Syntheses, X-ray structures, catalytic activity and magnetic properties of two new coordination polymers of Co(II) and Ni(II) based on benzenedicarboxylate and linear N,N′-donor Schiff base linkers†
Received
14th March 2014
, Accepted 22nd April 2014
First published on 24th April 2014
Abstract
Two isostructural coordination polymers based on Co(II) and Ni(II), {[M(azpy)3(p-bdc)2(H2O)4]·(CH3OH)2(H2O)3}n [where M = Co (1) and Ni (2); azpy = N,N′-bis-pyridin-4-ylmethylene-hydrazine and p-bdc = 1,4-benzenedicarboxylate], have been synthesized using mixed ligand systems at room temperature and characterized by single-crystal X-ray diffraction and other physicochemical methods. Structure determination reveals that both the complexes crystallize in the monoclinic space group C2/c and exhibit one-dimensional (1D) ladder like structures constructed by p-bdc and azpy ligands in which Schiff base (azpy) linkers serve as a bridging as well as pendent ligands. These pendent ligands are involved in H-bonding and π–π interactions with lattice water, methanol molecules and bridging azpy ligands, to form the 3D supramolecular structure. Notably, both the frameworks efficiently catalyze the Knoevenagel condensation reactions of a wide range of substituted benzaldehydes with active methylene compounds in heterogeneous medium under environmentally friendly conditions and the products were obtained in excellent yields. These catalysts were also found to exhibit excellent recyclability and re-usability without any significant loss of activity. A variable temperature magnetic study of both compounds was carried out and their magnetic properties justified on the basis of their crystal structure.
Introduction
Functional coordination polymer (CP) and/or metal–organic framework (MOF) is nowadays a very popular topic in the fields of inorganic and materials research due to its hybrid nature and synthetic tunability.1 Moreover, this class of molecules has versatile applications like gas storage,2 separation,3 proton conductance,4 sensing,5 drug delivery,6 magnetism,7 and heterogeneous catalysis.8 Out of the above applications of MOFs, heterogeneous catalysis was one of the earliest and efficiently demonstrated applications due to the catalysis-friendly features of MOFs such as poor solubility in common organic solvents, large surface areas and extraordinary porosity, flexibility to afford open channels and ordered pores with catalyst active functionalities.9
It has been noticed that several organic transformations along with many condensation reactions such as aldol condensation,10 aza-Michael condensation,11 Henry reaction,12 Pechmann reaction,13 Friedlander condensation14 and Knoevenagel condensation15etc. have been successfully carried out using conventional solid catalysts. Among these reactions, the Knoevenagel condensation of aldehydes with compounds containing activated methylene groups in the presence of a weak base has been widely employed in synthetic organic chemistry right from the syntheses of small molecules to the elegant intermediates of anti-hypertensive drugs and calcium antagonists.16 Different types of solid bases like inorganic salts (Al2O3, ZnCl2, KF–Al2O3),17 zeolite,18 calcite,19 basic MCM-41 silica,20 amine-functionalized mesoporous zirconia,21 amine-functionalized superparamagnetic nanoparticles22 and chitosan hydrogel23etc. are nowadays preferred very much as catalysts for the Knoevenagel condensation. The reaction protocol using a heterogeneous solid base catalyst, over a trivial organic base; is important from an environmental perspective as these can be easily separated by simple filtration. As stated before, MOFs having all the advantages of heterogeneous catalysts; can act as a nice catalyst if some basic sites could be functionalized in its surroundings.8,9 There are established strategies which have been adopted to make MOF based catalysts, for example; inclusion of open metal sites in the nodes, and incorporation of different active functional groups in the organic linkers to improve the catalytic activity of coordination polymers.8,9 The inclusion of base functionalized groups (such as, NH2, pyridine) in the framework has been well studied for heterogeneous catalysis,8,9 but the use of a milder Schiff base in such processes is not very common.
Recently, our group has made an extensive contribution to the synthesis of mixed ligand MOFs with Schiff base functionalized linkers.24 These linkers effectively decorated the pore walls with Schiff base which played a crucial role in the selective CO2 capture behavior of the aforesaid frameworks.24 Moreover, it is worth mentioning that the mixed ligand variety of MOFs have some inherent advantages like flexibility as well as stability in their framework structures. Thus, based on these facts, we were tempted to see if such Schiff base functionalized MOFs can be designed; these might be useful in base-catalyzed reactions, which would reinforce the chemical application of MOFs along with their all-around physical properties.
Here we have synthesized two isostructural mixed ligand coordination polymers containing pendent Schiff base functionalized ligands with the general formulae {[M(azpy)3(p-bdc)2(H2O)4]·(CH3OH)2(H2O)3}n [where M = Co (1) and Ni (2); azpy = N,N′-bis-pyridin-4-ylmethylene-hydrazine and p-bdc = 1,4-benzenedicarboxylate]. The single-crystal X-ray structure shows a one-dimensional (1D) ladder like structure. Both the compounds are found to achieve significant catalytic activity and reusability towards Knoevenagel condensation as a solid base catalyst. To the best of our knowledge, the reported compounds are the most efficient examples of coordination polymers that have been used heterogeneously to catalyze the Knoevenagel condensation reaction to date. A variable temperature magnetic study also corroborates the structure of both the complexes.
