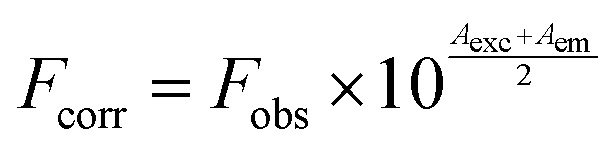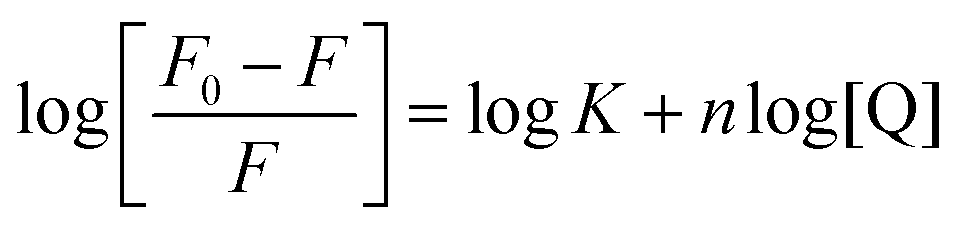Synthesis, structure, DNA/protein binding and in vitro cytotoxicity of new ruthenium metallates†
Received
10th October 2013
, Accepted 25th November 2013
First published on 24th January 2014
Abstract
The reaction of [RuHCl(CO)(PPh3)3] with an equimolar amount of salicylaldehyde-4(N)-methylthiosemicarbazone [H2-(Sal-mtsc)] resulted in two entities, namely [Ru(H-Sal-mtsc)Cl(CO) (PPh3)2] (1) and [Ru(Sal-mtsc)(CO)(PPh3)2] (2) from a single tub. The new complexes were characterized by various spectro (IR, absorption and NMR), analytical and single crystal X-ray diffraction studies. From the crystallographic studies, it is confirmed that in complex 1, the ligand coordinated through the thiolate sulfur and the deprotonated hydrazinic nitrogen N(2), resulting in the formation of an unusual strained four membered chelate ring. The third potential donor, phenolic oxygen, remained uncoordinated. In complex 2, the ligand coordinated as an ONS chelate with the formation of more common five and six membered chelate rings. Complexes 1 and 2 have been tested for their DNA/protein binding properties by taking CT-DNA/lysozyme as models. From the protein binding studies, the alterations in the secondary structure of lysozyme by the ruthenium(II) complexes (1 and 2) were confirmed with synchronous and three-dimensional fluorescence spectroscopic studies. The in vitro cytotoxicity of the newly-synthesized complexes was carried out in two different human tumour cell lines, A549 and HepG2. The cytotoxicity studies showed that complex 2 exhibited higher activity than 1.
Introduction
For the past few decades, thiosemicarbazones have been a center of attention and attraction for chemists and biologists because of their wide structural and pharmacological relevance.1 DNA and proteins are the main cellular targets for cytotoxicity due to the interaction between small molecules and DNA, which causes DNA damage in cancer cells by blocking the division of cancer cells and resulting in cell death.2–4 Current inorganic drugs have been successfully used in the treatment of many cancers.5,6 However, there are problems associated with their use, including the cumulative toxicities of nephrotoxicity, ototoxicity and peripheral neuropathy, which prevent the potential efficacy of them.7 In the search for new drugs with less side effects, ruthenium compounds are arguably the best candidates for cancer therapy and have attained recognition over the last 35 years.8–15 There are three main properties that make ruthenium compounds well suited to medicinal application.16 First, the rate of ligand exchange: the ligand exchange kinetics of metal complexes in aqueous solution, which seems to be crucial for anticancer activity and which varies for different metal cations by as much as fourteen orders of magnitude (rates from 10−6 to 108 s−1), are very similar for platinum(II) and ruthenium(II) complexes. In both cases, the ligand exchange processes are quite slow and may take hours (rates 10−3 to 10−2 s−1).17 Second, the range of accessible oxidation states (from 8 to −2): the redox accessible oxidation states occupied by ruthenium enables the body to catalyze oxidation and reduction reactions depending on the physiological environment, enabling ruthenium compounds to be selectively activated in diseased tissues. Third, the binding ability of ruthenium mimics iron binding by proteins in the plasma, making them ideally suited to metal-based drug design. These features combine to give ruthenium drugs a remarkably low toxicity compared to other metal complexes and a few ruthenium compounds have been shown to be quite selective for cancer cells.16,18 Due to the continuous search to derive a better biologically active ruthenium complex, herein, we report the synthesis of new ruthenium(II) complexes containing salicylaldehyde-4(N)-methylthiosemicarbazone. Furthermore, the interactions of the complexes with CT-DNA/protein, cytotoxic studies and cellular uptake against two different cancer cell lines were explored.
Experimental
Materials and measurements
The ligand [H2-(Sal-mtsc)] and the ruthenium precursor [RuHCl(CO)(PPh3)3] were synthesized according to the standard literature procedures.19–21 All the reagents used in this study were analar grade and the solvents were purified and dried according to the standard procedure.22 Infrared spectra were measured as KBr pellets with a Nicolet Avatar Model FT-IR spectrophotometer in the 400–4000 cm−1 range. Elemental analyses of carbon, hydrogen, nitrogen, and sulfur were determined using a Vario EL III CHNS at the Department of Chemistry, Bharathiar University, Coimbatore, India. The electronic spectra of the complexes were recorded in dichloromethane using a Jasco V-630 spectrophotometer in the 200–800 nm range. 1H NMR spectra were taken in DMSO at room temperature with a Bruker 400 MHz instrument with the chemical shift relative to tetramethylsilane. Cyclic voltammograms were recorded on a CH instrument using a platinum wire working electrode and a platinum disc counter electrode. All the potentials were referenced to the standard Ag/AgCl electrode and ferrocene was used as an external standard. Melting points were recorded using Lab India melting point apparatus.
X-ray crystallography
Single crystals of [Ru(H-Sal-mtsc)(CO)Cl(PPh3)2]·CH3CN (1) and [Ru(Sal-mtsc)(CO)(PPh3)2] (2) were obtained from a CH2Cl2–CH3CN mixture and DMF, respectively. The data sets for the single-crystal X-ray studies for the new complexes were collected with Mo Kα (λ = 0.71073 Å) radiation on a Bruker SMART 1000 CCD diffractometer. All the calculations were performed using SHELXTL.23
Preparation of the new ruthenium(II) complexes
A solution of [H2-(Sal-mtsc)] (0.022 g; 0.105 mmol) in 20 cm3 of benzene was added drop wise to a boiling solution of [RuHCl(CO)(PPh3)3] (0.100 g; 0.105 mmol) in benzene (20 cm3). The mixture was heated under reflux for 5 h. The resulting yellow coloured solution was concentrated. It was subjected to thin layer chromatography where two spots were identified and isolated by silica gel column chromatography using ethyl acetate–petroleum ether and benzene–methanol solvent mixtures. The first light yellow band was isolated using a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether (60–80 °C)–ethyl acetate mixture as the eluent and was crystallized using a CH2Cl2–CH3CN mixture, which afforded shiny yellow crystals of 1. Following the same procedure as 1, complex 2 was isolated from a thick yellow band, and afforded a dark yellow compound which upon crystallization in DMF yielded yellow-brown crystals.
:1 petroleum ether (60–80 °C)–ethyl acetate mixture as the eluent and was crystallized using a CH2Cl2–CH3CN mixture, which afforded shiny yellow crystals of 1. Following the same procedure as 1, complex 2 was isolated from a thick yellow band, and afforded a dark yellow compound which upon crystallization in DMF yielded yellow-brown crystals.
[Ru(H-Sal-mtsc)(CO)Cl(PPh3)2] (1).
Yield: 35%. M.p. 180 °C. Anal. Calcd for C46H40N3SO2ClP2Ru: C, 61.57; H, 4.49; N, 4.56; S, 3.57. Found: C, 61.46; H, 4.43; N, 4.56; S, 3.42%. FT-IR (cm−1) in KBr: 3430 (νO–H), 1580 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1265 (νC–O), 749 (νC–S), 1925 (νC
N), 1265 (νC–O), 749 (νC–S), 1925 (νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) 1429, 1087, 694 cm−1 (for PPh3); UV-vis (CH2Cl2), λmax: 252 (17226), 268 (16313), 292 (12721) nm (dm3 mol−1 cm−1) (intra-ligand transition); 311 (11720), 359 (9890), 379 (7021), nm (dm3 mol−1 cm−1) (LMCT s→d); 1H NMR (DMSO-d6, ppm): 10.05 (s, 1H, –O–H), 8.46 (s, 1H, –CHN), 4.82 (m, terminal –NH), 2.35 (d, (J = 5.04), –CH3), 5.3 (s, 1H, –NH imine), 6.69–7.70 (m, aromatic).
O) 1429, 1087, 694 cm−1 (for PPh3); UV-vis (CH2Cl2), λmax: 252 (17226), 268 (16313), 292 (12721) nm (dm3 mol−1 cm−1) (intra-ligand transition); 311 (11720), 359 (9890), 379 (7021), nm (dm3 mol−1 cm−1) (LMCT s→d); 1H NMR (DMSO-d6, ppm): 10.05 (s, 1H, –O–H), 8.46 (s, 1H, –CHN), 4.82 (m, terminal –NH), 2.35 (d, (J = 5.04), –CH3), 5.3 (s, 1H, –NH imine), 6.69–7.70 (m, aromatic).
[Ru(Sal-mtsc)(CO)(PPh3)2] (2).
Yield: 53%. M.p. 219 °C. Anal. Calcd for C46H39N3SO2P2Ru: C, 64.17; H, 4.54; N, 4.87; S, 3.72. Found: C, 64.06; H, 4.52; N, 4.80; S, 3.68%. FT-IR (cm−1) in KBr: 1592 (νCN), 1312 (νC–O), 745 (νC–S), 1945 (νCO) 1434, 1087, 694 cm−1 (for PPh3); UV-vis (CH2Cl2), λmax: 244 (16690), 280 (14432), 293 (13211) nm (dm3 mol−1 cm−1) (intra-ligand transition); 312 (11987), 384 (9059) nm (dm3 mol−1 cm−1) (LMCT s→d); 426 (7489) nm (dm3 mol−1 cm−1) forbidden (d→d) transition; 1H NMR (DMSO-d6, ppm): 8.01 (s, 1H, –CHN), 6.0 (m, terminal –NH), 2.53 (d, (J = 5.02), –CH3), 6.40–7.60 (m, aromatic).
DNA binding study
DNA binding studies have been done according to the method described in an earlier report.1n CT-DNA solutions of various concentrations (0.05–0.5 μM) dissolved in a tris-HCl buffer (pH 7) were added to the ruthenium complexes 1 and 2 (1 μM dissolved in a DMSO–H2O mixture). Absorption spectra were recorded after equilibrium at 20 °C for 10 min. The intrinsic binding constant, Kb, was determined using the Stern–Volmer eqn (1).| | | [DNA]/[εa − εf]) = [DNA]/[εb − εf] + 1/Kb[εb − εf] | (1) |
The absorption coefficients εa, εf, and εb correspond to Aobsd/[DNA], the extinction coefficient for the free complex and the extinction coefficient for the complex in the fully bound form, respectively. The slope and the intercept of the linear fit of the plot of [DNA]/[εa − εf] versus [DNA] give 1/[εa − εf] and 1/Kb[εb − εf], respectively. The intrinsic binding constant, Kb, can be obtained from the ratio of the slope to the intercept. Emission measurements were carried out using a JASCO FP-6600 spectrofluorometer. A tris-buffer was used as a blank to make preliminary adjustments. The excitation wavelength was fixed and the emission range was adjusted before the measurements. All measurements were made at 20 °C. For emission spectral titrations, the complex concentration remained constant at 1 μM and the concentration of DNA was varied from 0.05 to 0.5 μM. The emission enhancement factors were measured by comparing the intensities at the emission spectral maxima under similar conditions.
Ethidium bromide displacement study
In order to know the mode of attachment of CT-DNA to the ruthenium complexes, fluorescence quenching experiments of EB-DNA were carried out by adding 0–50 μM of the ruthenium(II) complexes to samples containing 10 μM EB, 10 μM DNA and a tris-HCl buffer (pH = 7). Before the measurements, the system was shaken and incubated at room temperature for ∼5 min. The emission was recorded at 530–750 nm.
Lysozyme binding studies
Lysozyme was purchased from Hi Media, India. The lysozyme was prepared in a phosphate buffer of pH 7.6 and stored in the dark at 4 °C before use. The concentration of lysozyme was determined spectrophotometrically using ε280 (lysozyme) = 37646 M−1 cm−1.24 Concentrated stock solutions of the complexes were prepared by dissolving the complex in phosphate buffer–DMSO (100:1) and suitably diluting with the phosphate buffer to the required concentrations for all the experiments. The steady-state fluorescence spectra of lysozyme were recorded under an excitation wavelength of 280 nm and the emission was monitored at 342 nm after irradiation with a xenon source. The excitation and emission slit widths (each 5 nm) remained constant for all the experiments. A scan rate of 500 nm min−1 was used. A 3 ml solution, containing the appropriate concentration of lysozyme (1 × 10−6 M), was titrated by successive additions of the complexes. In order to correct the inner filter effect the following eqn (2), was used| | 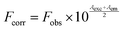 | (2) |
where Fcorr is the corrected fluorescence value, Fobs the measured fluorescence value, Aexc the absorption value at the excitation wavelength, and Aem the absorption value at the emission wavelength.25 Conformational changes were studied using synchronous fluorescence measurements. The spectra were measured at two different Δλ (the difference between the excitation and emission wavelengths of lysozyme) values, such as 15 and 60 nm. The three-dimensional fluorescence spectrum was performed under the following conditions: emission wavelengths at 290–450 nm, excitation at 280 nm and the other parameters were similar to the quenching experiments.
