = Si, Ge, P). | ||
In addition to the difference in reactivity of pincer metallylene ligands versus the well known phosphine ligands, X-ray structural (Fig. 3) and NMR spectroscopic analyses for complexes 14, 15, 17 and 18 revealed the markedly stronger σ-donor properties of bis-NHSi (Table 2). According to the Chatt–Dewar–Ducanson bonding model, when the electronic density on a metal center increases, the π-backbonding to the CC bond is more pronounced. This drives to an elongation on the olefinic bond as well as a high-field chemical shift in the NMR spectra for both, the protons and 13C nuclei involved. Therefore, the order of σ-donor strength on this series of pincer ligands could be drawn as follows: SiCSi > GeCGe > iPrN-PCP > tBu-PCP.
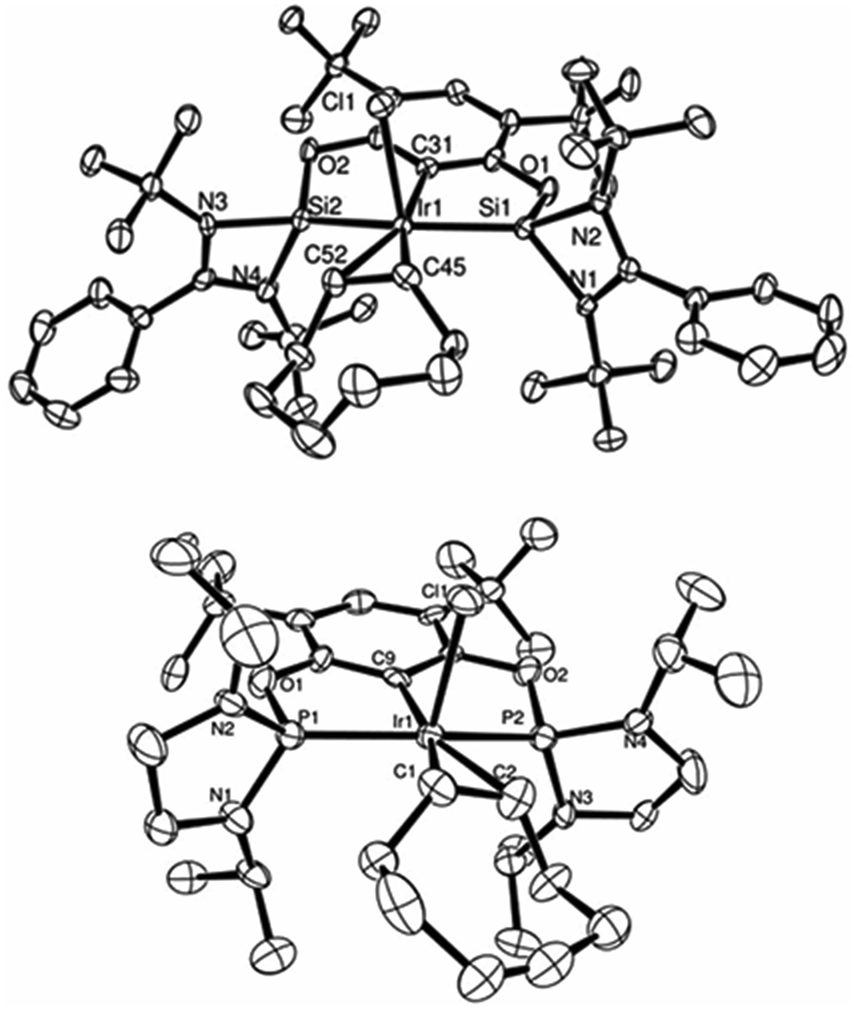 | ||
| Fig. 3 ORTEP representation of complexes 14 (top) and 18 (bottom): pre-catalysts for the C–H borylation of arenes using HBPin. (Thermal ellipsoids at 50% probability, H atoms omitted for clarity.) | ||
C on the [ECE]IrHCl(coe) complexes (E = Si, Ge, P)
| Analytic method | 14 | 15 | 18 |
|---|---|---|---|
| X-ray diffraction (XRD) d(CC) (Å) |
1.409(9) | — | 1.35(1) |
|
1H NMR CC–H |
3.38 | 4.06 | 4.31 |
|
31C NMR CC |
55.8 | 65.1 | 81.6 |
Based on the interesting properties presented by the complexes 14 and 15 with iridium as the metal centre, we expanded the scope to group 10, specifically nickel as a non-precious transition metal centre. The nickel pincer complexes with 7 as ligand the bis-germylene analogue and isoelectronic bis-phosphane ligands (19, 20 and 21 respectively) were obtained by a C–H activation with NiBr2(dme) as a precursor (Scheme 10 and Fig. 4) in high yields under an excess of a base.10
 | ||
| Scheme 10 Synthesis of complexes 19–21. | ||
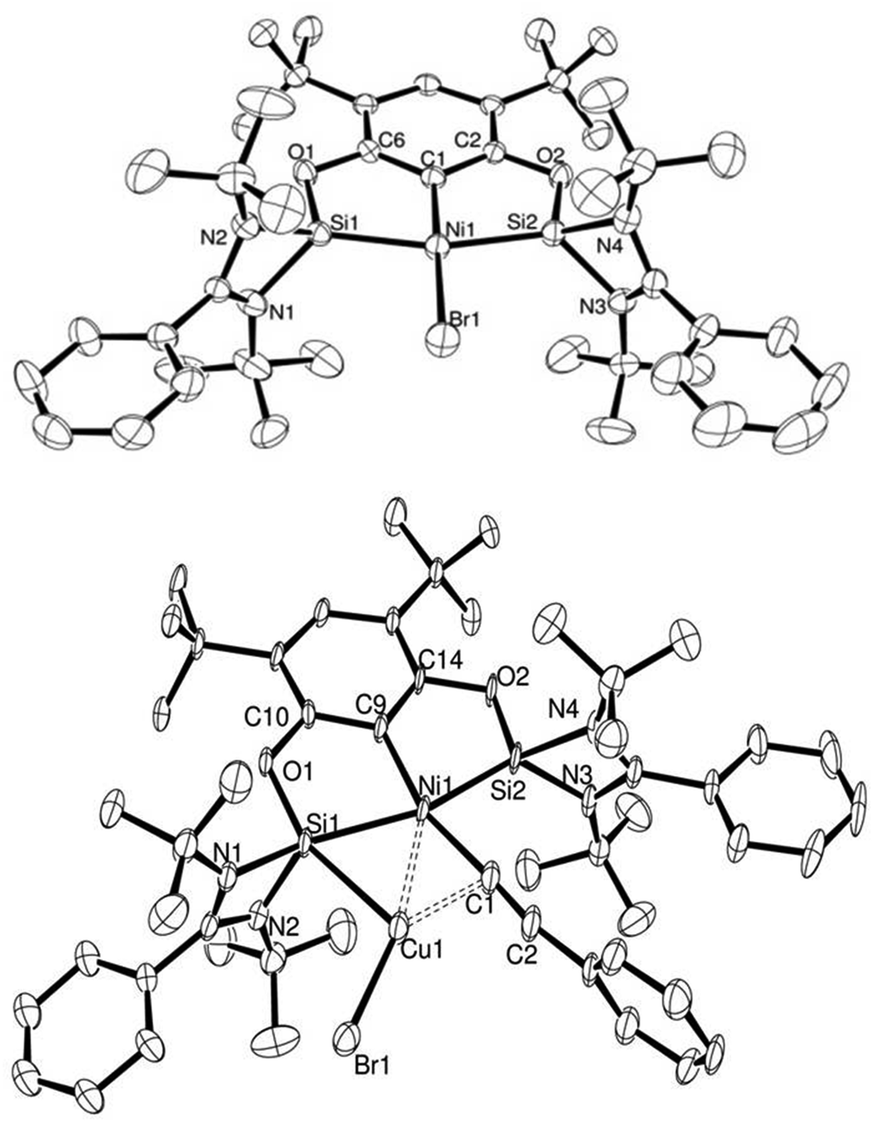 | ||
| Fig. 4 ORTEP representation of complex 19 (top) and 22 (bottom). Thermal ellipsoids set at 50% probability level, H atoms omitted for clarity. | ||
Interestingly, we observed that these complexes can act as a catalyst for the Sonogashira cross-coupling reaction between phenylacetylene and 1-octenyl iodide in moderate yields (Scheme 11). Direct comparison with a blank test, without any catalyst but with added CuI showed that indeed the nickel complexes are the active coupling catalyst. In this case just stoichiometric formation of the coupling product was induced by the Cu(I) salt.
 | ||
| Scheme 11 Catalytic evaluation of complexes 19–21 in the Sonogashira cross-coupling reaction. | ||
Variations on the catalytic conditions were tested without any improvement neither on the TON nor on the TOF. These pre-catalysts represent additional examples of the rather rare nickel-catalysed Sonogashira cross-coupling reaction,11 making them somewhat remarkable. Moreover, the catalytic activity is in the same range of the phosphine-based catalyst, suggesting that most probably the medium activity does not depend on the sensitivity of the catalytic system. We next sought to understand in more detail whether these complexes were stable under the different chemical transformations potentially occurring in the catalytic cycle, and thereby shed light on the mechanism.
We evaluated our system through the sequence of transmetallation→oxidative addition→reductive elimination in stoichiometric amounts and analysed, when possible, the reaction intermediates. Transmetallation with copper acetylides and the nickel complexes showed an equilibrium reaction between reagents and products. More importantly, we found that these ligands enabled the isolation and structural characterisation of the transmetallation product, in which the formed CuBr is bound to one of the NHSi arms and the acetylide ligand (22: {7:→Ni–CC–Ph→CuBr}) (Fig. 4).
We also evaluated this unusual structure through detailed DFT calculations focusing on the Electron Localization Function (ELF) and Mayer Bond Order (MBO). These results showed a two-electron three-centered system between Cu–Si–Ni (Scheme 12). In addition, the CuBr is coordinated in an end-on fashion to the acetylide coligand according to the MBO values.
 | ||
| Scheme 12 ELF plot in the main Ni coordination plane for [7:→Ni–CC–Ph→CuBr] (22) (adapted from ref. 10). | ||
