Catalytic Sonogashira couplings mediated by an amido pincer complex of palladium†
Yu-Ting
Hung‡
a,
Ming-Tsz
Chen
a,
Mei-Hui
Huang
a,
Ting-Yin
Kao
a,
Yu-Sheng
Liu
a and
Lan-Chang
Liang
*ab
aDepartment of Chemistry and Center for Nanoscience & Nanotechnology, National Sun Yat-sen University, Kaohsiung 80424, Taiwan, Republic of China. E-mail: lcliang@mail.nsysu.edu.tw
bDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 80708, Taiwan, Republic of China
First published on 9th April 2014
Abstract
This work describes the efficacy of [PNP]PdCl (1a), where [PNP]− = bis(2-diphenylphosphinophenyl)amide, as a catalyst precursor for Csp–Csp2 bond-forming cross-coupling reactions of terminal alkynes with aryl halides in the presence of copper bromide and aliphatic amines in ethereal solutions under mild conditions. This catalysis is compatible with acetylenes that are alkyl, alkenyl, (hetero)aryl, or silyl substituted and aryl iodides or bromides that are electronically activated, neutral, or deactivated. The low reaction constants of 0.82(6) and 0.97(7) obtained from Hammett plots of competitive reactions employing electronically distinct aryl iodides and terminal alkynes, respectively, are likely suggestive of irrelevance of the rate-determining step in these catalytic transformations to oxidative addition of aryl halides or generation of mono-substituted acetylides. In sharp contrast, reactions employing a phosphorus-bound isopropyl derived 1b gave rather unsatisfactory results, highlighting a profound phosphorus substituent effect on this aryl alkynylation catalysis.
Introduction
The palladium-catalyzed Csp–Csp2 bond-forming reactions of terminal alkynes with aryl or alkenyl (pseudo)halides, generally referred to as the Sonogashira coupling,1,2 are among the most versatile and valuable methods for the preparation of arylated alkynes and conjugated enynes. These compounds are important precursors or key intermediates for natural products, bioactive compounds, pharmaceuticals, liquid crystalline materials, conducting polymers, etc.3–12 To develop effective methods for these specific chemical transformations and to elucidate the reaction mechanisms involved therein are tasks of fundamental and practical importance.13–15It has been demonstrated that palladium complexes of amido phosphine ligands, e.g., 1a and 2 in Fig. 1, are highly active catalyst precursors for Heck and Suzuki couplings.16,17 These compounds may be regarded as electronic modifications of phosphine palladacycles, e.g., 3 and 4,18–21 which are popular constitutional motifs as pre-catalysts for cross-coupling reactions.22 Interestingly, while 3a is highly active for catalytic Heck reactions of aryl iodides and bromides but is almost inactive for chlorides,18 the oxygen-incorporated 3b is an efficient pre-catalyst for the olefination of these inherently inert yet industrially important substrates.19 The discrepancy in reactivity of 3b and 3a was ascribed to electronic factors. In a separate study, 3b was also found to be active for catalytic Sonogashira couplings of aryl chlorides with phenyl acetylene, though relatively high temperatures (160 °C) and participation of the co-catalyst ZnCl2 were required.23 Other precedents of pincer complexes active for catalytic Sonogashira couplings are also known.22,24–29 The high thermal stability of the palladium pincer complexes 1a and 3a–b is believed to be one of the keys to the success of these catalytic transformations.16,18,19,23 Though palladium complex 1b is unprecedented, nickel analogues of 1a–b have been known to facilitate catalytic Kumada couplings.30 In an effort to expand the reaction scope of these amido pincer complexes in cross-coupling catalysis,31–35 we became interested in the efficacy of 1 with respect to Sonogashira couplings. In this contribution, we aim to demonstrate that 1 is also a competent catalyst precursor for efficient alkynylation of aryl halides under relatively mild conditions. Mechanistic insights implied by reactivity discrepancy ascribed to distinct substituents at phosphorus in 1 and by competitive alkynylation of aryl iodides are discussed. To the best of our knowledge, this study represents the first report on Sonogashira couplings catalyzed by PNP complexes.
 | ||
| Fig. 1 Representative examples of phosphine palladacycles. | ||
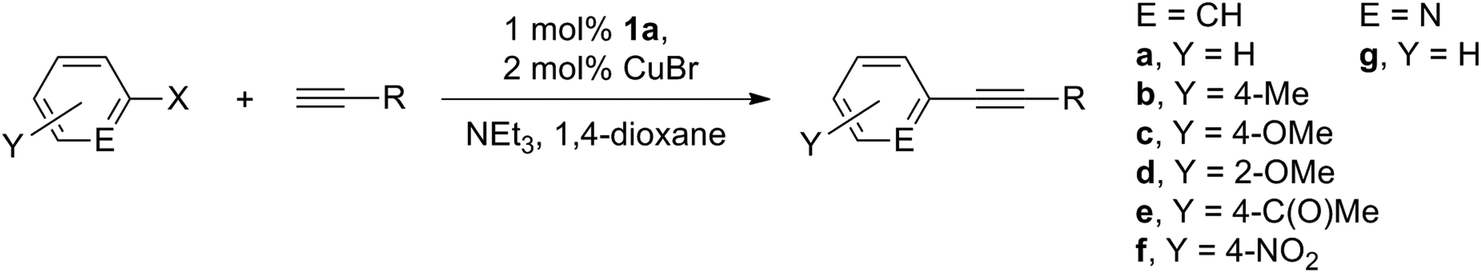
Results and discussion