Cytotoxicity
The effect of the complexes (1 and 2) on the viability of human lung cancer cells (A549) and liver cancer cells (HepG2) were assayed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay.26 The cells were seeded at a density of 10000 cells per well, in 200 μl of a RPMI 1640 medium and were kept overnight in a CO2 incubator. Then, the complexes dissolved in DMSO were added to the cells at a final concentration of 1, 10, 25 and 50 μM in the cell culture media. After 48 hours, the wells were treated with 20 μl of MTT (5 mg ml−1 PBS) and incubated at 37 °C for 4 hours. The purple formazan crystals which formed were dissolved in 200 μl of DMSO and read at 570 nm in a micro plate reader.
The LDH activity was determined by the linear region of a pyruvate standard graph using regression analysis and expressed as percentage (%) leakage, as described previously.27 Briefly, to a set of tubes, 1 ml of a buffered substrate (lithium lactate) and 0.1 ml of the media or cell extract were added and tubes were incubated at 37 °C for 30 min. After adding 0.2 ml of an NAD solution, the incubation was continued for another 30 min. The reaction was then arrested by adding 0.1 ml of a DNPH reagent and the tubes were incubated for a further period of 15 min at 37 °C. After this, 0.1 ml of the media or cell extract was added to blank tubes after arresting the reaction with DNPH. 3.5 ml of 0.4 N sodium hydroxide was added to all the tubes. The intensity of the colour which developed was measured at 420 nm in a Shimadzu UV-vis spectrophotometer. The amount of LDH released was expressed as a percentage.
With the continuation of the above studies, the amount of nitrite was determined by the method of Stuehr and Marletta.28 Nitrite reacts with Griess reagent and gives a coloured complex which was measured at 540 nm. To 100 μl of the medium, 50 μl of Griess reagent I was added, mixed and allowed to react for 10 min. This was followed by a 50 μl addition of Griess reagent II, then the reaction mixture was mixed well and incubated for another 10 min at room temperature. The intensity of the pink colour which developed was measured at 540 nm in a microquant plate reader (Biotek Instruments).
Cellular uptake study
The cellular uptake of the complexes (1 and 2) was quantified according to the literature method with a slight modification.29 Briefly, the lung (A549) and liver (HepG2) cancer cells were treated with the complexes for 2 hours. The medium was aspirated and the cells were washed thrice with ice cold PBS. Then, the cells were lysed with PBS containing 1% Triton X-100. The concentration of the complexes in the cell lysates was measured with a fluorescence spectrophotometer (Jasco FP 6600) at their maximum excitation/emission wavelengths of 492/530 and 426/495, respectively, for complexes 1 and 2. To offset the background fluorescence from the cellular components, separate standardization curves were prepared using cellular lysates containing a series of known concentrations of different complexes and the intracellular concentrations were found using the standard curve.
Results and discussion
Synthesis, characterization and structural analysis
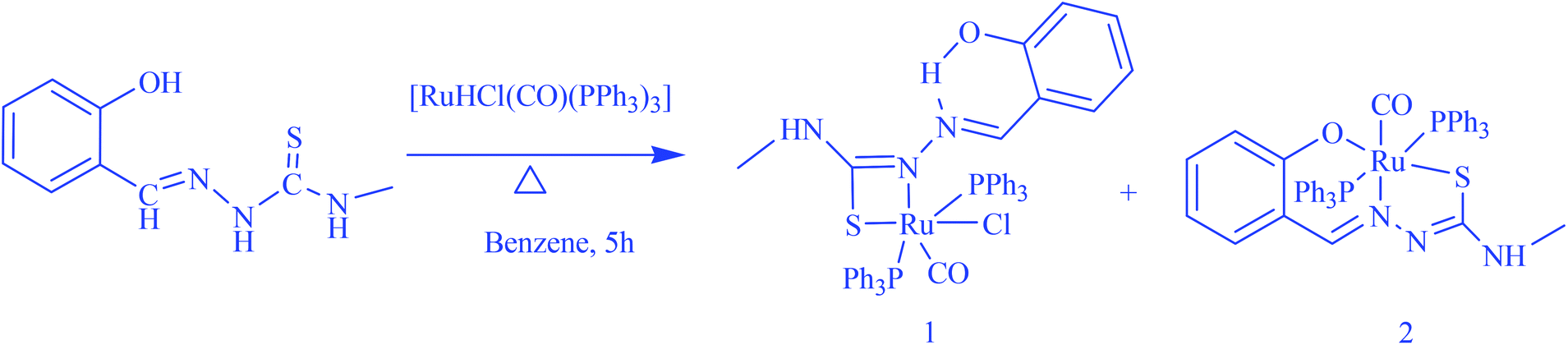
The reaction of [H2-(Sal-mtsc)] with an equimolar amount of [RuHCl(CO)(PPh3)3] resulted in the formation of two structurally different entities from a single tub (Scheme 1). The new complexes are soluble in common organic solvents, such as dichloromethane, chloroform, benzene, acetonitrile, ethanol, methanol, dimethylsulfoxide and dimethylformamide. The analytical data of the complexes agreed well with the proposed molecular formulae.
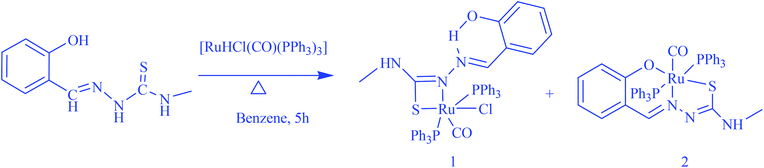 |
| | Scheme 1 Preparation of the new ruthenium(II) complexes. | |
The characteristic IR bands of the metal complexes compared with the free ligand provide significant indications regarding the coordination of thiosemicarbazone (Fig. S1–S3, ESI†). The IR spectrum of [H2-(Sal-mtsc)] showed a sharp band at 1609 cm−1 corresponding to the azomethine ν(CN) group, which has been lowered by 17–28 cm−1 in the complexes, indicating the coordination of the azomethine nitrogen to the ruthenium atom.30,31 A sharp band at 815 cm−1 corresponding to ν(CS) in the ligand completely disappeared in the new complexes and a new band appeared at 749 and 745 cm−1, respectively, corresponding to ν(C–S), indicating the tautomerism of the –NH–CS group and subsequent coordination of thiolate sulfur after deprotonation.32,33 A broad band corresponding to ν(OH) which appeared at 3358 cm−1 for the ligand was observed at 3430 cm−1 in complex 1, indicating the non-participation of phenolic oxygen in the coordination.34 However, this band completely disappeared in complex 2, indicating the involvement of phenolic oxygen in the coordination.31 A sharp band at 1925 and 1945 cm−1 accounts for the terminal carbonyl group in 1 and 2, respectively. In addition, vibrations corresponding to the presence of triphenylphosphine also appeared in the expected region.30 The electronic spectra of the new ruthenium(II) complexes recorded in dichloromethane showed intense absorptions in the ultraviolet and visible regions. The complexes displayed six bands in the region around 244–426 nm. The bands which appeared at the 244–293 nm region (16690–13211 dm3 mol−1 cm−1) have been assigned to intra ligand transitions35 and the bands at 311–384 nm (11720–9059 dm3 mol−1 cm−1) were assigned to LMCT (s→d). A band at 426 nm (7489 dm3 mol−1 cm−1) has been assigned to a forbidden (d→d) transition. The 1H-NMR spectrum of [H2-(Sal-mtsc)] showed three singlets at δ 11.4, 9.86 and 8.37 ppm, corresponding to –OH, –NH–CS and –CHN groups (Fig. S4–S6, ESI†).1i In addition, the terminal –NH and methyl group protons were found at δ 8.40 and δ 2.99 ppm, respectively. A complex overlap of signals as a multiplet at δ 6.6–7.9 ppm, corresponding to aromatic protons, were observed in the NMR spectra of the ligand and complexes.32 A sharp singlet appeared at δ 10.05 ppm in complex 1, corresponding to the phenolic –OH group, and a new sharp singlet observed at δ 5.3 ppm, corresponding to the imine –NH group, was formed through intramolecular hydrogen bonding between the phenolic –OH and N1 nitrogen atom, indicating the non-participation of phenolic oxygen in the bonding.36,37 However, the singlet corresponding to the –OH group completely disappeared in complex 2, indicating the involvement of phenolic oxygen in the coordination. A sharp singlet appeared at δ 8.46 ppm and 8.01 ppm in complexes 1 and 2 and was assigned to the azomethine proton of the Schiff base. In complex 1, a broad multiplet appeared at δ 4.82 ppm, which has been assigned to a terminal –NH proton, whereas the same signal was found at 6.0 ppm in complex 2. An additional doublet has been observed at δ 2.35–2.53 ppm corresponding to the methyl group protons of the ligands.38
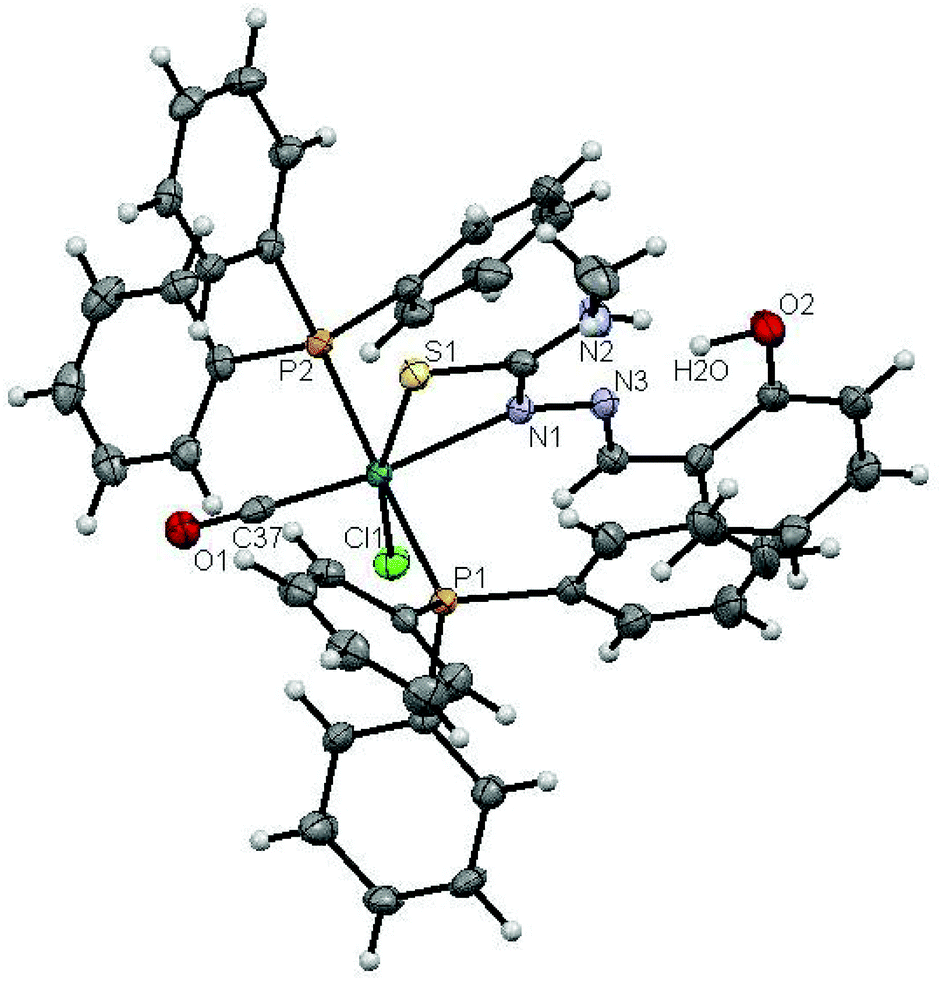
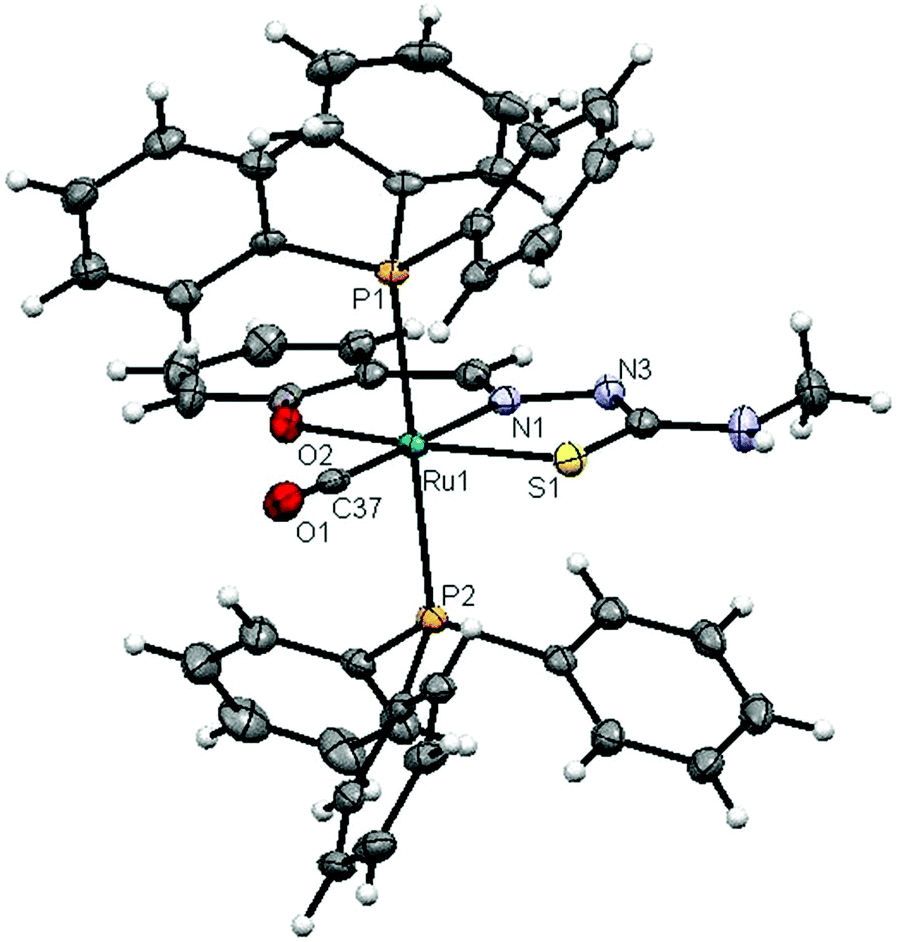
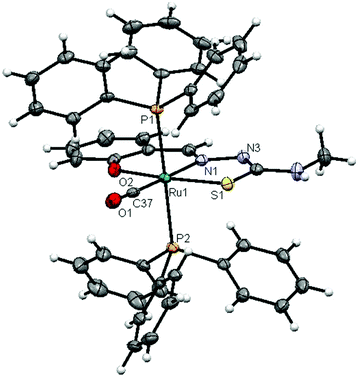