For the further analysis of the next step (oxidative addition→reductive elimination), we reacted the in situ generated 22 with three molar equiv. of E-1-octenyl iodide at 50 °C for 2 h. By NMR analysis we found the formation of the coupling product in high yields (>90%) and a compound with similar features to the starting catalyst. Independent synthesis of the expected [SiCSi]NiI complex (23) (see Scheme 13) showed that the species formed are in accord with an oxidative addition→reductive elimination pathway. The reaction intermediate for this chemical transformation was elusive, although the retention of the stereochemistry on the CC bond on the product suggested that this is the most likely pathway.
 | ||
| Scheme 13 Proposed catalytic pathway for the Sonogashira cross-coupling mediated by 19 and 20 (pincer ligand is represented as E–C–E, E = Si, Ge). | ||
Based on these stoichiometric studies a general reaction mechanism can be depicted as follows (Scheme 13): the [ECE]NiBr complexes react with copper phenylacetylide producing the adducts {[ECE]Ni–CC–Ph→CuBr} (22). The latter reacts further with the (E)-1-octenyl iodide either by (a) oxidative addition–reductive elimination or (b) radical pathway. At the end the product (E)-dec-3-en-1-ynylbenzene is liberated and the cycle is closed.
Around the same time as the aforementioned reports from our group, we also focussed our attention on iron, particularly given its abundance, low cost and recent burgeoning interest in its use in other catalytic processes not based on NHSis.12 To our surprise, only three iron-based NHSi complexes had been previously reported,13 all bearing the electron withdrawing {Fe(CO)4} moiety, and no catalytic transformation mediated by these complexes had been reported. This apparent gap in the literature encouraged us to explore a potentially more electron-rich iron fragment, other than {Fe(CO)4}, to which an NHSi could potentially coordinate, and thereby, due to the increase in electron density at Fe, increasing the reactivity of the emerging complex.
In this regard, we found the precursor complex [Fe(dmpe)2(PMe3)] (24),14 (dmpe = 1,2-bis(dimethylphosphino)ethane) a particularly attractive candidate since it has been shown to undergo facile PMe3 loss upon reaction with a variety of other unsaturated low-valent main group compounds, affording multiply bonded Fe–E (E = Ge or Sn) adducts.15 Inspired in part by this, we carried out the reaction of 24 with NHSi 4 which indeed affords by PMe3 loss the corresponding NHSi complex [(dmpe)2Fe←:4] (25) (Scheme 14) as the first example of an electron-rich iron NHSi complex. Moreover, this complex can readily be converted by hydride–halogen exchange to the hydrido silylene complex 26 (Scheme 14), representing a very rare example of a Si(II) hydride coordinated to a metal centre.16
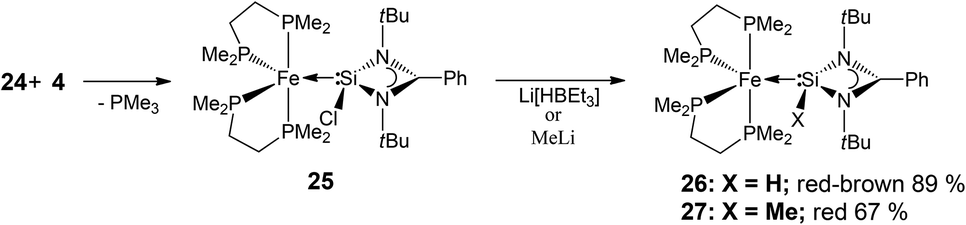 | ||
| Scheme 14 Synthesis of the NHSi iron complex 25, using a PMe3 elimination strategy, and subsequent H, Cl or Me exchange with Superhydride© and CH3Li affording the hydrido and methylated NHSi-complexes 26 and 27, respectively. | ||
Complexes 25 and 26 both exhibit trigonal bipyramidal geometries in the solid state, according to single crystal XRD studies (Fig. 5 and 6). The NHSi ligands reside in one of the equatorial positions which is a preferred position for a π-acidic ligand in a trigonal bipyramidal coordination sphere, suggesting possible π-back-bonding from Fe to Si in both complexes (Table 3). Moreover, the Fe–Si bond lengths in both complexes are comparatively short, and in fact comparable to other reported iron silylene complexes of the type LnFeSiR2 (2.154(1) Å, in complex Cp*COTMSFeSiMes2; Cp*: 1,2,3,4,5-pentamethylcyclopentadienyl; TMS: SiMe3; Mes: 2,4,6-trimethylphenyl).17
 | ||
| Fig. 5 ORTEP representation of complex 25: Thermal ellipsoids at 50% probability, H atoms omitted for clarity (adapted from ref. 16). | ||
 | ||
| Fig. 6 ORTEP representation of one of the stereoisomers found in the asymmetric unit of complex 26: thermal ellipsoids at 50% probability, H (except H1) atoms and carbon atoms of the dmpe ligands omitted for clarity (adapted from ref. 16). | ||
| d(Fe–Si)/Å | d(Si–X)/Å | d(Si–N1)/Å | d(Si–N2)/Å | |
|---|---|---|---|---|
| 25 | 2.1634(9) | 2.2281(11) | 1.920(2) | 1.931(2) |
| 26 | 2.184(2) | 1.50(2) | 1.930(6) | 1.915(7) |