Complex 1b could be readily prepared in high yield by treating a THF solution of bis(2-diisopropylphosphinophenyl)amine30 with either PdCl2 upon heating or PdCl2(PhCN)2 at room temperature. Similar to 1a,161b is a brick red crystalline solid that is not sensitive to oxygen or moisture and can be conveniently manipulated under aerobic conditions for subsequent reactions. The solution NMR data of 1b are all consistent with a structure having a square planar geometry, as evidenced by virtual triplet resonances ascribed to the o-phenylene carbon atoms in the 13C{1H} NMR spectrum. Complex 1a has previously been proved to be remarkably thermally stable even at temperatures as high as 200 °C (>100 hours).16 Similarly, 1b is also thermally stable; no decomposition was found when a toluene-d8 solution of 1b (63 mM) was heated to 110 °C for 4 days as indicated by 1H and 31P{1H} NMR studies. The high thermal stability of both 1a and 1b leads us to preclude their thermal degradation36,37 in catalysis under conditions milder than these prescribed examination parameters.Our initial Sonogashira studies focused on the survey of reaction parameters of 1 employing iodobenzene and trimethylsilyl acetylene as building blocks under various conditions. Table 1 summarizes the effects of phosphorus substituents in 1 and possible co-catalysts, bases, and solvents that are frequently employed in Sonogashira couplings. Notably, these building blocks may be effectively cross-coupled with a catalytic amount of 1a in the presence of CuBr as the co-catalyst and triethylamine as the base in 1,4-dioxane solutions under ambient conditions. This reaction proceeds smoothly and selectively, affording in an hour the desired cross-coupled product 5a in quantitative yield (entry 1); no Glaser coupling product38 was detectable. In contrast, the employment of catalytic 1b (entries 2 and 3) or other co-catalysts (entries 4–9) under otherwise identical conditions gave rather unsatisfactory results, highlighting the significance of the distinctive phosphorus substituents in PNP ligands39–41 and the identity of the co-catalysts employed, respectively. In particular, 1b hardly gave the desired cross-coupled product (entry 2) under the standard model reaction conditions. The reactivity of 1b, however, may be accelerated at elevated temperatures, e.g., 55 °C at entry 3. Though unsatisfactory, the turnover number (entry 3) of complex 1b is unambiguously indicative of its catalytic characteristics. The phenyl substituted 1a thus markedly outperforms the isopropyl derived 1b in terms of catalytic turnover frequencies in Sonogashira couplings under the conditions investigated. Such a discrepancy in catalytic activities is likely reflective of their inherent electronic nature, as ascribed to what was found for 3a and 3b.18,19 Herrmann and Beller et al.20 and Gibson and Cole-Hamilton et al.21 also independently demonstrated that phosphine palladacycles 4 that are P-arylated are superior catalyst precursors for Heck reactions to those containing ethyl or tert-butyl substituents at the phosphorus donors. The lower reactivity of 1b is tentatively attributed, at least in part, to its lesser electrophilicity that induces higher activation barriers for substrates, presumably the in situ generated nucleophilic acetylide, to coordinate and catalysis to proceed, in view of the established reactivity discrepancy found in nickel chemistry wherein these ligands are involved.40,41 In contrast to 3b,23 the participation of ZnCl2 did not promote the activities of 1 at all (entry 8). A number of aliphatic amines were surveyed as the base; triethylamine appears to be superior to the others (entries 10–13). Among the solvents investigated (entries 14–21), both polar and non-polar solvents are compatible, though THF and 1,4-dioxane solutions facilitate this catalysis most efficiently. Without the pre-catalyst 1a or the co-catalyst CuBr (entries 22–24), the cross-coupling reactions hardly proceed even with extended reaction time (entry 22), thereby underscoring clearly the synergistic role of these transition metal complexes in this catalysis. No induction period was found, as corroborated by a reaction profile established by product yields as a function of time (see ESI†).
|
|
||||
|---|---|---|---|---|
| Entry | Co-catalyst | Base | Solvent | Yieldb (%) |
| a Reaction conditions unless otherwise noted: 1.0 equiv. of iodobenzene, 1.1 equiv. of trimethylsilyl acetylene, 1.0 mol% of 1a (24 mM in specified solvent), 2.0 mol% of co-catalyst, 15 equiv. of base, 25 °C, 1 hour. b Determined by GC, based on iodobenzene using icosane as an internal standard; average of two runs. c Catalytic 1b instead of 1a was employed. d Reaction run at 55 °C for 36 hours. e DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. f Reaction run for 24 hours. g Reaction run without 1a. | ||||
| 1 | CuBr | NEt3 | 1,4-Dioxane | >99 |
| 2c | CuBr | NEt3 | 1,4-Dioxane | <1 |
| 3c | CuBr | NEt3 | 1,4-Dioxane | 17d |
| 4 | CuCl | NEt3 | 1,4-Dioxane | 15 |
| 5 | CuI | NEt3 | 1,4-Dioxane | 10 |
| 6 | CuOTf | NEt3 | 1,4-Dioxane | 12 |
| 7 | FeCl3 | NEt3 | 1,4-Dioxane | <1 |
| 8 | ZnCl2 | NEt3 | 1,4-Dioxane | <1 |
| 9 | AlCl3 | NEt3 | 1,4-Dioxane | <1 |
| 10 | CuBr | HNEt2 | 1,4-Dioxane | 19 |
| 11 | CuBr | HNiPr2 | 1,4-Dioxane | 66 |
| 12 | CuBr | MeNCy2 | 1,4-Dioxane | <1 |
| 13 | CuBr | DBUe | 1,4-Dioxane | 2 |
| 14 | CuBr | NEt3 | THF | >99 |
| 15 | CuBr | NEt3 | DMSO | 74 |
| 16 | CuBr | NEt3 | DMF | 64 |
| 17 | CuBr | NEt3 | DMA | 38 |
| 18 | CuBr | NEt3 | MeCN | 93 |
| 19 | CuBr | NEt3 | CH2Cl2 | 94 |
| 20 | CuBr | NEt3 | PhMe | 78 |
| 21 | CuBr | NEt3 | PhH | 92 |
| 22 | None | NEt3 | 1,4-Dioxane | 2f |
| 23g | CuBr | NEt3 | 1,4-Dioxane | 0 |
| 24g | None | NEt3 | 1,4-Dioxane | 0 |
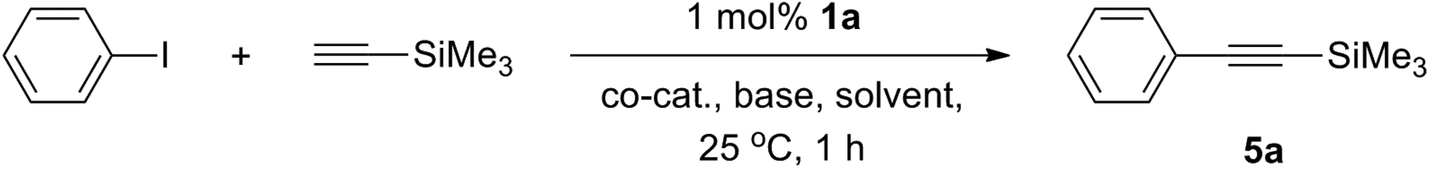