The molecular structures of complexes 1 and 2 with the numbering schemes are shown in Fig. 1 and 2. The crystallographic data, selected bond distances and angles are listed in Tables 1–3. Crystallographic analysis revealed that complex 1 crystallized in the monoclinic space group, P2(1)/n with selective inclusion of an acetonitrile moiety in the asymmetric unit. The ligand [H2-(Sal-mtsc)] coordinated to ruthenium in a bidentate mono-anionic form via alpha nitrogen and thiolate sulfur atoms, thus forming a strained four membered chelate ring with a N(1)–Ru(1)–S(1) bite angle of 66.26(6)° (Fig. 1). The Ru(1)–N(1) and Ru(1)–S(1) bond distances are 2.192(2) Å and 2.4012(7) Å, respectively. The other four sites are occupied by phosphorus atoms of two triphenylphosphine ligands, which are mutually trans to each other with Ru(1)–P(1) and Ru(1)–P(2) distances of 2.3815(7) and 2.3703(7) Å, and one chloride and a carbonyl group with Ru(1)–Cl(1) and Ru(1)–C(37) distances of 2.4247(7) and 1.845(3) Å, respectively. The observed bond distances are comparable with the values found in other reported ruthenium complexes.1m,36,37 The N(1)–Ru(1)–P(2) 88.91(6)°, N(1)–Ru(1)–P(1) 89.30(6)°, P(1)–Ru(1)–S(1) 87.40(2)° and P(2)–Ru(1)–Cl(1) 89.69(2)° cis angles are more acute than 90°, whereas the other cis angles, C(37)–Ru(1)–P(1) 91.62(8)°, C(37)–Ru(1)–P(2) 90.01(8)°, C(37)–Ru(1)–S(1) 100.49(8)°, P(2)–Ru(1)–S(1) 91.39(2)°, C(37)–Ru(1)–Cl(1) 96.54(8)°, N(1)–Ru(1)–Cl(1) 96.75(6)° and P(1)–Ru(1)–Cl(1) 91.04(2)° are more obtuse than 90°. The C(37)–Ru(1)–N(1) 166.67(10)°, P(2)–Ru–P(1) 178.13(3)° and S(1)–Ru(1)–Cl(1) 162.94(3)° trans angles significantly deviate from the linearity. The variation in the bond lengths and angles lead to a significant distortion from the ideal octahedral geometry of the complex. In addition, the complex contains one intramolecular hydrogen bond through the hydrogen atom of the hydroxy group with the hydrazinic nitrogen (N3) of the thiosemicarbazone moiety, with an O(1)–H(1)⋯N(3) distance of 2.62(9) Å (Table 3). Suitable crystals of complex 2 were obtained from dimethylformamide. It crystallized in the monoclinic space group, P2(1)/n. The thiosemicarbazone ligand coordinated to ruthenium in an ONS fashion by utilizing its phenolic oxygen, beta nitrogen and thiolate sulfur atoms, with the formation of one six membered and another five membered ring with a N(1)–Ru(1)–S(1) bite angle of 81.85(5)° (Fig. 2). However, in comparison with complex 1, the angular distortion observed in 2 is significantly less, which may be due to the strong ONS chelation of the ligand. The Ru(1)–N(1) and Ru(1)–S(1) bond distances were found to be 2.0865(18) Å and 2.3589(6) Å, respectively. The other three sites are occupied by phosphorus atoms of triphenylphosphine with Ru(1)–P(1) and Ru(1)–P(2) distances of 2.3934(5) and 2.4152(5) Å, respectively, and one carbonyl group with a Ru(1)–C(37) distance of 1.862(2) Å. These values are comparable with those found for complexes reported previously.1m,39 The remaining cis angles are given in Table 2. The trans angles O(2)–Ru(1)–S(1) 171.56(4)°, C(37)–Ru(1)–N(1) 178.06(8)° and P(1)–Ru–P(2) 179.26(2)° show some degree of deviation from the ideal geometry. The deviation in the bond angles and the variations in the bond lengths indicate distortion in the octahedral symmetry of the complexes. From the single crystal X-ray crystallographic results, it is interesting to note that the same donor atom (phenolic oxygen) played different roles in the two different entities from a single pot. In complex 1, it remains uncoordinated to the metal centre, whereas in complex 2 it is involved in bond formation. The non-participation of the phenolic oxygen in complex 1 may be due to the presence of intramolecular hydrogen bonding between the phenolic hydrogen and N1 hydrazinic nitrogen, which reduces the availability of the lone pair on nitrogen, possibly making these donor atoms unavailable for coordination and thus, giving a route to NS four membered ring formation by utilizing the N2 nitrogen and thiolate sulfur.
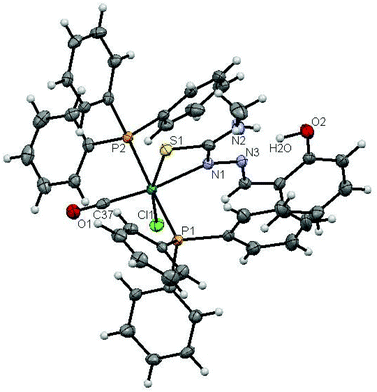 |
| | Fig. 1 ORTEP diagram of [Ru(H-Sal-mtsc)(CO)Cl(PPh3)2]·CH3CN (1) (the solvent moiety is removed for clarity and thermal ellipsoids are shown at 50% probability). All hydrogen atoms were placed in calculated positions and were refined with an isotropic displacement parameter 1.5 that of the adjacent carbon, nitrogen or oxygen atom. | |
 |
| | Fig. 2 ORTEP diagram of [Ru(Sal-mtsc)(CO)(PPh3)2] (2) (thermal ellipsoids are shown at 50% probability). All hydrogen atoms were placed in calculated positions and were refined with an isotropic displacement parameter 1.5 that of the adjacent carbon, nitrogen or oxygen atom. | |
Table 1 Crystal data and structure refinement of the new Ru(II) thiosemicarbazone complexes
| |
[Ru(H-Sal-mtsc)(CO)Cl(PPh3)2]·CH3CN (1) |
[Ru(Sal-mtsc)(CO)(PPh3)2] (2) |
| Empirical formula |
C48H43ClN4O2P2RuS |
C46H39N3O2P2RuS |
| Formula weight |
938.38 |
860.87 |
| Temperature |
110 K |
110 K |
| Wavelength |
0.71073 Å |
0.71073 Å |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P2(1)/n |
P2(1)/n |
| Unit cell dimensions |
|
|
|
a
|
14.2919(3) Å |
14.2779(5) Å |
|
b
|
16.3158(4) Å |
18.0273(6) Å |
|
c
|
19.0083(4) Å |
15.6331(6) Å |
| Alpha |
90° |
90° |
| Beta |
97.809(2)° |
103.194(4)° |
| Gamma |
90° |
90° |
| Volume |
4391.32(17) Å3 |
3917.6(2) Å3 |
|
Z
|
4 |
4 |
| Density (calculated) |
1.419 Mg m−3 |
1.460 Mg m−3 |
| Absorption coefficient |
0.582 mm−1 |
0.579 mm−1 |
|
F(000) |
1928 |
1768 |
| Theta range for data collection |
3.01 to 26.00° |
3.14 to 26.00° |
| Index ranges |
−17 ≤ h ≤ 13, −19 ≤ k ≤ 20, −21 ≤ l ≤ 23 |
−16 ≤ h ≤ 17, −22 = k ≤ 19, −19 ≤ l ≤ 19 |
| Reflections collected/unique |
20381/8553 [R(int) = 0.0378] |
20554/7648 [R(int) = 0.0307] |
| Completeness to theta= |
26.00 ° |
26.00 ° |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
8553/73/550 |
7648/0/500 |
| Goodness-of-fit on F2 |
0.919 |
0.920 |
| Final R indices [I > 2σ(I)] |
R
1 = 0.0345, wR2 = 0.0703 |
R
1 = 0.0282, wR2 = 0.0633 |
|
R indices (all data) |
R
1 = 0.0599, wR2 = 0.0737 |
R
1 = 0.0416, wR2 = 0.0652 |
| Largest diff. peak and hole |
0.668 and −0.465 e A−3 |
0.663 and −0.427 e A−3 |
Table 2 Selected bond lengths (Å) and angles (°) of the new Ru(II) thiosemicarbazone complexes
| [Ru(H-Sal-mtsc)(CO)Cl(PPh3)2]·CH3CN (1) |
[Ru(Sal-mtsc)(CO)(PPh3)2] (2) |
| Bond lengths |
Bond lengths |
| Ru(01)–N(1) |
2.192(2) |
Ru(1)–N(1) |
2.0865(18) |
| Ru(01)–P(1) |
2.3815(7) |
Ru(1)–P(1) |
2.3934(5) |
| Ru(01)–P(2) |
2.3703(7) |
Ru(1)–P(2) |
2.4152(5) |
| Ru(01)–S(1) |
2.4012(7) |
Ru(1)–S(1) |
2.3589(6) |
| Ru(01)–Cl(1) |
2.4247(7) |
Ru(1)–O(2) |
2.0863(14) |
| Ru(01)–C(37) |
1.845(3) |
Ru(1)–C(37) |
1.862(2) |
| Bond angles |
Bond angles |
| C(37)–Ru(01)–N(1) |
166.67(10) |
C(37)–Ru(1)–O(2) |
91.43(7) |
| C(37)–Ru(01)–P(2) |
90.01(8) |
C(37)–Ru(1)–N(1) |
178.06(8) |
| N(1)–Ru(01)–P(2) |
88.91(6) |
O(2)–Ru(1)–N(1) |
89.72(6) |
| C(37)–Ru(01)–P(1) |
91.62(8) |
C(37)–Ru(1)–S(1) |
97.00(6) |
| N(1)–Ru(01)–P(1) |
89.30(6) |
O(2)–Ru(1)–S(1) |
171.56(4) |
| P(2)–Ru(01)–P(1) |
178.13(3) |
N(1)–Ru(1)–S(1) |
81.85(5) |
| C(37)–Ru(01)–S(1) |
100.49(8) |
C(37)–Ru(1)–P(1) |
88.67(6) |
| N(1)–Ru(01)–S(1) |
66.26(6) |
N(1)–Ru(1)–P(1) |
89.85(5) |
| P(2)–Ru(01)–S(1) |
91.39(2) |