Complex 26 exhibits substantial statistical disorder in the X-ray structure solution, which was resolved as two independent stereoisomers in the asymmetric unit. This phenomenon is likely attributable to an increase in the energy barrier of the Berry pseudorotation (BSR), which affords two coexisting stereoisomers, under crystallisation conditions at low temperatures. This is not the case with its precursor, complex 25, highlighting a difference in the behaviour of the two complexes. The increase in the BSR barrier was additionally elucidated by 31P{1H} NMR spectra where at room temperature a relatively sharp singlet signal is observed for complex 25, suggesting fluxional exchange of the Paxial and Pequitorial atoms on the NMR time scale. Complex 26, on the other hand at the same measuring frequency and temperature, exhibits a very broad signal in the 31P{1H} NMR spectrum, pointing to a substantial increase in the energy requirements for the Paxial and Pequitorial exchange on NMR time scale.18 Moreover, progressive sharpening of this signal occurs on increasing the temperature as shown by variable temperature NMR investigations, which were also carried out (Fig. 7).
 | ||
| Fig. 7 Variable temperature 31P{1H} NMR spectra of 26. | ||
A further key spectroscopic feature of complex 26 is the existence of the silicon bound hydride atom. Due to the sluggish pseudorotation of the dmpe groups at room temperature, its chemical shift position could not obviously be located in the 1H NMR spectrum, due to its being hidden by aromatic resonance signals, as a complex multiplet (a triplet of triplets). A two dimensional Si,H spectrum, however showed a weak cross peak confirming its presence (Fig. 8). Moreover, on heating the sample, and with onset of pseudorotation (see above), a quintet multiplicity for the Si–H is observed, as a result of 3J(Si,P) coupling to four equivalent P atoms, further confirming its presence.
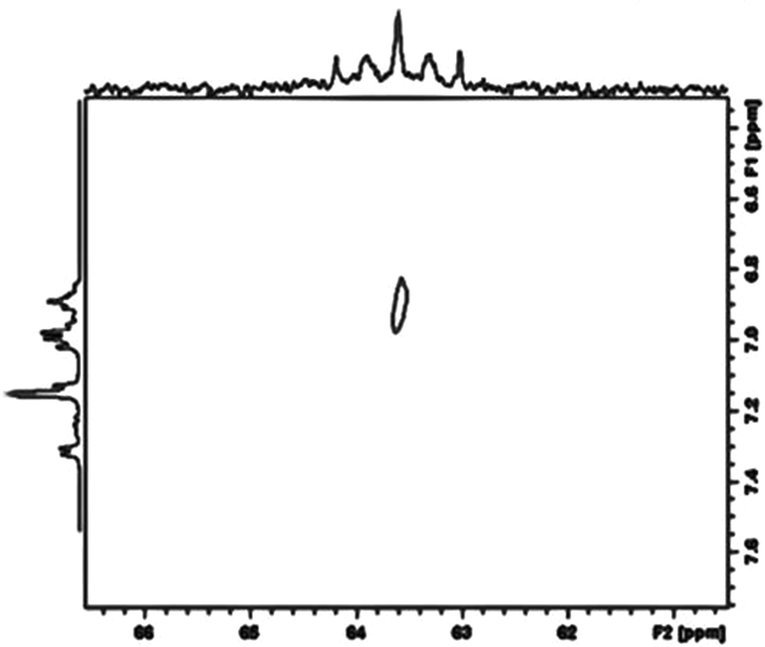 | ||
| Fig. 8 Two dimensional H, Si correlation spectrum (COSY) of complex 26 showing a weak cross peak for the Si signal and the Si bound hydride atom, confirming its connectivity to Si. | ||
The 29Si NMR shifts of complex 25 appear at δ = 43.1 and that of complex 26 at δ = 63.6 ppm. The related methylated complex 27, also prepared in this study (Scheme 14), exhibits a resonance signal at δ = 102.5 ppm. These values are related linearly to the respective Hammett-constants of the substituents at the Si centre,19 and can further be rationalised on the basis of Bent's rule.20 In all cases the signals appear as quintets, as a result of 2J(Si,P) coupling to P atoms, rendered equivalent, as a consequence of BSR.
Detailed DFT studies of complexes 25–27 were also reported (B3LYP/6-31G(d) for C, H, N, Cl and Si; LANL2DZ for Fe), to gain further insights into the bonding, in this study. Noteworthy is that in all cases the HOMO reflects π-back-bonding between the iron and silicon atoms, providing some explanation for the bond length contraction observed in the structural investigations. What is particularly of importance is that this orbital appears to form a much stronger overlap in the case of the hydride complex 26, compared to the other two complexes, explaining the increase in the Berry pseudorotation observed earlier. A better π-backbond should result in more rigidly coordinated dmpe groups, and concomitant increased energy requirements for their exchange facilitated by BSR (Fig. 9 and 10).
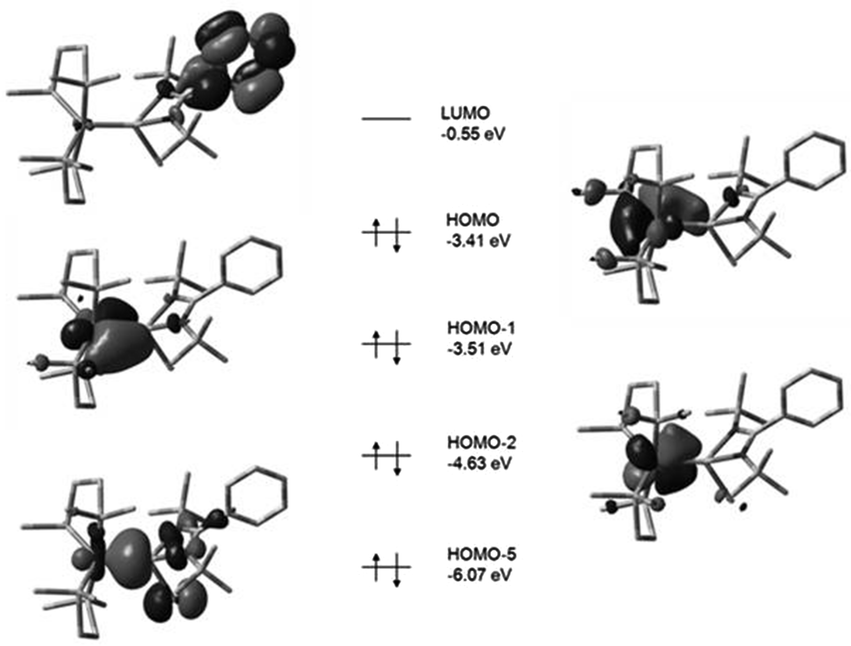 | ||
| Fig. 9 Calculated boundary surfaces of some key frontier MOs and their respective energies in complex 25. Complex 27 was found to exhibit analogous frontier MOs (adapted from ref. 16). | ||
 | ||
| Fig. 10 Calculated boundary surfaces of some key frontier MOs and their respective energies in hydride complex 26. The HOMO orbital clearly exhibits enhanced overlap compared to that observed in the related complexes 25 and 27 (adapted from ref. 16). | ||
The catalytic activity of complex 26 was investigated towards ketone hydrosilylation for a variety of substrates bearing different steric and electronic properties. What is noteworthy is that this is the first study showing the catalytic ability of a Si(II) hydride complex, and also the first such example for an Fe-based NHSi complex (Table 4).
| Entry | Substrate | Yield [%] |
|---|---|---|
| 1 |