In spite of the established virtually equal efficacy of catalysis run in both 1,4-dioxane and THF, we chose to investigate the reaction scope in the former due to its higher boiling point that offers the possibility of catalysis run at higher temperatures if necessary. As shown in Table 2 (accumulating 35 examples of products), a variety of functional groups are compatible, including (fluoro)alkyl, alkenyl, (hetero)aryl, silyl, fluoro, alkoxy, ketone, nitro, etc. Both aryl iodides and bromides are reactive, though the couplings involving the latter electrophiles require mild heating. Notably, aryl iodides may be effectively coupled in high yields with terminal alkynes at room temperature. Both ortho- and para-substituted substrates are suitable building blocks, though a slower rate is generally found for the former (entries 3 and 4, 17 and 18, 22 and 23, 27 and 28, 34 and 35), likely due to the steric congestion imposed by the 2-substituent and the amido pincer ligand. Nevertheless, an extension of the reaction time is sufficient to afford the cross-coupled products in reasonable yields (e.g., entry 4 versus 5). The reaction rates are also a function of the electronic characteristics of the halogenated electrophiles, following the order: electron-deficient > electron-neutral > electron-rich substituents. Consistent with what was found in Table 1, 1a is a superior pre-catalyst to 1b (entry 12 versus 13). In all attempts, no palladium black was observed, reflecting the robust nature of these palladium complexes even at elevated temperatures. In particular, catalysis runs in the presence of liquid mercury gave virtually identical results (e.g., entry 10 in Table 2), indicating that this catalysis is homogeneous and not a consequence of bulk or colloidal palladium(0).42–44 Whether a soluble palladium(0) species is produced from chemical degradation of the Pd[PNP] entity, however, is not clear at this stage. Attempts to identify possible intermediates in this regard by 31P{1H} NMR spectroscopy were inconclusive. It is worth noting that catalysis runs employing crude 1 without further purification generally led to significantly lower turnover frequencies, strongly implying the presence of some unidentified impurities and their incapability or poisoning effect in this catalysis. In contrast, purified 1, even with different synthetic batches, gave consistent catalysis results.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Temp (°C) | Time (h) | Product | Yieldb (%) | |
| a Reaction conditions unless otherwise noted: 1.0 equiv. of aryl halides (2.4 M in 1,4-dioxane), 1.1 equiv. of alkynes, 15 equiv. of NEt3. b Determined by GC, based on aryl halides using icosane as an internal standard; average of two runs. c Catalytic 1b instead of 1a was employed. | ||||||
| 1 | I | 25 | 1 |
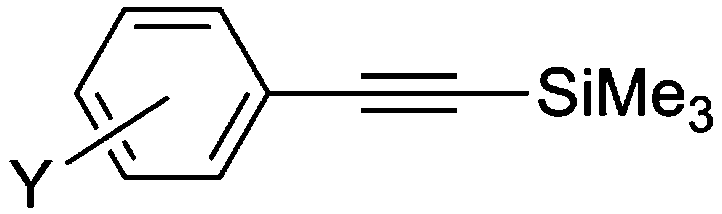
|
5a | >99 |
| 2 | I | 25 | 2 | 5b | 92 | |
| 3 | I | 25 | 2 | 5c | 76 | |
| 4 | I | 25 | 2 | 5d | 28 | |
| 5 | I | 25 | 6 | 5d | 77 | |
| 6 | I | 25 | 2 | 5e | >99 | |
| 7 | Br | 50 | 74 | 5e | 60 | |
| 8 | Br | 50 | 74 | 5f | 98 | |
| 9 | Br | 50 | 74 |
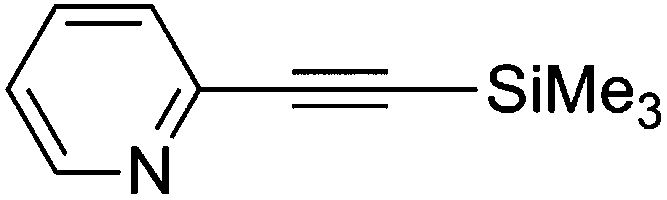
|
5g | >99 |
| 10 | I | 25 | 17 |
|
6a | >99 |
| 11 | I | 25 | 17 | 6b | 99 | |
| 12 | Br | 110 | 24 | 6b | 72 | |
| 13c | Br | 110 | 24 | 6b | 13 | |
| 14 | I | 25 | 17 | 6d | 97 | |
| 15 | I | 25 | 17 | 6e | >99 | |
| 16 | I | 25 | 24 |
|
7a | 94 |
| 17 | I | 25 | 24 | 7c | 92 | |
| 18 | I | 25 | 24 | 7d | 83 | |
| 19 | I | 25 | 24 | 7e | 98 | |
| 20 | I | 25 | 20 |
|
8a | 98 |
| 21 | I | 25 | 20 | 8b | 89 | |
| 22 | I | 25 | 20 | 8c | 57 | |
| 23 | I | 25 | 20 | 8d | 68 | |
| 24 | I | 25 | 20 | 8e | >99 | |
| 25 | I | 25 | 24 |
|
9a | 90 |
| 26 | I | 25 | 24 | 9b | >99 | |
| 27 | I | 25 | 24 | 9c | 92 | |
| 28 | I | 25 | 24 | 9d | 82 | |
| 29 | I | 25 | 24 | 9e | >99 | |
| 30 | Br | 110 | 24 | 9e | 96 | |
| 31 | Br | 110 | 24 | 9f | 93 | |
| 32 | Br | 110 | 24 |
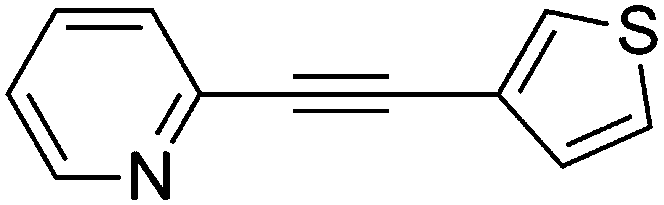
|
9g | >99 |
| 33 | I | 25 | 17 |
|
10a | 96 |
| 34 | I | 25 | 17 | 10c | 75 | |
| 35 | I | 25 | 17 | 10d | 30 | |
| 36 | I | 25 | 17 | 10e | 87 | |
| 37 | I | 25 | 17 |
|
11b | 73 |
| 38 | I | 25 | 17 | 11c | 53 | |
| 39 | I | 25 | 17 | 11e | 94 | |
| 40 | Br | 60 | 96 | 11f | 90 | |
In addition to electronic reasons (vide supra), the steric discrepancy of 1a and 1b was also considered to correlate with their reactivity differences as implied by the slightly higher reactivity of 4c than that of 4b in Heck reactions.20 To this end, we pursued evidence with crystallographic data of both 1a and 1b. Complex 1a has previously been structurally characterized.16 Red crystals of 1b suitable for X-ray diffraction analysis were grown from a concentrated diethyl ether solution at −35 °C. Fig. 2 depicts the solid state structure of 1b. These structures, however, resemble closely each other in view of their similar bond distances, angles, and dihedral angles about the palladium center (Table 3). The only notable difference is a slightly larger dihedral angle between two o-phenylene rings in 1b. The origin of such a discrepancy is tentatively attributed to the sterically distinct P-substituents incorporated. Other structural parameters are not exceptional.