S(1)–Ru(1)–P(1) |
93.39(19) |
| P(1)–Ru(01)–S(1) |
87.40(2) |
C(37)–Ru(1)–P(2) |
90.77(6) |
| C(37)–Ru(01)–Cl(1) |
96.54(8) |
O(2)–Ru(1)–P(2) |
93.27(4) |
| N(1)–Ru(01)–Cl(1) |
96.75(6) |
N(1)–Ru(1)–P(2) |
90.72(5) |
| P(2)–Ru(01)–Cl(1) |
89.69(2) |
S(1)–Ru(1)–P(2) |
87.16(19) |
| P(1)–Ru(01)–Cl(1) |
91.04(2) |
P(1)–Ru(1)–P(2) |
179.26(2) |
| S(1)–Ru(01)–Cl(1) |
162. 94(3) |
— |
— |
Table 3 Hydrogen bonds for complex 1 [Å and °]
| D–H⋯A |
d(D–H) |
d(H⋯A) |
d(D⋯A) |
<(DHA) |
| Symmetry operation: (x, y, z). |
| O(1)–H(1)⋯N(1) |
1.10(1) |
1.60(1) |
2.62(9) |
152.82 |
Electrochemistry
The redox properties of the new Ru(II) complexes were analyzed using cyclic voltammetry in a dichloromethane window with 0.1 M tetrabutylammonium perchlorate as the supporting electrolyte at a scan rate of 100 mV S−1. All the potentials were referenced to a Ag/AgCl electrode. Ferrocene was used as an external standard and the reversibility was found at 59 mV.40 The complexes were found to be electro-active in the sweep range of ±2.00 V. The cyclic voltammogram of 1 showed a quasi-reversible one electron oxidation response, E1/2(oxi), at 0.315 V with a peak-to-peak separation of 432 mV and an irreversible reduction at −0.450 V (Table 4). This quasi-reversible oxidation peak can be assigned to the Ru(II)/Ru(III) process.41 Complex 2 exhibited irreversible oxidation at 0.709 V and quasi-reversible reduction corresponding to Ru(II)/Ru(I) at −0.580 V, with a peak-to-peak separation of 120 mV. A quasi-reversible oxidation, E1/2, observed at 1.113 and 1.160 V with a peak-to-peak separation of 206 and 180 mV, respectively, in complexes 1 and 2 can be ascribed to the oxidation of the thiosemicarbazone ligand. The reason for the quasi-reversible electron transfer process may be due to the slow electron transfer or the adsorption of the complex onto the electrode surface.42
Table 4 Electrochemical data of the new Ru(II) complexes
| Complex |
Oxidation [Ru(II)–Ru(III)] |
Reduction [Ru(II)–Ru(I)] |
Ligand oxidation |
| |
E
pa (V) |
E
pc (V) |
E
½ (V) |
ΔEp (mV) |
E
pa (V) |
E
pc (V) |
E
½ (V) |
ΔEp (mV) |
E
pa (V) |
E
pc (V) |
E
½ (V) |
ΔEp (mV) |
| (1) |
0.099 |
0.531 |
0.315 |
432 |
— |
−0.450 |
— |
— |
1.010 |
1.216 |
1.113 |
206 |
| (2) |
— |
0.709 |
— |
— |
−0.640 |
−0.520 |
−0.580 |
120 |
1.070 |
1.250 |
1.160 |
180 |
DNA binding studies
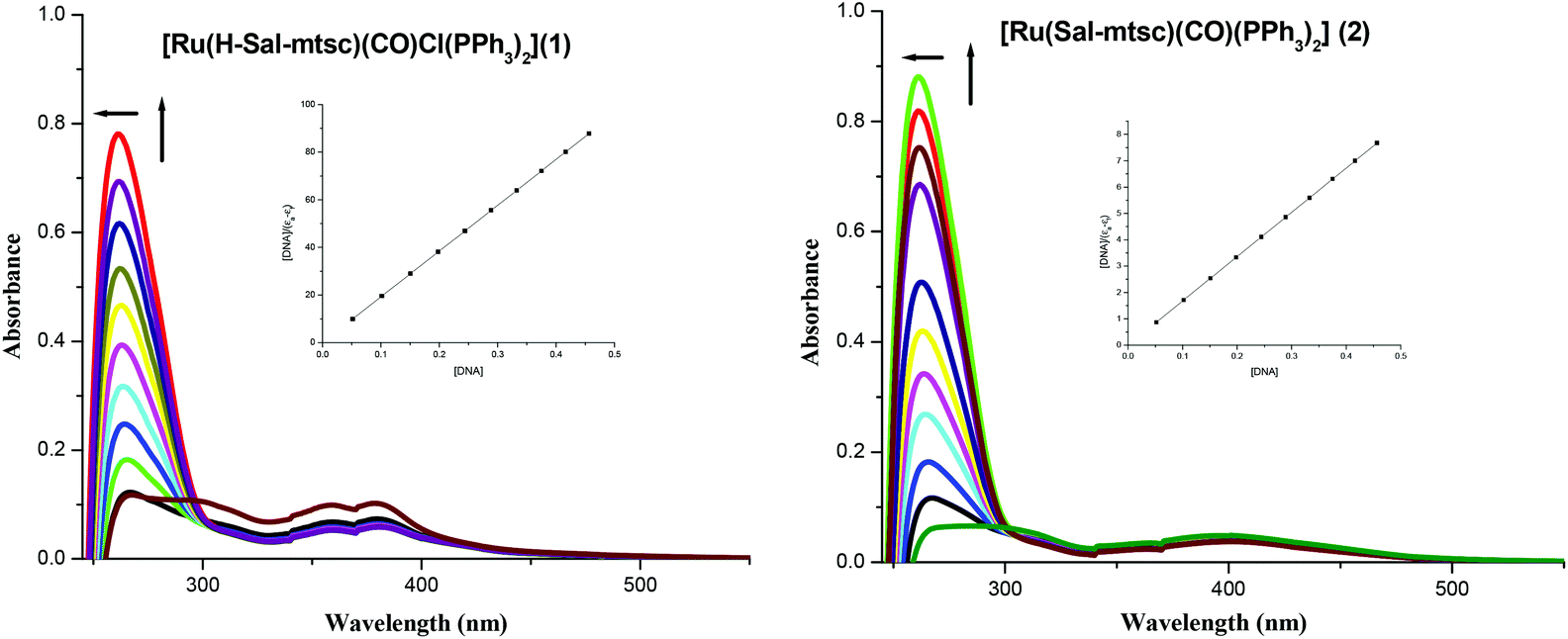
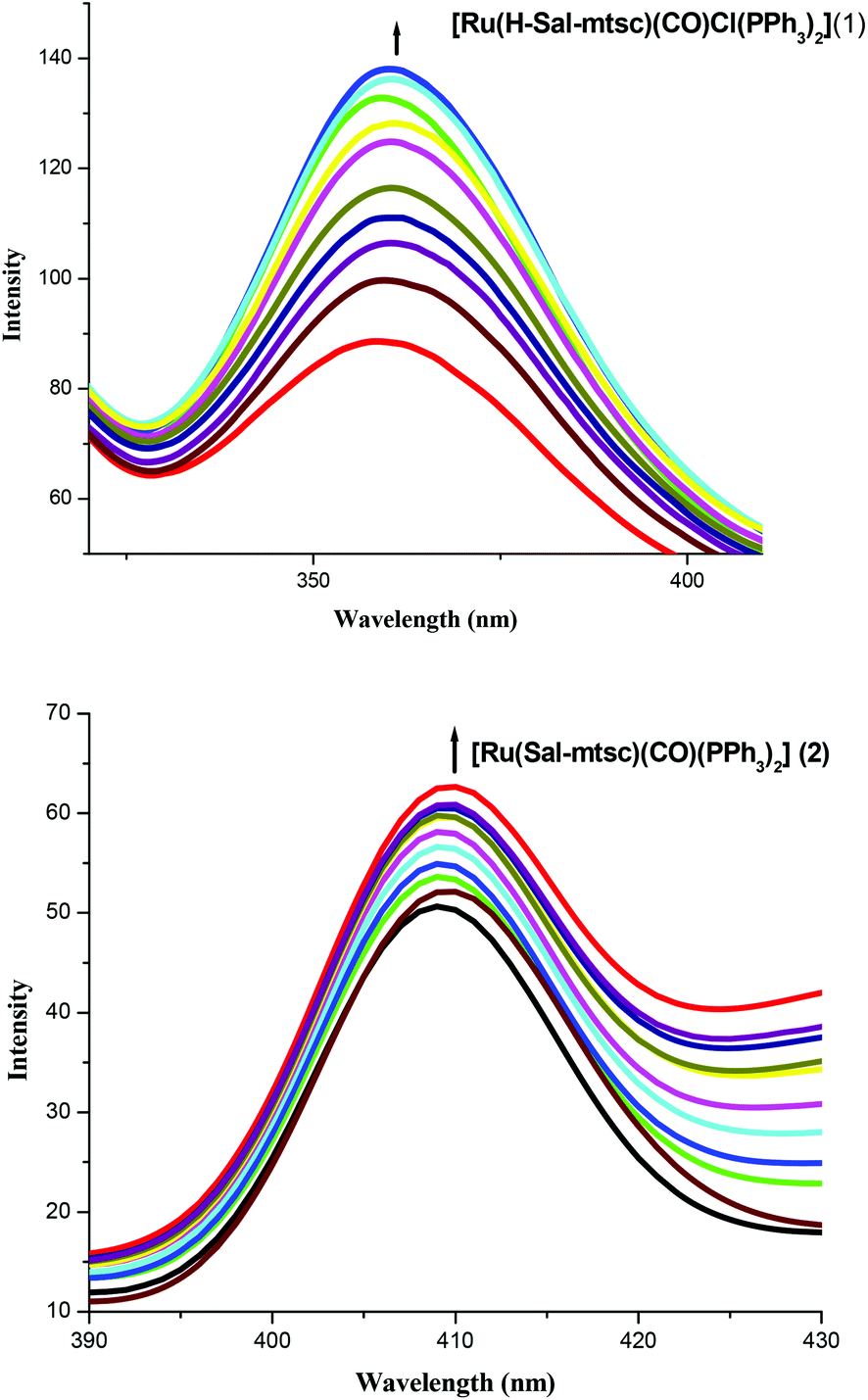
In order to study the binding affinity of the new ruthenium(II) complexes, they were subjected to DNA binding studies by taking CT-DNA as model DNA at room temperature. The interaction of transition metal complexes with DNA takes place via both covalent and/or non-covalent interactions.43 In the case of covalent binding, the labile ligand of the complexes is replaced by a nitrogen base of DNA, such as guanine N7, while the non-covalent DNA interactions include intercalative, electrostatic and groove binding of metal complexes outside the DNA helix. An absorption titration experiment was carried out to study the DNA binding properties of the new ruthenium complexes (1 and 2). The absorption spectra of the complexes at a constant concentration (1 μM) in the presence of different concentrations of CT-DNA (0.05–0.50 μM) are given in Fig. 3. The absorption spectra of complex 1 exhibited hyperchromism at 267 nm (A = 0.1101 to 0.7797) with a blue shift of 8 nm (up to 259 nm). The binding behaviour of complex 2 is also quite similar. It exhibited hyperchromism at 281 nm (A = 0.0659 to 0.8808) with a 7 nm blue shift. The observed hyperchromic effect with a blue shift suggests that the new complexes bind to CT-DNA by external contact, possibly due to electrostatic binding.44,45 The intrinsic binding constant, Kb, is a useful tool to monitor the magnitude of the binding strength of compounds with CT-DNA (Table 5). It can be determined by monitoring the changes in the absorbance in the IL band at the corresponding λmax with increasing concentration of DNA and is given by the ratio of the slope to the y intercept in plots of [DNA]/(εa − εf) versus [DNA] (insets in Fig. 3). From the binding constant values (Table 5), it is inferred that complex 2 exhibits better binding than 1. In the emission spectra, complex 1 had fluorescence at 362 nm (Fig. 4). When a CT-DNA solution was added to this complex solution, the fluorescence intensity was increased (I = 88.51 to 137.06) without any shift in the wavelength. Similarly, complex 2 showed hyperchromism (I = 48.58 to 61.65) at 410 nm. The enhanced fluorescence intensities observed for complexes 1 and 2 symbolize electrostatic binding of DNA and similar behaviour has already been observed for square planar palladium complexes.1k
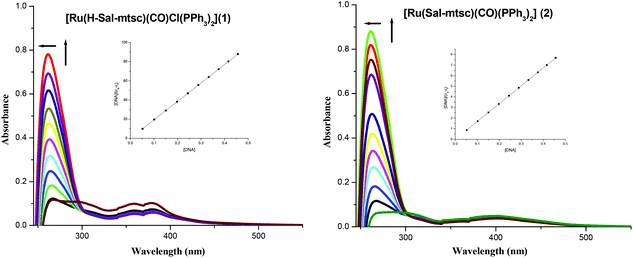 |
| | Fig. 3 Absorption titration spectra of 1 and 2 with increasing concentrations (0.05–0.5 μM) of CT-DNA (tris-HCl buffer, pH 7); the inset shows the binding isotherms with CT-DNA. | |
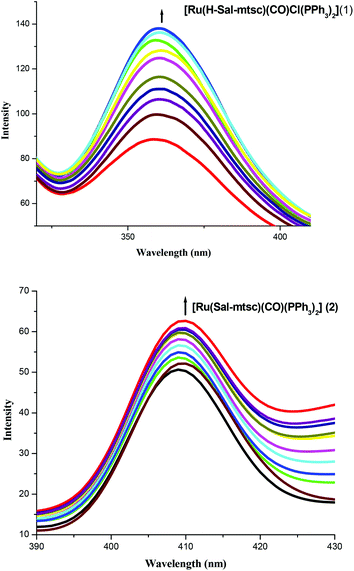 |
| | Fig. 4 Changes in the emission spectra of 1 and 2 with increasing concentrations (0.05–0.5 μM) of CT-DNA (tris-HCl buffer, pH 7). | |
Table 5 Binding constant for the interaction of the complexes with CT-DNA
| System |
K
b (×104 M−1) |
| CT-DNA + 1 |
1.68 ± 0.32 |
| CT-DNA + 2 |
1.92 ± 0.26 |
Competitive ethidium bromide (EB) binding studies were carried out in order to examine the binding mode of each complex with CT-DNA. The emission spectra of EB bound to CT-DNA in the absence and presence of each complex have been recorded at [EB] = 10 μM, [DNA] = 10 μM for increasing concentrations of each complex. The emission intensities of EB bound to CT-DNA at 621 nm showed an increasing trend with an increase in the concentration of the complexes (1 and 2), shown in Fig. S7 in the ESI,† indicating that they cannot effectively displace EB from the DNA-EB complex. This observation is often considered to be due to that they can bind weakly to the DNA, probably by electrostatic interactions. It is generally agreed that strong fluorescence enhancement accompanies intercalation of the dye into the double helix conformation of the nucleic acid, but there is also evidence for additional non-intercalative, less fluorescence-enhanced sites, which are presumed to involve electrostatic binding.46
Lysozyme binding studies
Understanding the protein–drug interaction plays a vital role in the fields of medicine, chemistry and biology.47,48 Lysozyme (14.4 kDa) is a small globular protein that contains structural elements commonly found in proteins.49 Lysozyme is a bacteriolytic agent with an ability to lyse the cell walls of bacteria by hydrolyzing the bond between N-acetylmuramic acid and N-acetylglucosamine of the peptidoglycan. It has many physiological and pharmaceutical functions, such as anti-inflammatory, anti-viral, immune modulatory, anti-histaminic and anti-tumor activities.50–54 However, interaction studies with lysozyme are less abundant than those with serum albumin.53 The possible binding interactions of the newly synthesized ruthenium(II) metal complexes 1 and 2 with lysozyme have been investigated by absorption and emission-titration experiments at room temperature.