|
92 |
| 2 |
|
73 |
| 3 |

|
>99 |
| 4 |
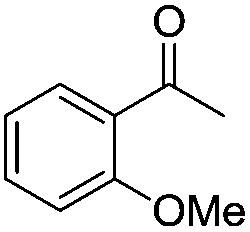
|
>99 |
| 5 |
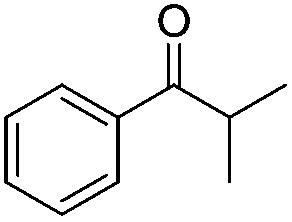
|
98 |
| 6 |

|
98 |
| 7 |
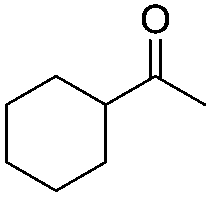
|
95 |
Some mechanistic investigations to gain insight into the elementary steps in the catalytic process were also carried out. In an attempt to isolate a plausible intermediate, a stoichiometric reaction of 25 with a ketone, in the absence of the hydride delivery agent, (EtO)3SiH, was carried out in the hope of isolating [(dmpe)2HFe←{:Si←[:OC(CH3)(napthyl)]L}] (L = PhC(NtBu)2) (28), which is a rational first intermediate in the catalytic process. Despite several efforts the product could not be isolated preparatively, owing to its instability. Instead, an NMR experiment was carried out to monitor changes in the 1H and 31P{1H} NMR spectra over time. Heating a sample of 25 with a stoichiometric amount of napthylmethyl acetone in toluene-d8, indeed revealed the selective formation of a new iron hydride species, featuring a broad multiplet signal at δ = −13.94 ppm in the 1H NMR spectrum, typical of an terminal iron hydride. Moreover, in the reaction, the 31P{1H} NMR spectra over time revealed the selective formation of two triplet signals at δ = 62.2 and 75.7 ppm (2J(P,P) = 27 Hz) along with consumption of 25. These results provide some, perhaps anecdotal, evidence of H migration from the silicon to the iron centre, mediated by the ketone coordination to the silicon centre. These results point to the fact that the NHSi ligand is not merely a spectator ligand in the catalytic process, but mediates it. In addition to these experimental results, DFT investigations were also carried out to study the nature of the likely intermediate 28. A slightly distorted octahedral structure could be located on the potential energy surface for a simplified model of 28 (28b), where benzaldehyde was employed (instead of the more demanding napthylmethyl acetone) as carbonyl (Fig. 11).
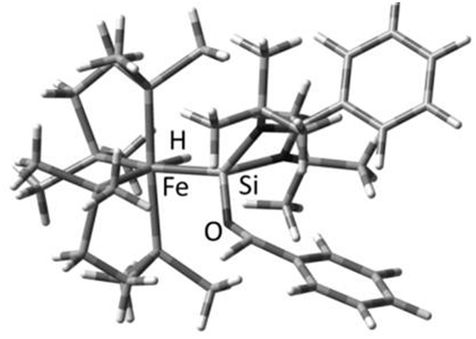 | ||
| Fig. 11 Optimized structure of a simplified model (benzaldehyde as organic carbonyl) of 28 (28b). (B3LYP/6-31G(d) for C, H, N, Cl and Si; LANL2DZ for Fe). | ||
A key feature of the structure of 28b is a slightly elongated Fe–Si bond length (2.342 Å) compared to the calculated structure of the parent complex 26 (2.1976 Å), indicating reduced π-back-bonding from the iron centre to Si. In addition, the bond order is substantially reduced as reflected by the calculated Wiberg bond index from 1.1585 in complex 26 to 0.864 in 28b. Noteworthy, the oxygen atom of the carbonyl is coordinated to the silicon centre and features a Si–O bond length of 1.6427 Å and a C–O bond length of 1.4495 Å. The latter parameter indicates elongation from a ‘free’ CO bond to a C–O single bond character, which is hence the likely source of the CO bond activation in the overall catalytic process. Moreover, the ketone oxygen atom forms a stabilising dative interaction with the even more electropositive silicon atom, following the H atom migration. What is noteworthy is that there is a substantial increase in the NBO charge on the Si atom in 28b compared to 26 (Table 5), which indeed suggests that 28b can be viewed as a “betaine-like” intermediate featuring a cationic Si atom, stabilised by the dative coordination of the ketone, along with an Fe(0) centre, bearing a negatively charged hydride ligand.
| Complex | NBO charge | WBI | d(Fe–Si) (Å) | |
|---|---|---|---|---|
| Fe | Si | |||
| 26 | 2.198 | +1.237 | 1.1585 | 2.1976 |
| 28b | 2.026 | +1.935 | 0.864 | 2.342 |
The yields of the isolated alcohols following work-up in all cases was excellent and even comparable to some existing benchmark systems for iron based ketone hydrosilylation.21
Taking these experimental and theoretical results into consideration, a possible catalytic mechanism was also presented by us in this report (Scheme 15).
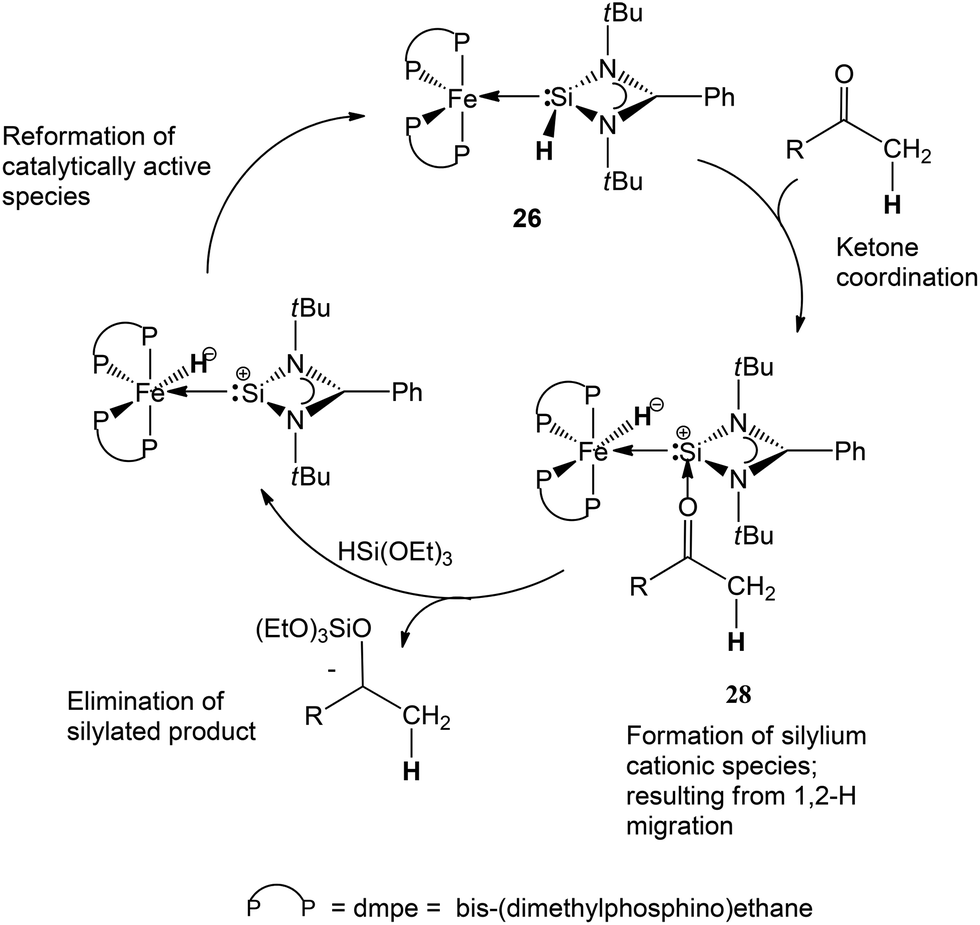 | ||
| Scheme 15 Proposed catalytic process for the catalytic hydrosilylation of ketones mediated by complex 26. | ||
The ability of transition metal complexes bearing our zwitterionic NHSi 3 to effective catalyse amide reduction, in the presence of silanes was also very recently reported by our group, shortly after the appearance of the iron-based hydrosilylation work.22
Numerous complexes bearing 3 as a ligand exist and the rich and varied reactivity of these complexes has been well documented, particularly by our group, in recent years.3 In contrast to the plethora of studies devoted to stoichiometric reactions with complexes bearing NHSi 3, no reports for catalytic transformations had been reported, which precipitated our interest.
Rhodium and iridium complexes bearing zwitterionic NHSi 3 were targeted, since the synthesis of NHC rhodium complexes from ‘free’ NHCs and [Rh(Cl)cod]2, for example, under mild conditions is well-established and the influence of different ligands on catalytic activity has been relatively thoroughly investigated.23 Initial attempts to employ the same synthetic strategy to access and NHSi complex of Rh, by simple reaction of the Rh dimer, [Rh(Cl)cod]2, with NHSi 3 however did not afford the desired NHSi complex, highlighting its poorer σ-donor strength (see Chart 3).
 | ||
| Chart 3 Comparison of different carbonyl stretching frequencies (A1 mode) of zwitterionic NHSi Ni(CO)3 complexes (adapted from ref. 23). | ||
However, the reaction of the ‘free’ NHSi 3 with ethereal HCl at low temperatures (−40 °C) leads to the formation of 1,4-addition product 3b as an initial product, in analogy to the reaction of 3 with Me3SiOTf.6b This 1,4-addition product exhibits increased σ-donor strength (Chart 3) and this chemical “trick” then enables coordination via Si resulting in the novel transition metal chlorosilylene complexes 29 and 30 respectively (Scheme 16).
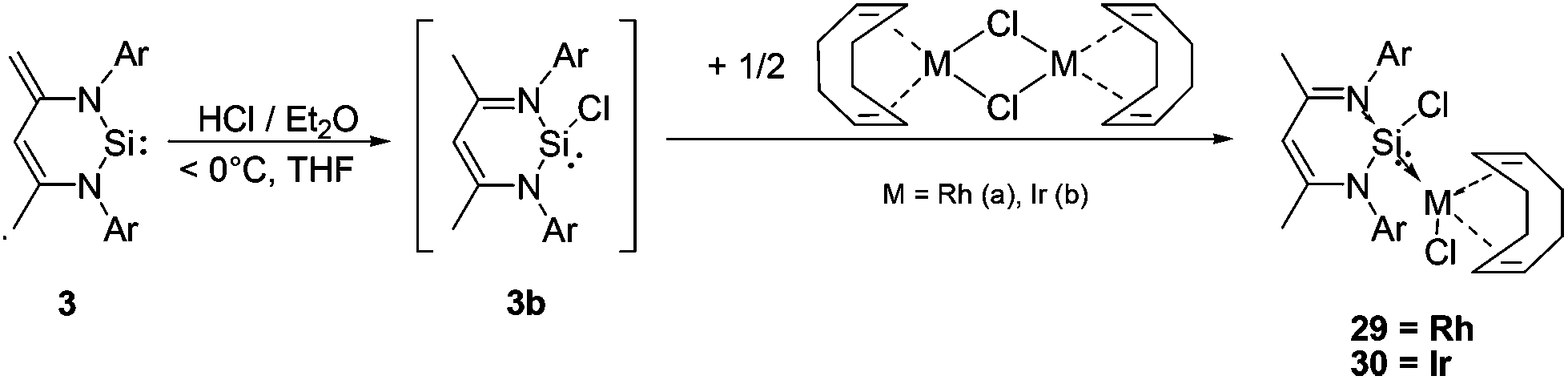 | ||
| Scheme 16 Synthesis of the novel NHSi Rh and Ir complexes 29 and 30via the in situ generated chlorosilylene 3b. | ||
The 1H NMR spectra of complexes 29 and 30 are consistent with the 1,4-addition of the HCl, shown by the protonation of the methylene group in the backbone of silylene 3, resulting in signals at δ = 1.53 (29) and 1.57 ppm (30), respectively. Both complexes exhibit similar 29Si{1H} chemical shifts of δ = −1.03 (29) and δ = 0.79 ppm (30), as expected, with the former resonance signal showing up as a doublet due to Rh coupling 1J(Si,Rh) = 128 Hz.