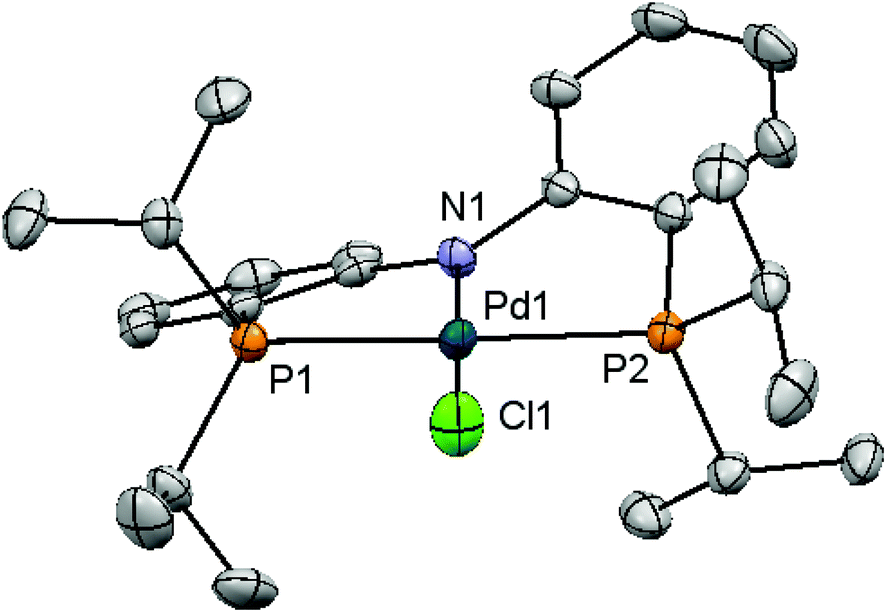 | ||
| Fig. 2 Molecular structure of 1b with thermal ellipsoids drawn at the 35% probability level. | ||
| Compound | 1a | 1b |
|---|---|---|
| a Data selected from ref. 16. | ||
| Pd–N | 2.056(11) | 2.029(3) |
| Pd–P | 2.3010(18), 2.3010(18) | 2.2860(12), 2.2898(11) |
| Pd–Cl | 2.313(5) | 2.3119(12) |
| P–Pd–P | 165.27(11) | 165.33(4) |
| N–Pd–Cl | 180.0000(10) | 177.39(11) |
| P–Pd–N | 82.63(6), 82.63(6) | 83.45(10), 82.87(10) |
| P–Pd–Cl | 97.37(6), 97.37(6) | 96.30(5), 97.60(5) |
| o-Phenylene/o-phenylene | 36.87 | 42.16 |
| P–N–P–Cl/C–N–C | 24.07 | 23.48 |
Structural resemblance of 1b to 1a does not necessarily lead us to conclude that sterics plays a negligible role in the reactivity discrepancy of these complexes, particularly taking into account the possibility of an increase in coordination number of palladium intermediates produced in the catalytic cycles. It is generally accepted that the catalytic cycle of Sonogashira couplings comprises successive oxidative additions of aryl/alkenyl halides, transmetallation of copper acetylides to palladium, and reductive elimination of the desired cross-coupled products.9 If this or similar mechanisms are operative in the current study, 1b might in principle outperform 1a in view of its more electron-releasing nature to facilitate oxidative addition and its sterically more demanding nature to accelerate reductive elimination, assuming that either one of these elementary steps is rate-determining. On the other hand, 1a that is phenyl instead of isopropyl substituted may sterically favor oxidative addition and electronically encourage reductive elimination. We have recently demonstrated that complexes with a phenyl substituted PNP ligand have much higher propensity to undergo reductive elimination than those containing isopropyl substituents.41 It has also been shown that phosphine dissociation from the metal center of group 10 PNP complexes is rather unlikely as deduced by their marked thermal stability.30,40,45,46 As a result, an increase in coordination number for palladium intermediates is highly possible in this catalysis, thereby sterically discouraging oxidative addition to occur for 1b or its subsequent derivatives.
Mechanistic possibilities involving radical pathways were considered. Addition of typical radical inhibitors47 such as 2,2,6,6-tetramethyl-1-piperidinyloxy or 2,6-di-tert-butyl-4-hydroxytoluene (1 equiv. to pre-catalyst) to the model reaction (entry 1, Table 1) appears not to affect much the activity and selectivity of 1a, thus implying that the radical participation is rather unlikely.
To gain more insights into the reaction mechanism, competitive experiments employing electronically deactivated, unactivated, and activated aryl iodides with 3-ethynylthiophene were conducted, leading to a Hammett plot (fitted with Hammett σp substituent constants) with a reaction constant ρ of 0.82(6) (Fig. 3). Similar reactions involving electronically distinct aryl acetylenes with phenyl iodide were also attempted, yielding ρ = 0.97(7) (Fig. 4). These results are unambiguously indicative of an increase in reaction rates with electron-withdrawing substituents at both aryl iodides and acetylenes, consistent with the development of a negative charge at the reaction center in the transition state. At first glance, the positive sign of these reaction constants appear to imply that the aryl iodide and acetylene building blocks are both electrophilic in character. This result is peculiar given the established concepts9 that acetylenes are typically regarded as nucleophiles in traditional Sonogashira couplings though aryl halides are indeed electrophilic. Lei et al. have recently demonstrated an elegant mechanistic study on traditional Sonogashira couplings, wherein the corresponding ρ values of 1.29 and −1.65 were found for electronically distinct aryl iodides and acetylenes, respectively.15 We reason that the positive ρ value (Fig. 4), in contrast to the negative value reported by Lei et al., highlights the ease of deprotonation of terminal acetylenes to generate mono-substituted acetylides in the presence of electron-withdrawing substituents. In this particular elementary step, acetylenes indeed act as electrophiles, in which the negative charge built in the transition state is more readily stabilized with electron-withdrawing groups. As a result, the opposite signs of ρ values corresponding to substituent constants in aryl acetylenes in the present and Lei's15 studies are likely suggestive of either inherently different reaction mechanisms or similar pathways with different rate-determining factors for these Sonogashira couplings, a discrepancy that is ascribed to the participation and the non-dissociative characteristic of the PNP ligand employed.