UV-vis absorption studies of lysozyme
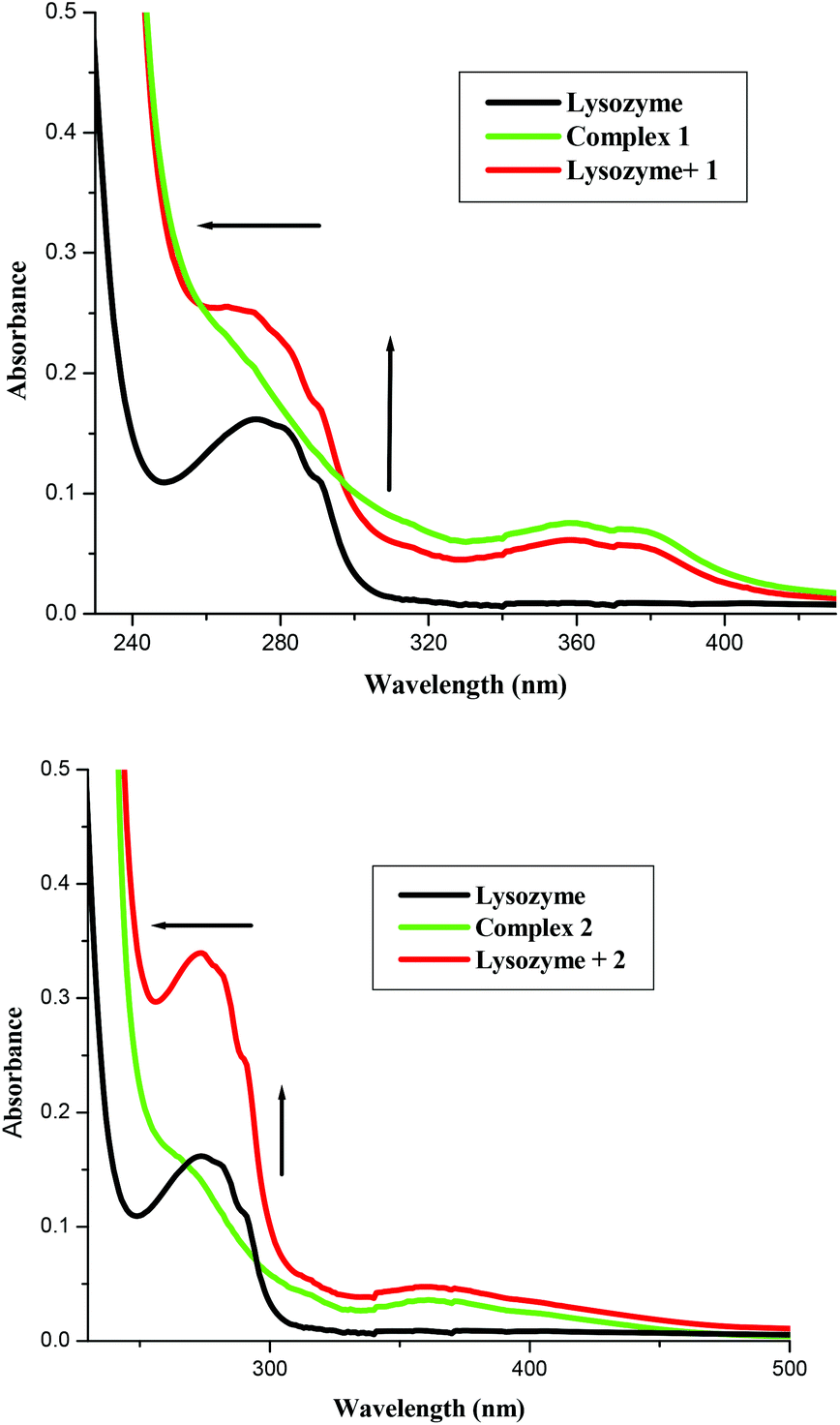
A UV-vis absorption measurement is a simple method to explore the structural changes in a protein and is useful to distinguish the type of quenching which exists i.e., static or dynamic quenching. Dynamic quenching only affects the excited states of the fluorophore and there are no changes in the absorption spectra, while static quenching refers to fluorophore–quencher complex formation in the ground state. Fig. 5 shows the absorption spectrum of lysozyme, 1 and Lyso + 1. The UV-vis spectra of lysozyme shows peaks at 280 nm whose intensities increased on increasing the concentrations of the complexes (Fig. 5) and clearly observed slight blue shifts in their absorbance maxima reveal that the microenvironment of the tryptophan residues on lysozyme are altered and the hydrophobicity is increased with the addition of the complex.1i,1k Similar changes in the absorption spectra resulted for complex 2. It is clear from the figure that in the presence of a metal complex, though the same concentration of lysozyme was used, the absorbance increased. This shows that the absorbance change is not additive but it could be due to the binding of the complex with the lysozyme and the resulting entity leading to significant enhancement, as reported in the literature involving lysozyme and other molecules.
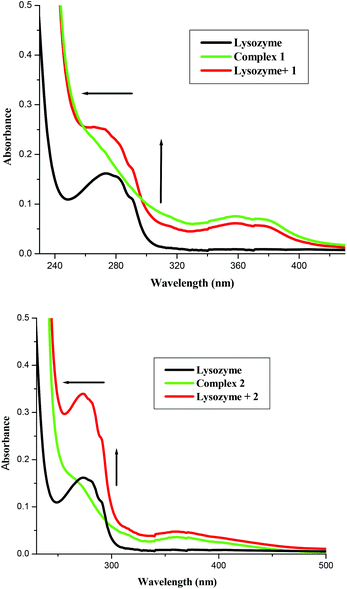 |
| | Fig. 5 Absorption spectra of lysozyme (1 × 10−5 M), Lyso-complex 1 and 2 [lysozyme = 1 × 10−6 M and 1 = 1 × 10−5 M] and complex 1 and 2, respectively. | |
Fluorescence quenching studies of lysozyme
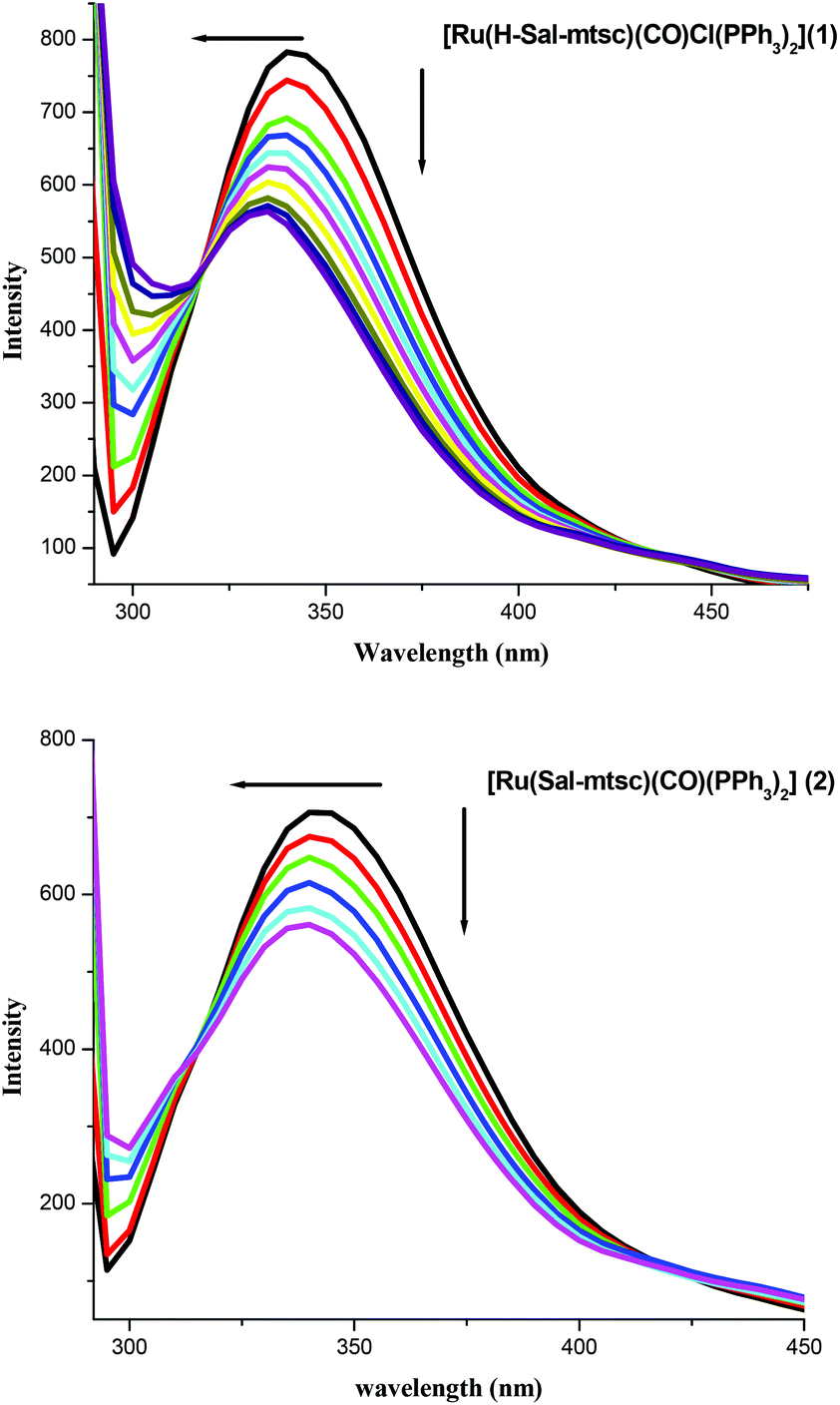
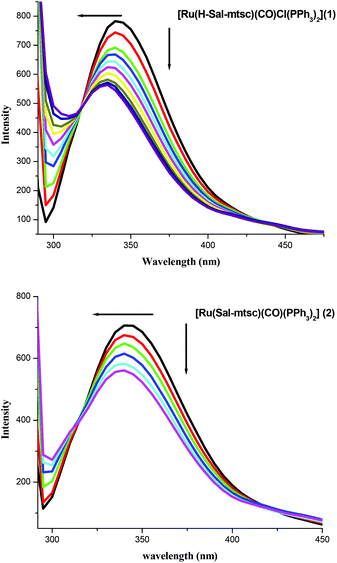
Fluorescence quenching experiments were carried out in order to illustrate the complex formation. The addition of the Ru(II) complexes to lysozyme produced a dramatic change to the emission profile (Fig. 6). The fluorescence of lysozyme was quenched effectively with a blue shift in its emission maximum and this may be due to the binding of the complexes with the active site in lysozyme.54 The mechanisms of quenching are usually classified as either dynamic quenching or static quenching. Static quenching refers to fluorophore–quencher complex formation and dynamic quenching refers to a process where the fluorophore and the quencher come into contact during the transient existence of the excited state. The possible quenching mechanism has been interpreted using the following Stern–Volmer eqn (3). The ratio of the fluorescence intensity in the absence of (I0) and in the presence of (Icorr-) the corrected fluorescence intensity of the quencher is related to the concentration of the quencher [Q] by a coefficient, KSV.
 |
| | Fig. 6 Fluorescence quenching of lysozyme by 1 and 2. | |
A linear curve is obtained within the concentrations studied (Fig. S8, ESI†) and KSV can be derived from the slope of the curve. Since the lifetime of lysozyme is in the order of 10−9 s, the calculated bimolecular quenching rate constant (kq) using KSV = kqτ0 was found to be higher than the maximum collisional quenching (kq) of various kinds of quenchers to biopolymers (2.0 × 1010 M−1 s−1). In addition, the percentage protein binding was calculated and found to be 17.42% and 27.14% for complexes 1 and 2, respectively. All the evidence suggests that the probable quenching mechanism of the Lyso-complex interaction was initiated by complex formation rather than dynamic quenching.
Binding constants and the number of binding sites
The binding constant, K, and the number of binding sites (n) can be obtained from the following Scatchard eqn (4).| | 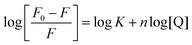 | (4) |
where, F0 and F are the fluorescence intensities in the absence and presence of the quencher. The value of K can be determined from the slope of log [(F0 − F)/F] versus log [Q], as shown in Fig. S9 in the ESI.† The calculated value of the binding constant (K) and the number of binding sites (n) are listed in Table 6. The binding behaviour decreases in the following order, 2 > 1. The values suggest that the binding occurs between the complexes (1 and 2) and lysozyme.
Table 6 Binding constant and number of binding sites for the interaction of the complexes with lysozyme
| S. No |
Complex |
K
SV × 104 M−1 |
k
q × 1013 M−1 s−1 |
K × 104 M−1 |
n
|
SEa |
SEb |
|
SE – Standard error for KSV.
SE – Standard error for K.
|
| 1 |
1
|
5.43 |
4.93 |
4.48 |
0.98 |
0.002 |
0.340 |
| 2 |
2
|
5.12 |
4.65 |
14.6 |
1.08 |
0.001 |
0.174 |
Conformational investigation
Synchronous fluorescence spectra.
Synchronous fluorescence spectra can give information about the conformational change of the protein molecular environment in the vicinity of fluorophore functional groups.55 The fluorescence of lysozyme is due to tryptophan (Trp) and tyrosine (Tyr) residues. Among them, tryptophan lies in the active site of the protein. When the D value (Δλ) between the excitation and emission wavelengths is stabilized at 60 nm, the synchronous fluorescence gives characteristic information of the tryptophan residues.56 The variation in the tryptophan emission is a consequence of the protein conformational changes. Therefore, to explore the structural change of lysozyme, we measured the synchronous fluorescence spectra at Δλ = 15 nm (Fig. S10, ESI†) and Δλ = 60 nm of lysozyme with complexes 1 and 2 (a representative figure is given in Fig. S11, ESI†). Both tryptophan and tyrosine shows a decrease in intensity but the tryptophan spectrum is accompanied by a blue shift. It reveals that the binding around Trp residues is strengthened. The results clearly indicate that both the complexes bind to the active site in the protein, which makes them potential molecules for biological applications.
Three-dimensional fluorescence spectra.