Complexes 29 and 30 were also the subject of a single crystal X-ray diffraction study, and the solid state structure of 30 is shown in Fig. 12. The complexes are essentially iso-structural and both feature slightly distorted tetrahedral Si(II) centres, coordinated to the metal centres.
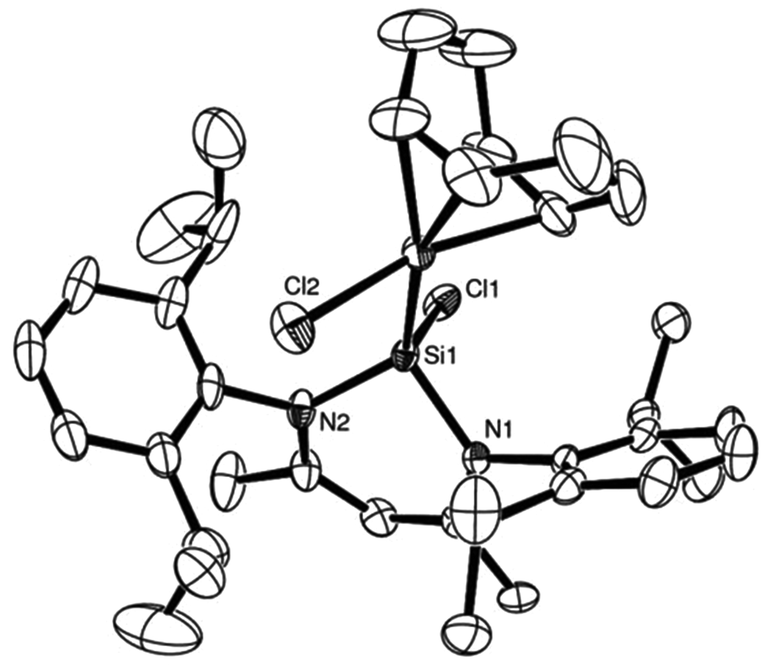 | ||
| Fig. 12 ORTEP representation of complex 30:29 is isostructural; thermal ellipsoids at 50% probability, H atoms omitted for clarity (adapted from ref. 22). | ||
The Si–Rh bond length in 29 of 2.295(1) Å is comparable to the Si–Rh bond lengths (2.29 and 2.32 Å) in the seminal rhodium based NHSi complexes by Neumann and Pfaltz.24
The Rh–Si bond lengths in 29 are slightly shorter when compared to silyl rhodium complexes (2.32–2.38 Å), indicating some possible π-back-donation in 29.25 In close analogy, the Ir–Si distance of 2.3164(8) Å in 30 is within the typical range of other previously reported silyl iridium complexes26 and nearly identical to another recently reported Ir based NHSi complex, from our group, bearing 3 as a ligand (2.3293(11) Å).27
We next explored the catalytic activity of complexes 29 and 30 towards the catalytic reduction of amides. We selected this transformation given its current interest, and numerous reports from several groups, most notably by Milstein and Beller.28
Based on these collective reports we decided to use the acetyl protected dibenzoazepine derivate 31 for our catalytic investigations with the novel NHSi Rh and Ir complexes 29 and 30 (Scheme 17). Two possible reductive products are a C–O cleavage product (32) and an N–C cleavage product (33).
 | ||
| Scheme 17 Reduction of a dibenzoazepine derivative 31 with complexes 29 or 30 as pre-catalysts. | ||
By employing complex 29 as a pre-catalyst, selective C–O cleavage was observed and a conversion of the starting material to 32 in 61% yield. In contrast, for complex 30 under analogous catalytic conditions, both reductive products 32 and 33 were observed. Moreover, with the pre-catalyst 30 the total conversion of 31 is higher (87%) compared to 29 (Fig. 13 and 14).
 | ||
| Fig. 13 Catalytic performance of complex 29 compared to [Rh(Cl)cod]2 as a function of time. | ||
 | ||
| Fig. 14 Catalytic performance of complex 30 compared to [Ir(Cl)cod]2 as a function of time. | ||
For comparative purposes, we also tested the catalytic activity of the complexes ([Rh(Cl)cod]2, [Ir(Cl)cod]2) towards the reduction of 31 to gain insights into the effects the presence of the NHSi has on the catalyst activity and selectivity.29 Indeed, the catalytic activity of [Rh(Cl)cod]2 is comparable to that of 29. After 24 h a selective C–O cleavage of 53% was observed. Strikingly, however, the [Ir(Cl)cod]2 dimer shows considerably less activity after 24 h (30%) as compared to 30 (total: 87%, C–O cleavage: 78%, C–N cleavage: 9%) and the dimer exhibits a different chemoselectivity compared to 30. The [Ir(Cl)cod]2 dimer as a pre-catalyst yields exclusively the C–O cleavage product, while 30 yields additionally the dibenzoazepine 33. Therefore the coordination of the silylene ligand 3b to the Ir centre increases the activity of the complex but at the same time affords both reductive products (Fig. 13 and 14).
Since hydride species have been implicated as important intermediates in the catalytic reduction of amides with other catalysts systems, we decided to probe the catalytic activity of complex 29 in the presence of a hydride source, Li[BHEt3] (Fig. 13). The idea was to potentially generate the hydride analogue of 29in situ by two hydride halide exchanges facilitated by Li[BHEt3], which we thought could improve the catalyst performance.
Surprisingly, this action seems to retard the catalyst performance (Fig. 15), and this apparent deactivation pathway was also further explored by attempting to isolate the hydride analogue of 29, compound 34, on preparative scale.
 | ||
| Fig. 15 ORTEP representation of complex 36, one of the decomposition products resulting from the attempted transformation of 29 to 34; H atoms omitted for clarity, thermal ellipsoids set at 50% probability (adapted from ref. 22). | ||
This was attempted by reaction of 29 with two molar equivalents of Li[BHEt3]. Several attempts at isolating the potentially reactive species 34 were however unsuccessful. Most likely, the inability to isolate this compound, is due to the migration of both H atoms (from Si and Rh, via 1,3 and 1,4H-shifts respectively) to the coordinated COD ligand, affording free COE, via an intermediate 35 (Scheme 18). This H migration was observed in repeated NMR scale experiments, where initial formation of Rh–H and Si–H signals is indeed observed, but then rapidly decays into the base line with concomitant formation of free COE and several other unidentifiable decomposed Rh complexes (Scheme 17, Fig. 15). One of these intractable decomposition products was characterized by single crystal X-ray diffraction studies (Fig. 15).
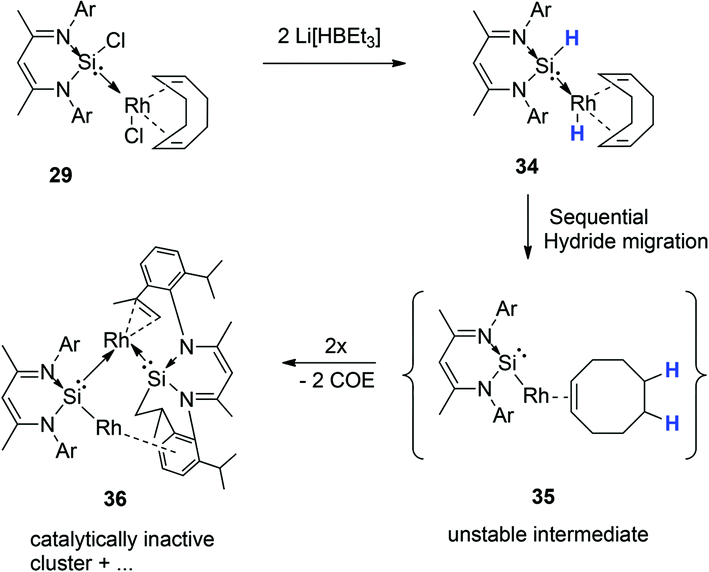 | ||
| Scheme 18 Attempted isolation of 34, leading through COE elimination (observed spectroscopically) to a range of decomposition products, one of which (36) was identified by single crystal X-ray diffraction analysis. | ||
These results provide some explanation for the observed deactivation/retardation in the catalysis in the presence of Li[BHEt3], since the in situ generated dihydride 34 is susceptible to COE loss and subsequent decomposition into intractable products which are most likely not catalytically active.
Finally, Inoue, Enthaler and co-workers have also recently reported the use of nickel complex [(cod)Ni←:6] (37) (cod = 1,5-cyclooctadiene)6f based on bis-NHSi ligand 6, as a pre-catalyst in C–C bond formation reactions (Fig. 16).30
 | ||
| Fig. 16 Structure of bis-NHSi nickel complex 37, capable of acting as a pre-catalyst in C–C cross-coupling reactions. | ||
A series of C–C cross-coupling reactions were reported in this study with aryl halides and organometallic zinc and Grignard reagents. In most cases, good to outstanding catalytic rates were observed with 37 as a pre-catalyst.
Some of these catalytic results are presented in Tables 6 and 7, respectively.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yield [%] |
| a Reaction conditions: substrate (0.72 mmol), benzyl zinc bromide (1.08 mmol, 0.5 M in THF), pre-catalyst 37 (2.0 mol%), 70 °C, 24 h. The yield was determined by GC-MS and 1H NMR. The yield of the homocoupling of the aryl halide is stated in parenthesis. | |||
| 1 |