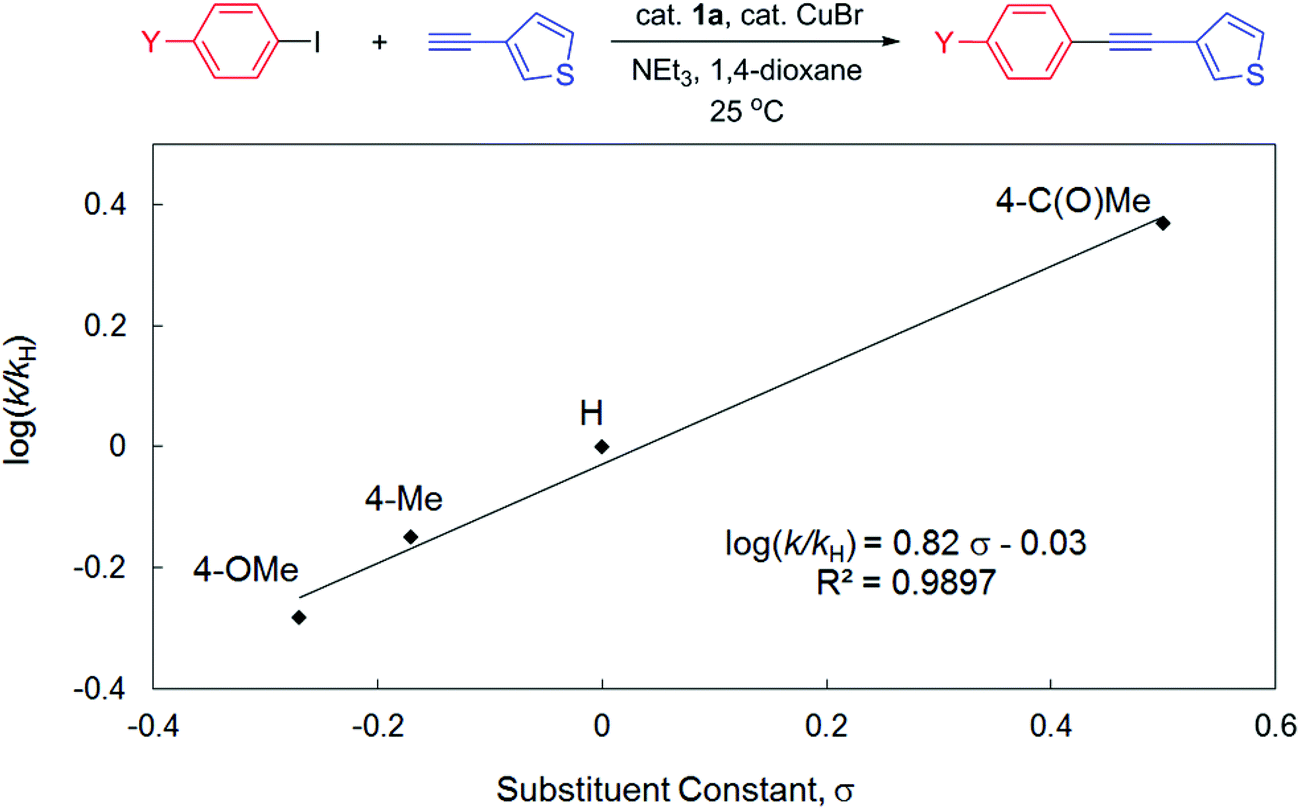 | ||
| Fig. 3 Hammett plot for the competitive couplings of 4-substituted phenyl iodides with 3-ethynylthiophene catalyzed by 1a in 1,4-dioxane at 25 °C. | ||
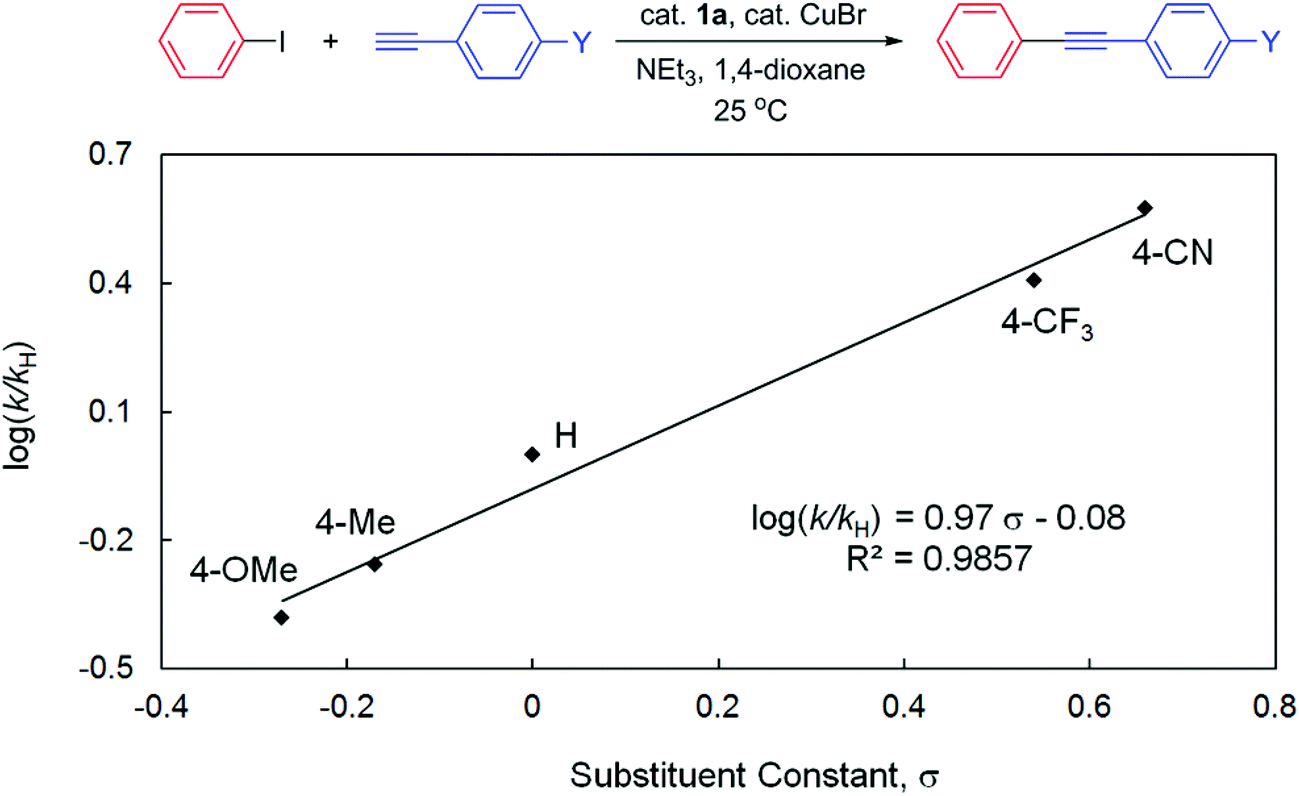 | ||
| Fig. 4 Hammett plot for the competitive couplings of 4-substituted phenyl acetylenes with phenyl iodide catalyzed by 1a in 1,4-dioxane at 25 °C. | ||
In contrast to the small ρ value derived in Fig. 3, several studies on oxidative addition of aryl halides to phosphine ligated zero-valent palladium have disclosed consistently larger reaction constants.48–51 For instance, Buchwald et al. demonstrated ρ = 2.3 for oxidative addition of aryl chlorides to Pd(XPhos)48 whereas Fauvarque et al. determined ρ = 2 for aryl iodides to Pd(PPh3)2.51 The notably smaller value found in the present study thus suggests that the involvement of oxidative addition of aryl halides in the rate-determining step is unlikely. This result is also consistent with the fact that the electronically more releasing 1b is an inferior pre-catalyst compared to 1a for this catalysis. Similar conclusions were also deduced in other studies with low reaction constants involving electronically distinct aryl halides in Heck olefination (e.g., ρ = 0.60 for 1a16 and 1.39 for 3a)18 or Suzuki couplings (e.g., ρ = 0.48 for 2),17 wherein either olefin coordination/insertion52 or transmetallation,53,54 respectively, is suggested to be slowest. In particular, Lei et al. demonstrated in their Sonogashira study with quantitative kinetic data that the transmetallation step is indeed rate-limiting.15 Given the facile reductive elimination rate found for phenyl substituted PNP complexes,41 we suggest that reductive elimination of the cross-coupled products is not rate-determining, either. We are currently conducting systematic, controlled experiments employing selected combinations of starting materials (in stoichiometric amount) of the model reaction, in an attempt to elucidate any possible reaction intermediates, particularly those informative to potentially distinguish Pd(0)/Pd(II),55,56 Pd(I)/Pd(III),57–60 or Pd(II)/Pd(IV)61,62 redox cycles.