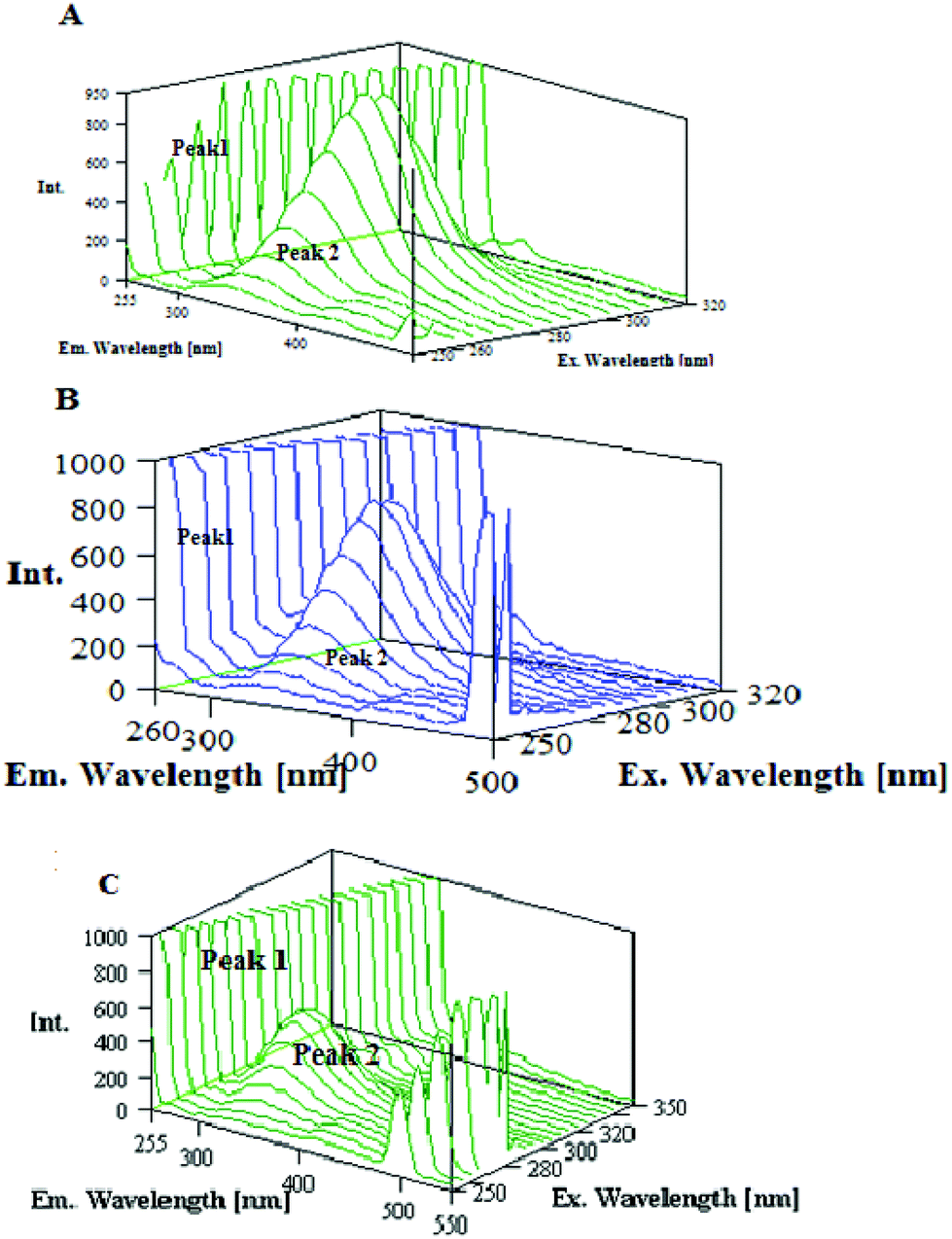
Three-dimensional fluorescence spectroscopy has become a popular fluorescence analysis technique in recent years.57,58 It can comprehensively show the fluorescence information of the sample, which makes the investigation of the characteristic conformational changes of a protein more scientific and credible. If there is a shift at the excitation or emission wavelength around the fluorescence peak, or the appearance of a new peak or the disappearance of an existing peak, it could be an important hint to suggest conformational changes to the protein. Fig. 7 presents the three dimensional fluorescence spectra and their characteristics of lysozyme (A), lysozyme–1 (B) and lysozyme–2 (C) with its contour map (Fig. S12, ESI†) and the corresponding parameters are presented in Table 7. In Fig. 7, peak 1 is the Rayleigh scattering peak (λex = λem), and peak 2 is the second-ordered scattering peak (λem = 2λex). Peak 1 mainly reveals the spectral behavior of the Trp and Tyr residues. The reason is that when lysozyme is excited at 280 nm, it mainly reveals the intrinsic fluorescence of the Trp and Tyr residues. Beside peak 1, there is another strong fluorescence peak 2, which mainly exhibits the fluorescence characteristics of polypeptide backbone structures.59 The fluorescence intensity of peak 2 decreased significantly after interacting with the new ruthenium(II) complexes (1 and 2), which implies that the peptide strand structure of lysozyme is changed. Analysis of the intensity changes of peaks 1 and 2 reveals that they both obviously decreased for both complexes but to different degrees: the fluorescence intensity of peak 2 has been quenched by 7.48% for complex 1 while by 33.43% for complex 2. The decrease in the fluorescence intensity of the two peaks in combination with the synchronous fluorescence results indicates that there is a specific interaction occurring between the lysozyme and new ruthenium(II) complexes (1 and 2), and the binding of the lysozyme–ruthenium(II) complexes (1 and 2) induced the slight unfolding of the polypeptides of lysozyme, which resulted in conformational changes of lysozyme to increase the exposure of some hydrophobic regions that had been buried.60 All these phenomena and analyses show that the binding of the new ruthenium(II) complexes (1 and 2) to lysozyme induced secondary structure changes in lysozyme.
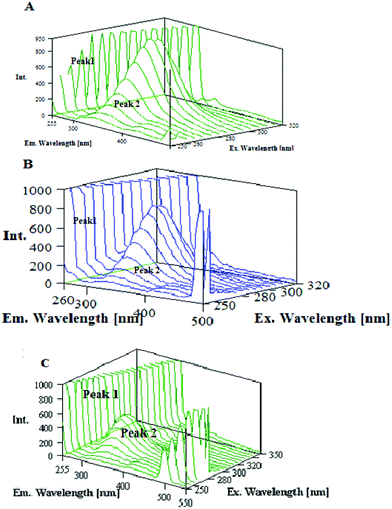 |
| | Fig. 7 Three dimensional fluorescence spectra for (A) Lyso, (B) Lyso-1, (C) Lyso-2. | |
Table 7 Three dimensional fluorescence spectral characteristics of (A) Lyso, (B) Lyso-1, (C) Lyso-2
| System |
Peak 2 λex/λem |
Δλ |
Intensity |
SE |
| Lyso (A) |
280/340 |
60 |
797.9 |
4.57 |
| Lyso + 1(B) |
280/335 |
55 |
738.2 |
3.50 |
| Lyso + 2(C) |
280/338 |
58 |
531.1 |
4.15 |
Cytotoxicity (MTT, LDH and NO)
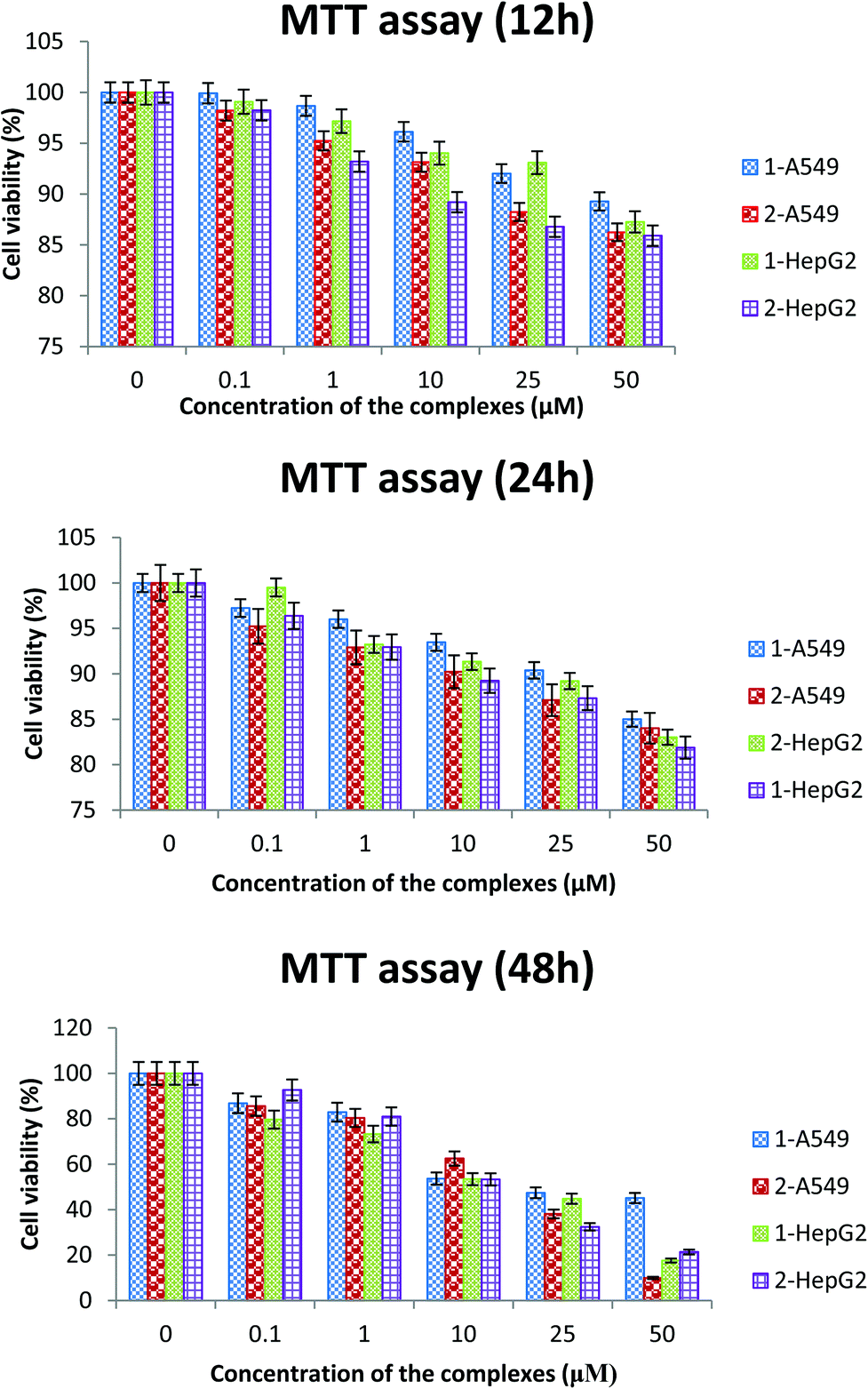
Preliminary screening of the anticancer activity of the new ruthenium(II) complexes in two different human cancer cell lines (A549 and HepG2) was performed by means of a colorimetric assay (MTT assay), which measures mitochondrial succinate dehydrogenase activity as an indication of cell viability. The cell viability after treating the cancer cells with different concentrations of the complexes for 12 h, 24 h and 48 h indicates that the cell viability was significantly reduced after the 48 h treatment than the other time points (Fig. 8). Hence, a 48 h time period was used for further cytotoxicity studies. Both the complexes (1 and 2) showed cytotoxicity and their IC50 values show inhibition of cancer cell growth at a 50% level. Earlier reports suggest that ruthenium complexes exhibit higher IC50 values on A549 and HepG2 cancer cells.61,62 The IC50 values of the two complexes fall in low micro molar concentrations. The results of the present study reveal that complex 2 is more active than 1 in the tested cell lines (Table 8).
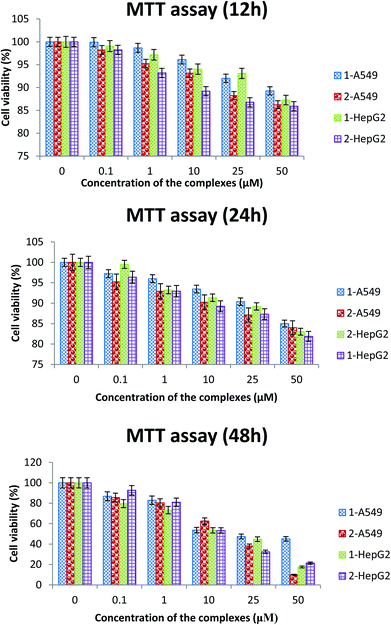 |
| | Fig. 8 The cell viability after treating the cancer cells with different concentrations of the complexes for 12 h, 24 h and 48 h. | |
Table 8
In vitro cytotoxicity of complexes 1 and 2 for the human tumour cell lines A549 and HepG2. The cells were exposed to different concentrations of the complexes for 48 h and the cell viability was assessed using an MTT assay. The IC50 values were calculated and tabulated. The results shown are the mean ± SEM, which are three separate experiments performed in triplicate
| Complexes |
IC50 values (μM) |
| |
A549 |
HepG2 |
|
1
|
20 ± 0.51 |
18 ± 1.01 |
|
2
|
15 ± 0.97 |
12 ± 0.84 |
| Cisplatin |
25 ± 0.46 |
9 ± 0.09 |
It is interesting to note that complexes 1 and 2 exhibited better activity than cisplatin in the A549 cell line. The IC50 values for complexes 1 and 2 were determined as 20 ± 0.51 and 15 ± 0.97 μM, respectively, and for cisplatin it was found to be 25 ± 0.46 μM for the A549 cells, whereas in the HepG2 cell line, the IC50 values for the complexes were determined as 18 ± 1.01 and 12 ± 0.84 μM, respectively, which are high when compared with cisplatin whose IC50 value is 9 ± 0.09 μM. With lower IC50 values, the complexes indicate their potential to inhibit cancer cell growth even at lower concentrations. In addition, there are reports available for complexes exhibiting higher IC50 values with the least activity in lower concentrations.61,62 The present data shows that complexes with a lower incubation period and with higher cytotoxic effects can replace compounds which need longer time periods. A longer incubation period may result in the development of drug resistance and lead to a deteriorating effect on non-target sites in the human body. Hence, compounds which show activity in shorter incubation time periods are preferred for clinical purposes.