|

|
98 (2) |
| 2 |
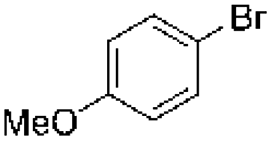
|

|
29 (11) |
| 3 |

|
|
8 (<1) |
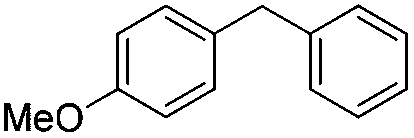
| 4 |

|
|
97 (<1) |
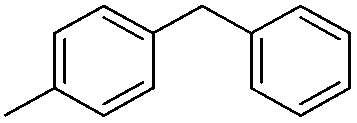
| 5 |

|
|
74 (19) |
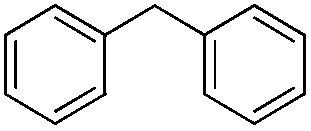
| 6 |

|
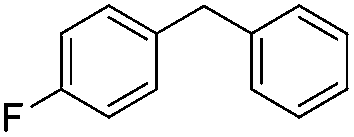
|
49 (11) |
| 7 |

|

|
85 (15) |
| 8 |
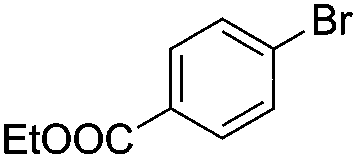
|

|
>99 (<1) |
| 9 |

|
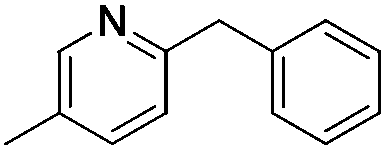
|
>99 (<1) |
| 10 |
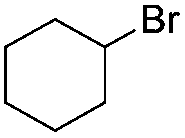
|

|
<1 |
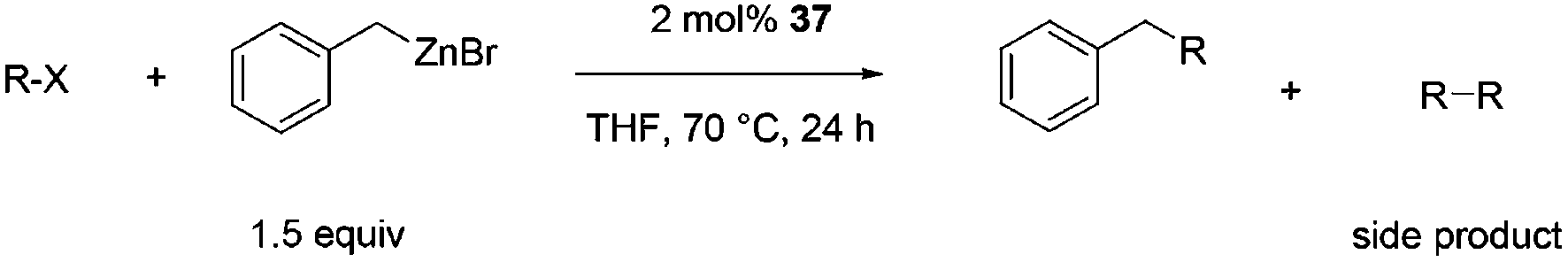
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yield [%] |
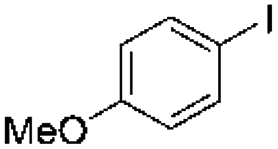
| a Reaction conditions: substrate (0.72 mmol), Grignard reagent (1.08 mmol, 1.0 M in THF), precatalyst 37 (2.0 mol%), 70 °C, 24 h. The yield was determined by GC-MS and 1H NMR. (Yields of dehalogenated products and homocoupling products are omitted.) | |||
| 1 |
|