Conclusions
We have demonstrated that the amido pincer complex 1a is a competent catalyst precursor for the Sonogashira couplings of aryl halides with terminal alkynes under mild conditions. This catalysis is compatible with a variety of functional groups incorporated into these building blocks, including (fluoro)alkyl, alkenyl, (hetero)aryl, silyl, fluoro, alkoxy, ketone, nitro, etc. The fact that 1a outperforms 1b in catalytic activities and low reaction constants obtained from Hammett plots suggests that oxidative addition of aryl halides, in this case aryl iodides, is not rate-determining. Studies directed to identify reaction intermediates and to elucidate the reaction mechanism of this catalysis are currently under way.Experimental section
General procedures
Unless otherwise specified, all experiments were performed under nitrogen using standard Schlenk or glovebox techniques. All solvents were reagent grade or better and purified by standard methods. Compound 1a was prepared following the procedures reported previously.16 All other chemicals were used as received from commercial vendors. All NMR spectra were recorded at room temperature in specified solvents using Varian Unity or Bruker AV instruments. Chemical shifts (δ) are listed as parts per million downfield from tetramethylsilane and coupling constants (J) are in hertz. Routine coupling constants are not listed. 1H NMR spectra are referenced using the residual solvent peak at δ 7.16 for C6D6 and δ 7.27 for CDCl3. 13C NMR spectra are referenced using the internal solvent peak at δ 128.39 for C6D6 and δ 77.23 for CDCl3. The assignment of the carbon atoms for all compounds is based on the DEPT 13C NMR spectroscopy. 31P and 19F NMR spectra are referenced externally using 85% H3PO4 at δ 0 and CFCl3 in CHCl3 at δ 0, respectively. The Sonogashira coupling reactions were analyzed by GC on a Varian chrompack CP-3800 instrument equipped with a CP-Sil 5 CB chrompack capillary column or by GC/MS on a Varian 450-GC/240-MS instrument equipped with a Restek MXT-5 column. The identity of the cross-coupling products was confirmed by comparison with authentic samples. GC yields were quantified by signal integral relative to those of prescribed amounts of icosane as an internal standard. MALDI mass spectra were recorded with an α-cyano-4-hydroxycinnamic acid (generally abbreviated as CHCA) matrix on a Bruker Autoflex Mass Spectrometer. Elemental analysis was performed on a Heraeus CHN-O Rapid analyzer.X-ray crystallography
Crystallographic data for 1b (CCDC reference number 956287) are available in ESI.† Data were collected on a diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.7107 Å). Structures were solved by direct methods and refined by full matrix least squares procedures against F2 using SHELXL-97.63 All full-weight non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions.Synthesis of 1b
Method 1: To a suspension of PdCl2 (77 mg, 0.44 mmol) in THF (6 mL) was added a THF solution (4 mL) of bis(2-diisopropylphosphinophenyl)amine (175 mg, 0.44 mmol). The reaction mixture was transferred to a Teflon-capped vessel and heated in an oil bath to 110 °C for 1 h. All volatiles were removed in vacuo. Diethyl ether (10 mL) was added. The diethyl ether solution was filtered through a pad of Celite and evaporated to dryness under reduced pressure to afford the product as a red solid; yield 223 mg (94%).Method 2: To a solid mixture of bis(2-diisopropylphosphinophenyl)amine (150 mg, 0.37 mmol) and PdCl2(PhCN)2 (144 mg, 0.37 mmol) was added THF (5 mL) at room temperature. The solution was stirred at room temperature for 10 min and evaporated to dryness under reduced pressure. Diethyl ether (8 mL) was added. The ether solution was filtered through a pad of Celite and evaporated to dryness under reduced pressure. The solid residue thus obtained was washed with pentane (2 mL × 2) and dried in vacuo to afford the product as a red solid; yield 187 mg (92%). 1H NMR (C6D6, 500 MHz) δ 7.69 (d, 2, Ar), 6.93 (m, 4, Ar), 6.48 (m, 2, Ar), 2.28 (br m, 4, CHMe2), 1.40 (dd, 12, CHMe2), 1.09 (dd, 12, CHMe2). 31P{1H} NMR (C6D6, 202.3 MHz) δ 49.0. 13C{1H} NMR (C6D6, 125.5 MHz) δ 164.3 (t, JCP = 11.0, C), 133.4 (s, CH), 131.9 (s, CH), 120.2 (t, JCP = 18.3, C), 117.8 (t, CH), 117.0 (t, JCP = 5.9, CH), 25.3 (t, CHMe2), 18.9 (s, CHMe2), 18.2 (s, CHMe2). Anal. Calcd for C24H36ClNP2Pd: C, 53.13; H, 6.69; N, 2.58. Found: C, 53.18; H, 6.29; N, 2.16.
General procedures for the Sonogashira couplings outlined in Table 2
A Schlenk flask was sequentially charged with 1a (24 mM in 1,4-dioxane for each single experiment, corresponding to 1.0 mol%) along with an appropriate amount of CuBr (2.0 mol%), aryl halide (1.0 equiv.), alkyne (1.1 equiv.), NEt3 (15 equiv.), and a magnetic stir bar. The flask was capped with a stopper and the solution was stirred either at 25 °C or heated in an oil bath at prescribed temperatures for a specified period of time. After the reaction mixture was cooled to room temperature, aqueous hydrochloric acid (1 M, 6 mL) was added and the product was extracted with diethyl ether (15 mL × 3). The aqueous solution was separated from the organic layer. The diethyl ether solution was washed with deionized water (15 mL × 3), dried over MgSO4, and evaporated to dryness under reduced pressure to afford the desired product, which, if necessary, was subjected to flash column chromatography on silica gel using hexane or diethyl ether as the eluent.4-(Phenylethynyl)acetophenone (6e)64
1H NMR (CDCl3, 300 MHz) δ 7.94 (d, 2, J = 8.4, Ar), 7.61 (d, 2, J = 8.4, Ar), 7.55 (d, 2, J = 3.9, Ar), 7.37 (m, 3, Ar), 2.60 (s, 3, C(O)Me). 13C NMR (CDCl3, 75 MHz) δ 197.4 (C(O)Me), 136.3 (Ar), 132.6 (Ar), 131.9 (ArH), 131.8 (ArH), 129.3 (ArH), 128.6 (ArH), 128.4 (ArH), 122.8 (Ar), 92.8 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 88.7 (CC), 26.7 (C(O)Me). MS (MALDI) Calcd for C16H12O m/z 220.089 ([M]+), found m/z 220.877 ([M + H]+).
C), 88.7 (CC), 26.7 (C(O)Me). MS (MALDI) Calcd for C16H12O m/z 220.089 ([M]+), found m/z 220.877 ([M + H]+).
4-(4-Fluorophenylethynyl)acetophenone (7e)64
1H NMR (CDCl3, 300 MHz) δ 7.92 (d, 2, J = 7.8, Ar), 7.58 (d, 2, J = 7.5, Ar), 7.52 (m, 2, Ar), 7.05 (m, 2, Ar), 2.60 (s, 3, C(O)Me). 13C NMR (CDCl3, 75 MHz) δ 197.3 (C(O)Me), 162.9 (JCF = 248.5, CF), 136.4 (Ar), 133.8 (JCF = 8.6, ArH), 131.7 (ArH), 128.4 (ArH), 128.1 (Ar), 118.9 (Ar), 115.9 (JCF = 21.8, ArH), 91.7 (CC), 88.4 (CC), 26.7 (C(O)Me). 19F NMR (CDCl3, 282 MHz) δ −109.9. MS (MALDI) Calcd for C16H11FO m/z 238.079 ([M]+), found m/z 238.887 ([M + H]+).