Lactate dehydrogenase (LDH) leakage is an indicator of cell membrane integrity.63 When the cell membrane is disrupted, the membrane-bound LDH leaks into the medium and hence, LDH leakage is considered a hallmark of cytotoxicity. Cell death leads to a collapse in membrane integrity, thereby releasing LDH into the medium. In the present study, a significant level of LDH leakage was observed in the cell culture medium of the A549 and HepG2 cell lines when they were treated with the respective IC50 concentrations of the complexes for a period of 48 hours (Table 9). We have previously reported that ruthenium and other complexes with significant membrane-damaging effects showed selectivity towards cancer cells while exhibiting minimal/no toxicity towards normal cells.1j,o
Table 9 Percentage of lactate dehydrogenase released by the human cancer cell lines A549 and HepG2 after an incubation period of 48 hours with complexes 1 (20 μM for A549 and 18 μM for HepG2) and 2 (15 μM for A549 and 12 μM for HepG2). The results shown are the mean ± SEM, which are three separate experiments performed in triplicate
| Complexes |
LDH release (%) |
| |
A549 |
HepG2 |
| Control |
0 |
0 |
|
1
|
24.52 ± 1.69 |
26.35 ± 2.03 |
|
2
|
29.25 ± 2.59 |
31.63 ± 2.88 |
A major challenge in the treatment of lung and liver cancer is improving the response to chemotherapy. Nitric oxide (NO) has great potential as an agent to augment the effects of radiation therapy in several cancers.64–67 Excess NO production by tumor cells may be detrimental to their own survival due to NO-induced apoptosis.68 Given the remarkable array of bioeffector roles that nitric oxide has been discovered to play, it is critical to design improved agents which enhance the nitric oxide production of cancer cells and thereby inducing them to culminate their life cycle. Consequently, we have been attempting to exploit the chemical versatility of the new ruthenium complexes to achieve such a role. There was a significant increase in the level of nitric oxide in the culture medium of the cells treated with the ruthenium complexes when compared to the control cells of both A549 and HepG2 cells, in which complex 2 is more potent (Table 10). At higher concentrations, NO induces oxidative stress, thereby damaging biomolecules, such as proteins, lipids and nucleic acids, which in turn induces apoptosis.67 Our results indicate that the complexes may exhibit their cytotoxicity through the induction of oxidative stress, as indicated by the significant release of nitric oxide in the culture medium, which was measured as the stable product nitrite.
Table 10 nmoles of nitrite released by the human cancer cell lines A549 and HepG2 after an incubation period of 48 hours with complexes 1 (20 μM for A549 and 18 μM for HepG2) and 2 (15 μM for A549 and 12 μM for HepG2). The results shown are the mean ± SEM, which are three separate experiments performed in triplicate
| Complexes |
NO release (nmoles of nitrite released) |
| |
A549 |
HepG2 |
| Control |
2.35 ± 0.12 |
2.71 ± 0.10 |
|
1
|
4.94 ± 0.09 |
5.31 ± 0.12 |
|
2
|
5.24 ± 0.14 |
5.87 ± 0.08 |
Cellular uptake study
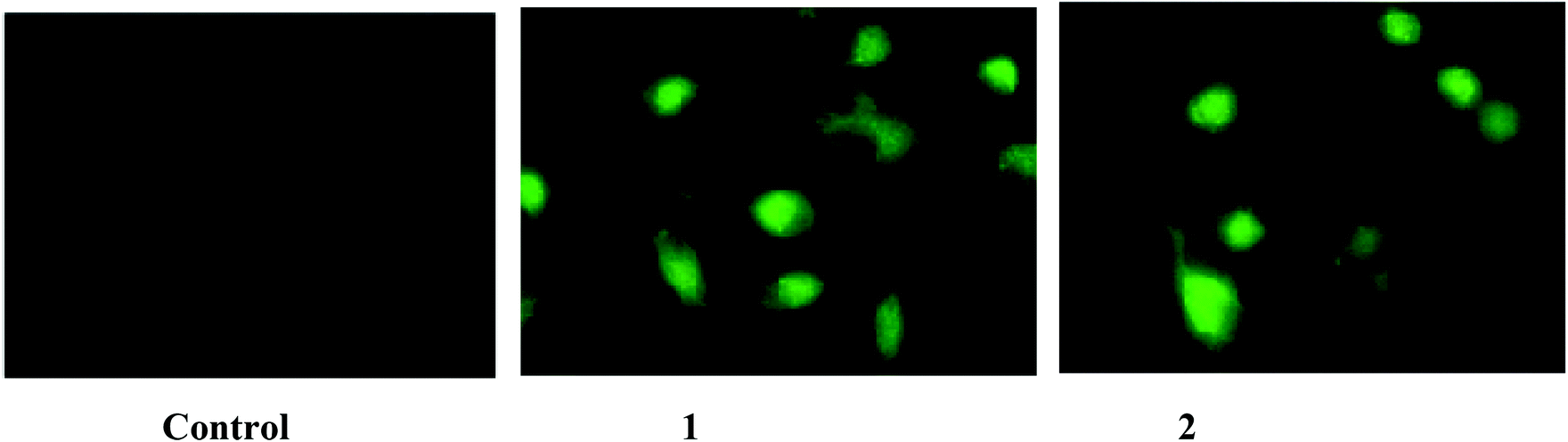
The intracellular uptake of a specific drug plays a vital role in ameliorating several diseases. Since the IC50 values are critical when compared to those of the normal cells in the human body, the present study focused on the concentrations which showed 50% inhibition for the A549 and HepG2 cell lines. The intracellular concentrations of complexes 1 and 2 were found using the method described in the methodology section. The intracellular uptake of the complexes will be in the early stages and hence, the study was carried out after a 2 h treatment. After a 48 h incubation time, the fluorescence emitted by the cells will be due to the cellular remnants and not due to the complexes alone. The percentage of cellular uptake was found to be higher in complex 1 followed by complex 2 (Table 11). The uptake of these complexes was more than 25% for complex 1 and more than 15% for complex 2. Since the anticancer drug doxorubicin has red fluorescence, it is used as a positive control. It has been observed that the potential of the cells to internalize the complexes was less than doxorubicin, which is used in lower concentrations (5 μM) than the tested complexes. From the results, it is clear that even low concentrations of the complexes are cytotoxic to lung cancer cells when they are completely absorbed by the cells. To visualize the accumulation of the complexes in to the cancer cells, the complex-treated A549 cells were observed under a fluorescent microscope (Fig. 9). The results confirmed that the intracellular accumulation of complex 1 was more than the complex 2-treated group. These results indicate that the uptake levels are dependent on the dose of each complex used and also indicate that their cytotoxicities, as determined by the MTT assay, were not disproportionately influenced by the complexes having different cellular uptake levels.
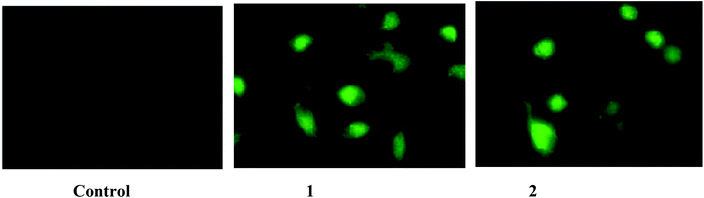 |
| | Fig. 9 Intracellular fluorescence of complexes 1 (20 μM) and 2 (15 μM) with A549 after an incubation period of 2 h were photographed. The figures are representatives of three independent experiments with similar results (magnification 400×). | |
Table 11 Percentage of cellular uptake of complexes 1, 2 and doxorubicin by human cancer cell lines A549 and HepG2 after an incubation period of 2 hours with complex 1 (20 μM for A549 and 18 μM for HepG2) and 2 (15 μM for A549 and 12 μM for HepG2). The results shown are the mean ± SEM, which are three separate experiments performed in triplicate
| Complexes |
Cellular uptake (%) |
| |
A549 |
HepG2 |
| Control |
0 |
0 |
|
1
|
24.89 ± 0.68 |
28.02 ± 0.83 |
|
2
|
15.01 ± 0.48 |
19.12 ± 0.76 |
| Doxorubicin |
25.9 ± 0.71 |
18.85 ± 0.73 |
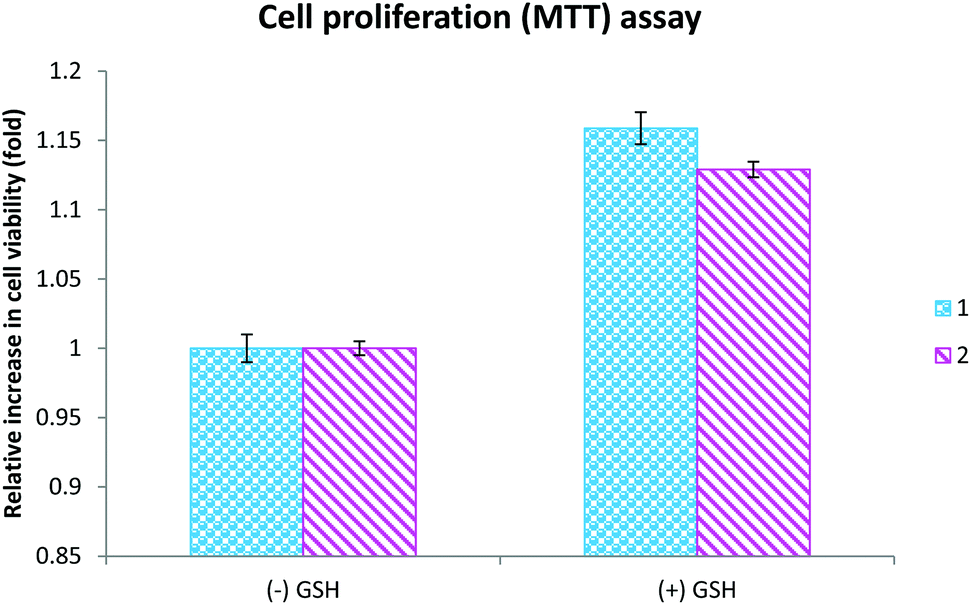
Oxidative stress occurs as a result of an imbalance between the production of reactive oxygen species (ROS) and their neutralization by antioxidants.69 Irrespective of their origin, ROS induce cell death via mechanisms including lipid peroxidation, alteration of cellular proteins, and initiation of diverse stress-signaling pathways. These potentially more destructive molecules could induce cellular damage rapidly if an antioxidant is not present in a sufficient concentration in the cells.70 Among antioxidants, glutathione is a nonprotein thiol that neutralizes intracellular reactive oxygen species generation.71 Reduced GSH, which is present in the cytosol (cGSH, 80% of total cellular GSH) and the mitochondria (mGSH, 10–15% of total GSH), acts as a hydrogen donor in the neutralization of ROS and is itself converted to the inactive glutathione disulfide.72 Moreover, GSH function in cell protection is related largely to the neutralization of reactive oxygen intermediates produced from H2O2.70 Besides, there are possible chances for the complexes to be conjugated with the extracellular GSH and be less cytotoxic in its presence. To check whether the complex-induced the cytotoxicity through the induction of oxidative stress, we analyzed the cell viability in the presence of glutathione. The results indicated that complex-induced cytotoxicity was inhibited by glutathione, revealing the fact that the complexes exhibited their cytotoxicity through the induction of reactive oxygen species generation (Fig. 10). In addition, the results of the cell viability assay indicates that the concentration required to cause 50% cell death of both lung and liver cancer cells is less than the already reported ruthenium(II) complexes containing salicylaldehydethiosemicarbazone.1m Even though the levels of LDH, NO and cellular uptake were lower for the newly synthesized ruthenium(II) complexes containing 4(N)-methyl substituted thiosemicarbazone than the already reported complexes,1m the corresponding lower IC50 values for the newly synthesized complexes shows their higher efficacy.
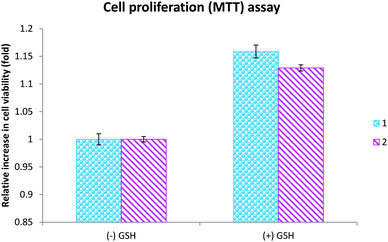 |
| | Fig. 10 The cells were exposed to complexes 1 (20 μM) and 2 (15 μM) for 48 h in the presence and absence of glutathione and the cell viability was assessed using an MTT assay. The results shown are the mean ± SEM, which are three separate experiments performed in triplicate. | |
Conclusions
The reaction of salicylaldehyde-4(N)-methylthiosemicarbazone [H2-(Sal-mtsc)] with [RuHCl(CO)(PPh3)3] was carried out and the stoichiometric reaction afforded two different products with different structural features and were characterized by various spectral, analytical, X-ray crystallography and cyclic voltammetric techniques. It is interesting to note that the ligand coordinated as an NS bidentate chelate in 1 by forming a strained four membered ring and leaving the phenolic oxygen uncoordinated. However, in 2, the ligand coordinated as an ONS dibasic tridentate chelate by forming more common five and six membered rings. These new complexes have been used to study their binding property with CT-DNA/lysozyme. From the results obtained through UV absorption titrations, fluorescence spectroscopy and ethidium bromide competitive studies, it is inferred that both complexes non-intercalatively bind with CT-DNA. When the complexes interacted with lysozyme, the fluorescence intensity of lysozyme was quenched and the quenching was found to be static. In addition, the synchronous and three dimensional fluorescence spectral investigations revealed that complexes 1 and 2 induced a small change in the secondary structure of lysozyme at the studied conditions. From the in vitro cytotoxicity studies in two human tumor cell lines, A549 and HepG2, and cellular uptake studies, it is inferred that complex 2 exhibited higher activity than complex 1 and this may be due to the strong chelation and increased electron delocalization in six and five membered chelate rings, which increases the permeability of the ruthenium ion into the cells.
Acknowledgements
The financial support received from the Department of Science and Technology (DST), India and the Council of Science and Industrial Research (CSIR), New Delhi is gratefully acknowledged.