|
63 |
| 2 |

|
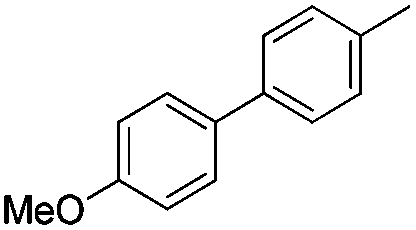
|
74 |
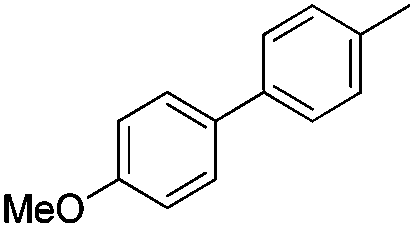
| 3 |

|
|
79 |
| 4 |
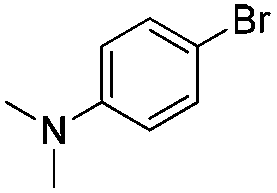
|

|
72 |
| 5 |

|
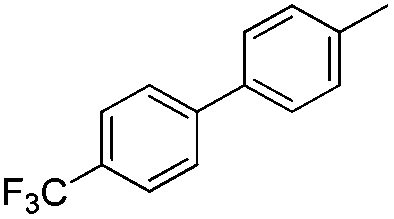
|
93 |
| 6 |

|

|
61 |
| 7 |
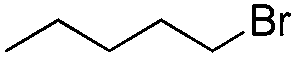
|

|
<1 |

The authors also present a mechanistic proposal for the coupling reactions based on previous mechanistic studies for other Ni-based catalysts. A reasonable mechanistic pathway potentially involves de-coordination of the cod from the pre-catalyst 37, as the first key elementary step, followed by oxidative addition of the arene halide over the Ni(0) centre. This affords a Ni(II) intermediate of the type 6:→Ni(R)X. Subsequent transmetallation with the Grignard or arylzinc reagent then generates the Ni(II) diaryl species 6:→NiRR′ as the next step. Reductive elimination then affords the coupled product and regenerates the active Ni(0) species, closing the catalytic cycle (Scheme 19).
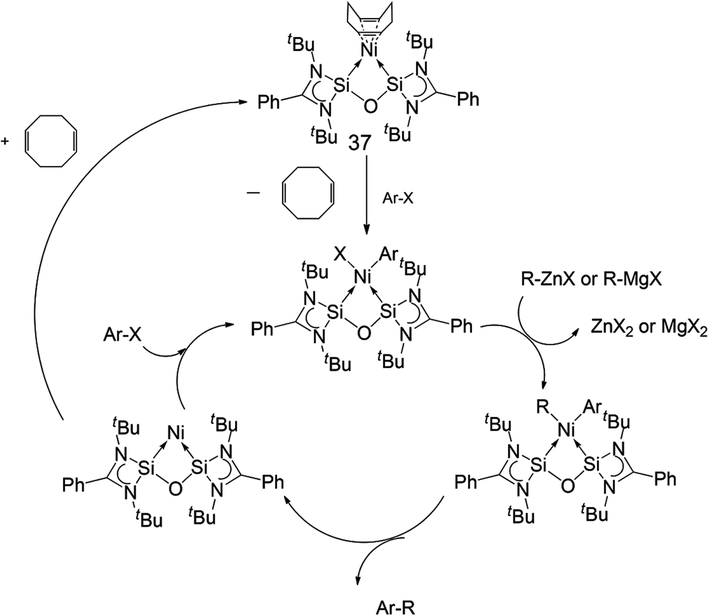 | ||
| Scheme 19 Mechanistic proposal for C–C cross-coupling mediated by 37, following a sequence of oxidative–addition/transmetallation/reductive elimination steps. | ||
Conclusions
It is apparent from this short review that the use of N-heterocyclic silylene (NHSi) stabilised complexes in catalysis is still in its infancy but led already to a number of remarkable examples. The early stage of development of this research is somewhat surprising, given that NHSi ligands are isoelectronic to N-heterocyclic carbene (NHC) ligands, where literally thousands of reports detailing their use as supporting ligands in a plethora of catalytic processes across the periodic table have been reported.The few reports that do exist in this context, from the seminal work of Fürstner and Roesky, and later from our efforts, do however show that NHSi ligands are not merely passive spectators, or isoelectronic replacements for NHCs or phosphines, but in some cases play a pivotal role in the catalyst cycle, activity and even selectivity. They have the ability to fine-tune the electronic situation at the metal centre, through stronger σ-donation, compared for example to traditional phosphines, and thereby improve catalytic activity, as exemplified by our contribution on iridium-based NHSi complexes. Some NHSis can even mediate processes through cooperative effects, as seems to be the case with our NHSi hydridoiron complex.
A systematic study of NHSi complexes across the transition metals and their capacity to perform novel catalytic transformations or perhaps improve existing transformations is certainly a worthwhile aim. We are convinced that the coming years will witness more developments in this new and invigorating research area. Future endeavours might include the exploration of early transition metal (group III or IV) NHSi complexes, and their suitability in homogeneous catalysis; or even f-block NHSi complexes, which in themselves are a rarity in the literature.31
Acknowledgements
We are grateful to the Deutsche Forschungsgemeinschaft (DR 226-17/2) and the Cluster of Excellence UniCat (sponsored by the Deutsche Forschungsgemeinschaft and administered by the TU Berlin) for financial support of this research. We thank M. Stoelzel, Dr S. Enthaler, and Prof. Dr S. Inoue for fruitful discussions in the preparation of this review.Notes and references
- G. Schmid and E. Welz, Angew. Chem., 1977, 89, 823 ( Angew. Chem., Int. Ed. Engl. , 1977 , 16 , 785 ) CrossRef CAS.
- See as selected recent reviews, with references therein: (a) L. Zhang and Z. Hou, Pure Appl. Chem., 2012, 84, 1705 CrossRef CAS; (b) S. Budagumpi, R. A. Haque and A. W. Salman, Coord. Chem. Rev., 2012, 256, 1787 CrossRef CAS PubMed; (c) C. Valente, S. Calimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef CAS PubMed; (d) A. Correa, S. P. Nolan and L. Cavallo, Top. Curr. Chem., 2011, 302, 131 CrossRef CAS; (e) A. Poulain, M. Iglesias and M. Albrecht, Curr. Org. Chem., 2011, 15, 3325 CrossRef CAS; (f) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776 RSC; (g) T. Strassner, Top. Organomet. Chem., 2007, 22, 125 CrossRef CAS; (h) W. J. Sommer and M. Weck, Coord. Chem. Rev., 2007, 251, 860 CrossRef CAS PubMed; (i) H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032 RSC.
- B. Blom, M. Stoelzel and M. Driess, Chem.–Eur. J., 2013, 19, 40 CrossRef CAS PubMed.
- These are in fact the first examples of base-free silylene complexes. See: (a) S. D. Grumbine, T. D. Tilley and A. L. Rheingold, J. Am. Chem. Soc., 1993, 115, 358 CrossRef CAS; (b) S. D. Grumbine, T. D. Tilley, F. P. Arnold and A. L. Rheingold, J. Am. Chem. Soc., 1993, 115, 7884 CrossRef CAS; (c) S. D. Grumbine, T. D. Tilley, F. P. Arnold and A. L. Rheingold, J. Am. Chem. Soc., 1994, 116, 5495 CrossRef CAS.
- C. Zybill and G. Muller, Angew. Chem., Int. Ed. Engl., 1987, 26, 669 CrossRef.
- (a) M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belaykov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691 CrossRef CAS; (b) M. Driess, S. Yao, M. Brym, C. van Wüllen and D. Lentz, J. Am. Chem. Soc., 2006, 128, 9628 CrossRef CAS PubMed; (c) C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., 2006, 118, 4052 ( Angew. Chem., Int. Ed. , 2006 , 45 , 3948 ) CrossRef; (d) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123 CrossRef CAS PubMed; (e) W. Wang, S. Inoue, E. Irran and M. Driess, Angew. Chem., 2012, 124, 3751 ( Angew. Chem., Int. Ed. , 2012 , 51 , 3691 ) CrossRef; (f) W. Wang, S. Inoue, S. Yao and M. Driess, J. Am. Chem. Soc., 2010, 132, 15890 CrossRef CAS PubMed; (g) W. Wang, S. Inoue, S. Enthaler and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 6167 CrossRef CAS PubMed; (h) L. Kong, J. Zhang, H. Song and C. Cui, Dalton Trans., 2009, 5444 RSC; (i) P. Zark, A. Schäfer, A. Mitra, D. Haase, W. Saak, R. West and T. Müller, J. Organomet. Chem., 2010, 695, 398 CrossRef CAS PubMed; (j) For more details about reactivity and properties of NHSis see: M. Assay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354 CrossRef PubMed.