1-(Phenylethynyl)-4-(trifluoromethyl)benzene (8a)64,65
1H NMR (CDCl3, 300 MHz) δ 7.64 (m, 4, Ar), 7.55 (m, 2, Ar), 7.38 (m, 3, Ar). 13C NMR (CDCl3, 75 MHz) δ 132.0 (ArH), 131.9 (ArH), 129.0 (ArH), 128.6 (ArH), 127.3 (Ar), 125.9 (Ar), 125.4 (ArH), 122.7 (Ar), 91.9 (CC), 88.1 (CC). 19F NMR (CDCl3, 282 MHz) δ −62.8. MS (MALDI) Calcd for C15H9F3m/z 246.066 ([M]+), found m/z 247.077 ([M + H]+).
4-(4-Trifluoromethylphenylethynyl)acetophenone (8e)66
1H NMR (CDCl3, 300 MHz) δ 7.95 (d, 2, J = 6, Ar), 7.62 (m, 6, Ar), 2.61 (s, 3, C(O)Me). 13C NMR (CDCl3, 75 MHz) δ 197.3 (C(O)Me), 136.8 (Ar), 132.3 (ArH), 131.9 (ArH), 128.4 (ArH), 127.5 (Ar), 126.6 (Ar), 125.5 (ArH), 122.2 (Ar), 91.1 (CC), 90.9 (CC), 26.7 (C(O)Me). 19F NMR (CDCl3, 282 MHz) δ −62.9. MS (MALDI) Calcd for C17H11F3O m/z 288.076 ([M]+), found m/z 289.051 ([M + H]+).
3-(4-Acetylphenylethynyl)thiophene (9e)66
1H NMR (CDCl3, 300 MHz) δ 7.93 (d, 2, Ar), 7.59 (m, 3, Ar), 7.32 (dd, 1, Ar), 7.21 (m, 1, Ar), 2.60 (s, C(O)Me). 13C NMR (CDCl3, 75 MHz) δ 197.4 (C(O)Me), 136.3 (Ar), 131.7 (ArH), 130.1 (Ar), 129.9 (ArH), 129.6 (ArH), 128.4 (ArH), 125.7 (ArH), 121.9 (Ar), 88.3 (CC), 88.0 (CC), 26.7 (C(O)Me). MS (MALDI) Calcd for C14H10OS m/z 226.045 ([M]+), found m/z 226.858 ([M + H]+).
3-(2-Pyridylethynyl)thiophene (9g)67
1H NMR (CDCl3, 300 MHz) δ 8.59 (d, 1, J = 4.5, Ar), 7.64 (m, 2, Ar), 7.48 (d, 1, J = 7.8, Ar), 7.29 (m, 1, Ar), 7.24 (m, 2, Ar). 13C NMR (CDCl3, 75 MHz) δ 150.1 (ArH), 143.6 (Ar), 136.2 (ArH), 130.3 (ArH), 130.1 (ArH), 127.1 (ArH), 125.6 (ArH), 122.8 (ArH), 121.5 (Ar), 88.4 (CC), 84.6 (CC). MS (MALDI) Calcd for C11H7NS m/z 185.030 ([M]+), found m/z 185.804 ([M + H]+).
4-(Isobutylethynyl)acetophenone (11e)
1H NMR (CDCl3, 300 MHz) δ 7.86 (d, 2, J = 8.1, Ar), 7.45 (d, 2, J = 8.1, Ar), 2.56 (s, 3, C(O)Me), 2.32 (d, 2, J = 6.6, CH2), 1.93 (m, 1, CH), 1.04 (d, 6, J = 6.6, (CH3)2). 13C NMR (CDCl3, 75 MHz) δ 197.4 (C(O)Me), 135.9 (Ar), 131.7 (ArH), 129.3 (Ar), 128.3 (ArH), 93.4 (CC), 81.1 (CC), 28.8 (CH2), 28.2(CH), 26.6 (C(O)Me), 22.1 (CH(CH3)2). MS (MALDI) Calcd for C14H16O m/z 200.120 ([M]+), found m/z 200.992 ([M + H]+).
Representative procedures for the competition reactions (Fig. 3)
A vial was charged with 1a (3 μmol), CuBr (6 μmol), aryl iodides (0.13 mmol each), 3-ethynylthiophene (0.58 mmol), NEt3 (7.8 mmol), 1,4-dioxane (4 mL), and a magnetic stir bar. The reaction mixture was stirred at 25 °C for 12 h. An aliquot was taken and quenched with aqueous hydrochloric acid as described above. The product identity and yield were analyzed by GC/MS, from which the relative rate constants were derived and employed for the Hammett plot (see ESI†).Mercury drop experiment
The catalytic solutions were generated using the procedures described above and mercury (0.35 g) was added. The reaction temperature, time, and work-up procedures were all identical to those conducted without mercury. The product's identity and yield were examined by GC/MS.Acknowledgements
We thank the Ministry of Science and Technology of Taiwan for financial support (NSC 99-2113-M-110-003-MY3 and NSC 102-2113-M-110-002-MY3), Mr Ting-Shen Kuo (NTNU) for assistance with X-ray crystallography, and the National Center for High-performance Computing (NCHC) for access to chemical databases.References
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- J. A. Marsden and M. M. Haley, in Metal-catalyzed Cross-coupling Reactions, ed. A. de Mejeire and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004, pp. 317–394 Search PubMed.
- A. L. K. S. Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034–1057 CrossRef CAS PubMed.
- E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2017 CrossRef CAS PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- H. Doucet and J. C. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834–871 CrossRef CAS PubMed.
- R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- U. H. F. Bunz, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS PubMed.
- S.-C. Chien, H.-H. Chen, H.-C. Chen, Y.-L. Yang, H.-F. Hsu, T.-L. Shih and J.-J. Lee, Adv. Funct. Mater., 2007, 17, 1896–1902 CrossRef CAS.
- H.-H. Chen, H.-A. Lin, Y.-H. Lai, S.-Y. Lin, C.-H. Chiang, H.-F. Hsu, T.-L. Shih, J.-J. Lee, C.-C. Lai and T.-S. Kuo, Chem. – Eur. J., 2012, 18, 9543–9551 CrossRef CAS PubMed.
- Y. Nishihara, K. Ikegashira, K. Hirabayashi, J.-I. Ando, A. Mori and T. Hiyama, J. Org. Chem., 2000, 65, 1780–1787 CrossRef CAS PubMed.
- O. Vechorkin, D. Barmaz, V. Proust and X. L. Hu, J. Am. Chem. Soc., 2009, 131, 12078–12079 CrossRef CAS PubMed.