References
-
(a) S. Padhye and G. B. Kauffman, Coord. Chem. Rev., 1985, 63, 127–160 CrossRef CAS;
(b) D. X. West, A. E. Liberta, S. B. Padhye, R. C. Chikate, P. B. Sonawane, A. S. Kumbhar and R. G. Yerande, Coord. Chem. Rev., 1993, 123, 49–71 CrossRef CAS;
(c) J. S. Casas, M. S. G. Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef CAS;
(d) M. J. M. Campbell, Coord. Chem. Rev., 1975, 15, 279–319 CrossRef CAS;
(e) D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS PubMed;
(f) H. Beraldo and D. Gambino, Mini-Rev. Med. Chem., 2004, 4, 31–39 CrossRef CAS;
(g) M. J. Clarke, F. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511–2533 CrossRef CAS PubMed;
(h) A. G. Quiroga and C. N. Ranninger, Coord. Chem. Rev., 2004, 248, 119–133 CrossRef CAS PubMed;
(i) P. Kalaivani, R. Prabhakaran, E. Ramachandran, F. Dallemer, G. Paramaguru, R. Renganathan, P. Poornima, V. Vijaya Padma and K. Natarajan, Dalton Trans., 2012, 41, 2486–2499 RSC;
(j) R. Prabhakaran, P. Kalaivani, P. Poornima, F. Dallemer, G. Paramaguru, V. Vijaya Padma, R. Renganathan and K. Natarajan, Dalton Trans., 2012, 41, 9323–9336 RSC;
(k) P. Kalaivani, R. Prabhakaran, F. Dallemer, P. Poornima, E. Vaishnavi, E. Ramachandran, V. Vijaya Padma, R. Renganathan and K. Natarajan, Metallomics, 2012, 4, 101–113 RSC;
(l) P. Kalaivani, R. Prabhakaran, M. V. Kaveri, R. Huang, R. J. Staples and K. Natarajan, Inorg. Chim. Acta, 2013, 405, 415–426 CrossRef CAS PubMed;
(m) P. Kalaivani, R. Prabhakaran, P. Poornima, F. Dallemer, K. Vijayalakshmi, V. Vijaya Padma and K. Natarajan, Organometallics, 2012, 31, 8323–8332 CrossRef CAS;
(n) R. Prabhakaran, P. Kalaivani, R. Huang, P. Poornima, V. Vijaya Padma, F. Dallemer and K. Natarajan, JBIC, J. Biol. Inorg. Chem., 2013, 18, 233–247 CrossRef CAS PubMed;
(o) P. Kalaivani, R. Prabhakaran, P. Poornima, R. Huang, V. Hornebecq, F. Dallemer, V. Vijaya Padma and K. Natarajan, RSC Adv., 2013, 3, 20363–20378 RSC.
- V. S. Li, D. Choi, Z. Wang, L. S. Jimenez, M. S. Tang and H. Kohn, J. Am. Chem. Soc., 1996, 18, 2326–2331 CrossRef.
- G. Zuber, J. C. Quada and S. M. Hecht, J. Am. Chem. Soc., 1998, 120, 9368–9369 CrossRef CAS.
- S. M. Hecht, J. Nat. Prod., 2000, 63, 158–168 CrossRef CAS PubMed.
- J. Reedijk, Chem. Commun., 1996, 801–806 RSC.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed , and references cited therein.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS.
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS PubMed.
- M. R. Gill, H. Derrat, C. G. W. Smythe, G. Battaglia and J. A. Thomas, ChemBioChem, 2011, 12, 877–880 CrossRef CAS PubMed.
- G. S. Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- A. M. Pizarro and P. J. Sadler, Biochimie, 2009, 91, 1198–1211 CrossRef CAS PubMed.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458–470 RSC.
- I. Bratsos, S. Jedner, T. Gianferrara and E. Alessio, Chimia, 2007, 61, 692–697 CrossRef CAS.
- M. J. Hannon, Pure Appl. Chem., 2007, 79, 2243–2263 CrossRef CAS.
- G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069–9075 RSC.
- C. S. Allardyce and P. J. Dyson, Platinum Met. Rev., 2001, 45, 62–69 CAS.
- J. Reedijk, Platinum Met. Rev., 2008, 52, 2–11 CrossRef CAS.
- J. Kostova, Curr. Med. Chem., 2006, 13, 1085–1107 CrossRef.
- S. Purohit, A. P. Kolay, L. S. Prasad, P. T. Manoharan and S. Ghosh, Inorg. Chem., 1989, 28, 3735–3742 CrossRef CAS.
- D. L. Klayman, J. F. Brtosevich, T. S. Griffin, C. J. Mason and J. P. Scovill, J. Med. Chem., 1997, 22, 855–862 CrossRef.
- N. Ahmed, J. J. Levision, S. D. Robinson and M. F. Uttley, Inorg. Synth., 1974, 15, 45–64 CrossRef.
-
A. I. Vogel, Textbook of Practical Organic Chemistry, 5h edn, Longman, London, 1989, p. 268 Search PubMed.
-
G. M. Sheldrick, SHELXTL Version 5.1, An Integrated System for Solving, Refining and Displaying Crystal Structures from Diffraction Data, Siemens Analytical X-ray Instruments, Madison, WI, 1990 Search PubMed.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS PubMed.
- M. Van de Weert and L. Stella, J. Fluoresc., 2010, 20, 625–629 CrossRef CAS PubMed.
- T. Mossman, J. Immunol. Methods, 1983, 65, 55–63 CrossRef.
- W. E. C. Wacker, D. D. Ulmer and B. L. Valee, J. Med., 1956, 255, 449–456 CAS.
- D. J. Stuehr and M. A. Marletta, J. Immunol., 1987, 139, 518–525 CAS.
- X. B. Xiong, Z. Ma, R. Lai and A. Lavasanifar, Biomaterials, 2010, 31, 757–768 CrossRef CAS PubMed.
- R. Prabhakaran, C. Jayabalakrishnan, V. Krishnan, K. Pasumpon, D. Sukanya, H. Bertagnolli and K. Natarajan, Appl. Organomet. Chem., 2006, 20, 203–213 CrossRef CAS.
- R. Prabhakaran, R. Karvembu, T. Hashimoto, K. Shimizu and K. Natarajan, Inorg. Chim. Acta, 2005, 358, 2093–2096 CrossRef CAS PubMed.
- R. Prabhakaran, S. V. Renukadevi, R. Karvembu, R. Huang, J. Mautz, G. Huttner, R. Subhaskumar and K. Natarajan, Eur. J. Med. Chem., 2008, 43, 268–273 CrossRef CAS PubMed.
- R. Prabhakaran, R. Huang, S. V. Renukadevi, R. Karvembu, M. Zeller and K. Natarajan, Inorg. Chim. Acta, 2008, 361, 2547–2552 CrossRef CAS PubMed.
- R. Prabhakaran, P. Kalaivani, R. Jayakumar, M. Zeller, A. D. Hunter, S. V. Renukadevi, E. Ramachandran and K. Natarajan, Metallomics, 2011, 3, 42–48 RSC.
- J. G. Tojal, J. L. Dizarro, A. G. Orad, A. R. P. Sanz, M. Ugalda, A. A. Diaz, J. L. Serra, M. I. Arriortua and T. Rojo, J. Inorg. Biochem., 2001, 86, 627–633 CrossRef.
- F. Basuli, M. Ruf, C. G. Pierpont and S. Bhattacharya, Inorg. Chem., 1998, 37, 6113–6116 CrossRef CAS.
- R. Prabhakaran, P. Kalaivani, R. Huang, M. Sieger, W. Kaim, P. Viswanathamurthi, F. Dallemer and K. Natarajan, Inorg. Chim. Acta, 2011, 376, 317–324 CrossRef CAS PubMed.
- D. L. Klayman, J. P. Scovill, J. F. Brtosevich and J. Bruce, J. Med. Chem., 1983, 26, 35–39 CrossRef CAS.
- R. Prabhakaran, R. Huang, R. Karvembu, C. Jayabalakrishnan and K. Natarajan, Inorg. Chim. Acta, 2007, 360, 691–694 CrossRef CAS PubMed.
- R. R. Gagne, C. A. Koval and G. C. Lisensky, Inorg. Chem., 1980, 19, 2854–2855 CrossRef CAS.
- M. Murali and M. Palaniandavar, Dalton Trans., 2006, 730–743 RSC.
- A. W. Wallace, W. R. Murphy and J. D. Peterson, Inorg. Chim. Acta, 1989, 166, 47–54 CrossRef CAS.
- Q. L. Zhang, J. G. Liu, H. Chao, G. Q. Xue and L. N. Ji, J. Inorg. Biochem., 2001, 83, 49–55 CrossRef CAS.
-
(a) E. C. Long and J. K. Barton, Acc. Chem. Res., 1990, 23, 271–273 CrossRef CAS;
(b) R. F. Pasternack, E. J. Gibbs and J. J. Villafranca, Biochemistry, 1983, 22, 2406–2414 CrossRef CAS.
- J. Olmsted and D. R. Kearns, Biochemistry, 1977, 16, 3647–3654 CrossRef CAS and references cited therein.
-
(a) J. M. Ruso, D. Attwood, M. Garcίa, P. Taboada, L. M. Varela and V. Mosquera, Langmuir, 2001, 17, 5189–5195 CrossRef CAS;
(b) T. Banerjee, S. K. Singh and N. Kishore, J. Phys. Chem. B, 2006, 110, 24147–24156 CrossRef CAS PubMed.
- G. Paramaguru, A. Kathiravan, S. Selvaraj, P. Venuvanalingam and R. Renganathan, J. Hazard. Mater., 2010, 175, 985–991 CrossRef CAS PubMed.
- J. L. Parrot and G. Nicot, Nature, 1963, 197, 496–497 CrossRef CAS.
- S. L. Huang, V. Maiorov, P. L. Huang, A. Ng, H. C. Lee, Y. T. Chang, N. Kallenbach, P. L. Huang and H. C. Chen, Biochemistry, 2005, 44, 4648–4655 CrossRef PubMed.
- S. L. Huang, P. L. Huang, Y. T. Sun, P. L. Huang, H. F. Kung, D. L. Blithe and H. C. Chen, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2678–2681 CrossRef.
- Y. Mine, F. P. Ma and S. Lauriau, J. Agric. Food Chem., 2004, 52, 1088–1094 CrossRef CAS PubMed.
- G. P. Gorbenko, V. M. Loffe and P. K. J. Kinnunen, Biophys. J., 2007, 93, 140–153 CrossRef CAS PubMed.
- L. T. Lemiesz and H. Kozlowski, Bioorg. Med. Chem., 1996, 4, 1709–1713 CrossRef.
- Y. J. Hu, Y. O. Yang, C. M. Dai, Y. Liu and X. H. Xiao, Biomacromolecules, 2010, 11, 106–112 CrossRef CAS PubMed.
-
G. Z. Chen, X. Z. Huang, J. G. Xu, Z. B. Wang and Z. Z. Zhang, Method of Fluorescent Analysis, 2nd edn, Science Press, Beijing, 1990, ch. 4, p. 123, 126 Search PubMed.
- J. N. Miller, Proc. Anal. Div. Chem. Soc., 1979, 16, 203–208 CAS.
- G. Weber, Nature, 1961, 190, 27–29 CrossRef CAS.
- F. C. Wu, R. B. Mills, R. D. Evans and P. J. Dillon, Anal. Chem., 2004, 76, 110–113 CrossRef CAS.
- Q. Xiao, S. Huang, Y. Liu, F. F. Tian and J. C. Zhu, J. Fluoresc., 2009, 19, 317–326 CrossRef CAS PubMed.
- J. N. Tian, J. Q. Liu, W. Y. He, Z. D. Hu, X. J. Yao and X. G. Chen, Biomacromolecules, 2004, 5, 1956–1961 CrossRef CAS PubMed.
- S. Grguric-Sipka, I. N. Stepanenko, J. M. Lazic, C. Bartel, M. A. Jakupec, V. B. Arion and B. K. Keppler, Dalton Trans., 2009, 3334–3339 RSC.
- X. L. Zhao, Z. S. Li, Z. B. Zheng, A. G. Zhanga and K. Z. Wang, Dalton Trans., 2013, 42, 5764–5777 RSC.
- G. Belmadani, A. M. Tramu, P. S. Betbeder and E. E. Steyn, Arch. Toxicol., 1998, 72, 656–662 CrossRef.
- J. B. Mitchell, D. A. Wink, W. DeGraff, J. Gamson, L. K. Keefer and M. C. Krishna, Cancer Res., 1993, 53, 5845–5848 CAS.
- R. J. Griffin, C. M. Makepeace, W. J. Hur and C. W. Song, Int. J. Radiat. Oncol., Biol., Phys., 1996, 36, 377–383 CrossRef CAS.
- M. Kurimoto, S. Endo, H. Hirashima, T. Ogiichi and A. Takaku, J. Neuro-Oncol., 1999, 42, 35–44 CrossRef CAS.
- H. T. Chung, H. O. Pae, B. M. Choi, T. R. Billiar and Y. M. Kim, Biochem. Biophys. Res. Commun., 2001, 282, 1075–1079 CrossRef CAS PubMed.
- K. Xie, S. Huang, Z. Dong, S. H. Juang, M. Gutman, Q. W. Xie, C. Nathan and I. J. Fidler, J. Exp. Med., 1995, 181, 1333–1343 CrossRef CAS.
- D. Bonnefont-Rousselot, Curr. Opin. Clin. Nutr. Metab. Care, 2002, 5, 561–568 CrossRef CAS PubMed.
- V. H. Guaiquil, J. C. Vera and D. W. Golde, J. Biol. Chem., 2001, 276, 40955–40961 CrossRef CAS PubMed.
- S. Li, X. Li and G. J. Rozanski, J. Mol. Cell. Cardiol., 2003, 35, 1145–1152 CrossRef CAS.
- J. C. Fernandez-Checa, N. Kaplowitz, C. Garcia-Ruiz, A. Colell, M. Miranda, M. Mari, E. Ardite and A. Morales, Am. J. Physiol. Gastrointest. Liver Physiol., 1997, 273, G7–G17 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 821302 for [Ru(H-Sal-mtsc)(CO)Cl(PPh3)2]·CH3CN (1) and 821303 for [Ru(Sal-mtsc)(CO)(PPh3)2] (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qi00070b |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.