- A. Fürstner, H. Krause and C. W. Lehmann, Chem. Commun., 2001, 2372 RSC.
- M. Zhang, X. Liu, C. Shi, C. Ren, Y. Ding and H. W. Roesky, Z. Anorg. Allg. Chem., 2008, 634, 1755 CrossRef CAS.
- A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 11478 CrossRef PubMed.
- D. Gallego, A. Brück, E. Irran, F. Meier, M. Kaupp, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 15617 CrossRef CAS PubMed.
- Reviews for Sonogashira cross-coupling reaction: (a) R. R. Tykwinski, Angew. Chem., 2003, 115, 1604 ( Angew. Chem. Int. Ed. , 2003 , 42 , 1566 ) CrossRef; (b) E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979 CrossRef CAS PubMed; (c) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed; (d) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084 RSC . Examples using Ni-based catalysts: ; (e) L. Wang, P. Li and Y. Zhang, Chem. Commun., 2004, 514 RSC; (f) I. P. Beletskaya, G. V. Latyshev, A. V. Tsvetkov and N. V. Lukashev, Tetrahedron Lett., 2003, 44, 5011 CrossRef CAS.
- See as representative and recent examples (and references therein): (a) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (b) Iron Catalysis in Organic Chemistry, ed. B. Plietker, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) S. Gaillard and J.-L. Renaud, ChemSusChem, 2008, 1, 505 CrossRef CAS PubMed; (d) A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC; (e) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS PubMed; (f) R. M. Bullock, Angew. Chem., 2007, 119, 7504 ( Angew. Chem. Int. Ed. , 2007 , 46 , 7360 ) CrossRef; (g) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS PubMed; (h) E. B. Bauer, Curr. Org. Chem., 2008, 12, 1341 CrossRef CAS; (i) W. M. Czaplik, M. Mayer, J. Cvengros and A. Jacobi von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS PubMed; (j) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 120, 3363 ( Angew. Chem. Int. Ed. , 2008 , 47 , 3317 ) CrossRef; (k) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794 CrossRef CAS PubMed; (l) M. Haberberger, E. Irran and S. Enthaler, Eur. J. Inorg. Chem., 2011, 2797 CrossRef CAS; (m) S. Enthaler, M. Haberberger and E. Irran, Chem.–Asian J., 2011, 6, 1613 CrossRef CAS PubMed.
- Only three examples exist: (a) Ref. 1; ; (b) W. Yang, H. Fu, H. Wang, M. Chen, Y. Ding, H. W. Roesky and A. Jana, Inorg. Chem., 2009, 48, 5058 CrossRef CAS PubMed; (c) T. A. Schmedake, M. Haaf, B. J. Paradise, A. J. Millevolte, D. R. Powell and R. West, J. Organomet. Chem., 2001, 636, 17 CrossRef CAS.
- This complex was first prepared in the laboratory of Prof. Dr A. C. Filippou (University of Bonn), by the first author of this publication in 2009. For the synthesis and full characterization see: B. Blom, Reactivity of Ylenes at Late Transition Metal Centres, Doctoral thesis, University of Bonn, Cuvillier Verlag, 2011 Search PubMed.
- [Fe(dmpe)2(PMe3)] reacts with germylenes and stannylenes of the type RECl (E = Ge or Sn, R = 2,6-Mes2-C6H3 or 2,6-Trip2-C6H3; Mes = 2,4,6-Me3-C6H2, Trip = 2,4,6-iPr3-C6H2) affording by PMe3 elimination complexes of the type [(dmpe)2FeECl(R)], which can also be converted to triply bonded complexes: [(dmpe)2Fe
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) E–R]+A−.
E–R]+A−. - B. Blom, S. Enthaler, S. Inoue, E. Irran and M. Driess, J. Am. Chem. Soc., 2013, 135, 6703 CrossRef CAS PubMed.
- H. Tobita, A. Matsuda, H. Hashimoto, K. Ueno and H. Ogino, Angew. Chem., Int. Ed., 2004, 43, 221 CrossRef CAS PubMed.
- R. G. Bryant, J. Chem. Educ., 1983, 60, 933 CrossRef CAS.
- For a leading reference on the Hammett constant see: C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- H. A. Bent, Chem. Rev., 1961, 61, 275 CrossRef CAS.
- As a leading reference see: K. Junge, K. Schröder and M. Beller, Chem. Commun., 2011, 47, 4849 RSC.
- M. Stoelzel, C. Praesang, B. Blom and M. Driess, Aust. J. Chem., 2013, 66, 1163 CAS.
- M. Ahmed, C. Buch, L. Routaboul, R. Jackstell, H. Klein, A. Spannenberg and M. Beller, Chem.–Eur. J., 2007, 13, 1594 CrossRef CAS PubMed.
- E. Neumann and A. Pfaltz, Organometallics, 2005, 24, 2008 CrossRef CAS.
- M. Aizenberg, J. Ott, C. J. Elsevier and D. Milstein, J. Organomet. Chem., 1998, 551, 81 CrossRef CAS.
- J. Y. Corey and J. Braddock-Wilking, Chem. Rev., 1998, 99, 175 CrossRef PubMed.
- A.-K. Jungton, A. Meltzer, C. Präsang, T. Braun, M. Driess and A. Penner, Dalton Trans., 2010, 39, 5436 RSC.
- (a) G. W. Gribble, Chem. Soc. Rev., 1998, 27, 395 RSC; (b) J. Seyden-Penne, Reductions by the Alumino- and Borohydrides in Organic Synthesis, Wiley, New York, 2nd edn, 1997 Search PubMed; (c) M. Igarashi and T. Fuchikami, Tetrahedron Lett., 2001, 42, 1945 CrossRef CAS; (d) S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770 CrossRef CAS PubMed; (e) R. Kuwano, M. Takahashi and Y. Ito, Tetrahedron Lett., 1998, 39, 1017 CrossRef CAS; (f) R. Goikhman and D. Milstein, Chem.–Eur. J., 2005, 11, 2983 CrossRef CAS PubMed; (g) N. Schneider, M. Finger, C. Haferkemper, S. Bellemin-Laponnaz, P. Hofmann and L. H. Gade, Angew. Chem., Int. Ed., 2009, 48, 1609 CrossRef CAS PubMed; (h) E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756 CrossRef CAS PubMed; (i) S. Krackl, C. I. Someya and S. Enthaler, Chem.–Eur. J., 2012, 18, 15267 CrossRef CAS PubMed.
- In fact this has been reported by Brookhart: C. Cheng and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11304 CrossRef CAS PubMed.
- C. I. Someya, M. Haberberger, W. Wang, S. Enthaler and S. Inoue, Chem. Lett., 2013, 42, 286 CrossRef CAS.
- See as a seminal example: W. J. Evans, J. M. Perotti, J. W. Ziller, D. F. Moser and R. West, Organometallics, 2003, 22, 1160 CrossRef CAS.
| This journal is © the Partner Organisations 2014 |