- C. He, J. Ke, H. Xu and A. W. Lei, Angew. Chem., Int. Ed., 2013, 52, 1527–1530 CrossRef CAS PubMed.
- M.-H. Huang and L.-C. Liang, Organometallics, 2004, 23, 2813–2816 CrossRef CAS.
- L.-C. Liang, P.-S. Chien and M.-H. Huang, Organometallics, 2005, 24, 353–357 CrossRef CAS.
- M. Ohff, A. Ohff, M. E. van der Boom and D. Milstein, J. Am. Chem. Soc., 1997, 119, 11687–11688 CrossRef CAS.
- D. Morales-Morales, R. Redon, C. Yung and C. M. Jensen, Chem. Commun., 2000, 1619–1620 RSC.
- W. A. Herrmann, C. Brossmer, C. P. Reisinger, T. H. Riermeier, K. Ofele and M. Beller, Chem. – Eur. J., 1997, 3, 1357–1364 CrossRef CAS.
- S. Gibson, D. F. Foster, G. R. Eastham, R. P. Tooze and D. J. Cole-Hamilton, Chem. Commun., 2001, 779–780 RSC.
- M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed.
- M. R. Eberhard, Z. Wang and C. M. Jensen, Chem. Commun., 2002, 818–819 RSC.
- N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed.
- J. L. Bolliger and C. M. Frech, Adv. Synth. Catal., 2009, 351, 891–902 CrossRef CAS.
- B. Ines, R. SanMartin, F. Churruca, E. Dominguez, M. K. Urtiaga and M. I. Arriortua, Organometallics, 2008, 27, 2833–2839 CrossRef CAS.
- S. J. Gu and W. Z. Chen, Organometallics, 2009, 28, 909–914 CrossRef CAS.
- H. V. Huynh and C.-S. Lee, Dalton Trans., 2013, 42, 6803–6809 RSC.
- S. J. Gu, D. C. Xu and W. Z. Chen, Dalton Trans., 2011, 40, 1576–1583 RSC.
- L.-C. Liang, P.-S. Chien, J.-M. Lin, M.-H. Huang, Y.-L. Huang and J.-H. Liao, Organometallics, 2006, 25, 1399–1411 CrossRef CAS.
- L.-C. Liang, Coord. Chem. Rev., 2006, 250, 1152–1177 CrossRef CAS.
- O. V. Ozerov, in The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. M. Jensen, Elsevier, Amsterdam, 2007, pp. 287–309 Search PubMed.
- L. Fan, B. M. Foxman and O. V. Ozerov, Organometallics, 2004, 23, 326–328 CrossRef CAS.
- B. L. Tran, M. Pink and D. J. Mindiola, Organometallics, 2009, 28, 2234–2243 CrossRef CAS.
- E. Calimano and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 11161–11173 CrossRef CAS PubMed.
- L.-C. Liang, C.-C. Chien, M.-T. Chen and S.-T. Lin, Inorg. Chem., 2013, 52, 7709–7716 CrossRef CAS PubMed.
- L.-C. Liang, S.-T. Lin, C.-C. Chien and M.-T. Chen, Dalton Trans., 2013, 42, 9286–9293 RSC.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2632–2657 CrossRef CAS.
- L.-C. Liang, P.-S. Chien and Y.-L. Huang, J. Am. Chem. Soc., 2006, 128, 15562–15563 CrossRef CAS PubMed.
- L.-C. Liang, P.-S. Chien and P.-Y. Lee, Organometallics, 2008, 27, 3082–3093 CrossRef CAS.
- L.-C. Liang, Y.-T. Hung, Y.-L. Huang, P.-S. Chien, P.-Y. Lee and W.-C. Chen, Organometallics, 2012, 31, 700–708 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2011, 112, 1536–1554 CrossRef PubMed.
- D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 855–859 CrossRef CAS.
- P. Foley, R. DiCosimo and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 6713–6725 CrossRef CAS.
- L.-C. Liang, J.-M. Lin and C.-H. Hung, Organometallics, 2003, 22, 3007–3009 CrossRef CAS.
- L.-C. Liang, J.-M. Lin and W.-Y. Lee, Chem. Commun., 2005, 2462–2464 RSC.
- L.-C. Liang, W.-Y. Lee, Y.-T. Hung, Y.-C. Hsiao, L.-C. Cheng and W.-C. Chen, Dalton Trans., 2012, 41, 1381–1388 RSC.
- M. R. Biscoe, B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 6686–6687 CrossRef CAS PubMed.
- M. Portnoy and D. Milstein, Organometallics, 1993, 12, 1665–1673 CrossRef CAS.
- C. S. Consorti, G. Ebeling, F. R. Flores, F. Rominger and J. Dupont, Adv. Synth. Catal., 2004, 346, 617–624 CrossRef CAS.
- J.-F. Fauvarque, F. Pflüger and M. Troupel, J. Organomet. Chem., 1981, 208, 419–427 CrossRef CAS.
- F. Miyazaki, K. Yamaguchi and M. Shibasaki, Tetrahedron Lett., 1999, 40, 7379–7383 CrossRef CAS.
- H. Weissman and D. Milstein, Chem. Commun., 1999, 1901–1902 RSC.
- D. Zim, V. R. Lando, J. Dupont and A. L. Monteiro, Org. Lett., 2001, 3, 3049–3051 CrossRef CAS PubMed.
- A. de Mejeire and F. Diederich, Metal-catalyzed Cross-coupling Reactions, 2nd edn, Wiley-VCH, Weinheim, 2004 Search PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 CrossRef CAS PubMed.
- K. J. Bonney, F. Proutiere and F. Schoenebeck, Chem. Sci., 2013, 4, 4434–4439 RSC.
- M. Aufiero, F. Proutiere and F. Schoenebeck, Angew. Chem., Int. Ed., 2012, 51, 7226–7230 CrossRef CAS PubMed.
- L. M. Mirica and J. R. Khusnutdinova, Coord. Chem. Rev., 2013, 257, 299–314 CrossRef CAS.
- A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS PubMed.
- A. J. Canty, Dalton Trans., 2009, 10409–10417 RSC.
- G. M. Sheldrick, SHELXTL, Version 5.1, Bruker AXA Inc., Madison, WI, 1998 Search PubMed.
- R. Thorwirth, A. Stolle and B. Ondruschka, Green Chem., 2010, 12, 985–991 RSC.
- P. Li, L. Wang, M. Wang and F. You, Eur. J. Org. Chem., 2008, 5946–5951 CrossRef CAS.
- S. Park, M. Kim, D. Koo and S. Chang, Adv. Synth. Catal., 2004, 346, 1638–1640 CrossRef CAS.
- R. E. Beveridge and B. A. Arndtsen, Synthesis, 2010, 1000–1008 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data for 1b, a catalytic reaction profile, details of competition reactions to construct Hammett plots, and NMR spectra of products isolated. CCDC 956287. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qi00086a |
| ‡ Current address: Department of Chemistry & Biochemistry, University of Delaware, Newark, Delaware 19716, USA. |
| This journal is © the Partner Organisations 2014 |