Experimental
Materials
N,N′-Bis-pyridin-4-ylmethylene-hydrazine (azpy) was synthesized by the procedures reported earlier.25 High purity cobalt(II) nitrate hexahydratate, nickel(II) nitrate hexahydratate, terephthalic acid, substituted benzaldehydes, malononitrile and ethyl cyanoacetate were purchased from the Sigma-Aldrich Chemical Company Inc. and used as received. All other chemicals were of AR grade and were used as received.
Physical measurements
Elemental analyses (carbon, hydrogen, and nitrogen) were performed using a Perkin-Elmer 240C elemental analyzer. Infrared spectra (4000–400 cm−1) were taken on KBr pellets, using a Perkin-Elmer Spectrum BX-II IR spectrometer. Thermal analysis (TGA) was carried out on a METTLER TOLEDO TGA 850 thermal analyzer under nitrogen atmosphere (flow rate: 50 cm3 min−1) at the temperature range 30–600 °C with a heating rate of 2 °C min−1. X-ray powder diffraction (PXRD) patterns in different states of the sample were recorded on a Bruker D8 Discover instrument using Cu-Kα radiation. All low pressure gas adsorption experiments (up to 1 bar) were performed on a Quantachrome Quadrasorb automatic volumetric instrument. 1H NMR spectra were recorded at ambient temperature in CDCl3 with tetramethylsilane as internal standard. The chemical shifts (δ) and coupling constants (J) are expressed in ppm and Hz, respectively on a Bruker Avance 300 instrument. Variable temperature (2–300 K) magnetic susceptibility measurements on polycrystalline samples were carried out with a Quantum Design SQUID MPMS XL-5 device operating at different magnetic fields.
Synthesis of {[Co2(azpy)3(p-bdc)2(H2O)4]·(CH3OH)2(H2O)3}n (1)
An aqueous solution (20 ml) of disodium 1,4-benzenedicarboxylate (Na2p-bdc) (1 mmol, 0.210 g) was mixed with a methanolic solution (20 ml) of N,N′-bis-pyridin-4-ylmethylene-hydrazine (azpy) (1 mmol, 0.210 g) and stirred for 20 min to mix it well. Co(NO3)2·6H2O (1 mmol, 0.281 g) was dissolved in 20 ml of water. Then in a crystal tube 4 ml of Co(II) solution was slowly and carefully layered with the 8 ml of the abovementioned mixed-ligand solution using 2 ml of buffer solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of water and MeOH) in between the two solutions. The tube was sealed and kept undisturbed at room temperature and after one week red colored block shaped single crystals suitable for X-ray diffraction analysis were obtained at the wall of the tube. Crystals were separated and washed with MeOH–water (1:1) mixture and air dried (yield 79%). Anal. Calc. for C54H60Co2N12O17 (Mr = 1266.99): C, 51.19; H, 4.77; N, 13.26. Found: C, 51.29; H, 4.92; N, 13.11. IR spectra (in cm−1): ν(C
:1 of water and MeOH) in between the two solutions. The tube was sealed and kept undisturbed at room temperature and after one week red colored block shaped single crystals suitable for X-ray diffraction analysis were obtained at the wall of the tube. Crystals were separated and washed with MeOH–water (1:1) mixture and air dried (yield 79%). Anal. Calc. for C54H60Co2N12O17 (Mr = 1266.99): C, 51.19; H, 4.77; N, 13.26. Found: C, 51.29; H, 4.92; N, 13.11. IR spectra (in cm−1): ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1569; ν(C–O), 1310–1230; ν(CH–Ar), 3100–2900 and ν(CC), 1600–1440.
N), 1569; ν(C–O), 1310–1230; ν(CH–Ar), 3100–2900 and ν(CC), 1600–1440.
Synthesis of {[Ni2(azpy)3(p-bdc)2(H2O)4]·(CH3OH)2(H2O)3}n (2)
This complex was synthesized by following the same procedure as for 1 using Ni(NO3)2·6H2O (1 mmol, 0.280 g) instead of Co(NO3)2·6H2O. Shiny green single crystals suitable for X-ray diffraction analysis were obtained after one month (yield 64%). Anal. Calc. for C54H60Ni2N12O17 (Mr = 1266.52): C, 51.21; H, 4.78; N, 13.27. Found: C, 51.35; H, 4.63; N, 13.41. IR spectra (in cm−1): ν(CN), 1568; ν(C–O), 1320–1210; ν(CH–Ar), 3100–2900 and ν(CC), 1600–1440.
Crystallographic data collection and refinement
Suitable single crystals of 1 and 2 were mounted on the tips of glass fibers with commercially available super glue. X-ray single crystal data collection for both the crystals was performed at room temperature using a Bruker APEX II diffractometer, equipped with a normal focus, sealed tube X-ray source with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The data were integrated using the SAINT26 program and the absorption corrections were made with SADABS.27 Both the structures were solved by SHELXS 9728 using the Patterson method and followed by successive Fourier and difference Fourier synthesis. Full matrix least-squares refinements were performed on F2 using SHELXL-9728 with anisotropic displacement parameters for all non-hydrogen atoms. For 1 and 2, the occupancy of one lattice water molecule (O4W) was fixed at 0.5 before final refinement. All the hydrogen atoms except the hydrogen of one lattice water (O4W) were fixed geometrically by HFIX command and placed in ideal positions for both structures. In 1, one of the H atoms on O1W is found in two fold disorder and this is fixed considering both 0.5 occupancies from the Fourier map. All calculations were carried out using SHELXL 97, SHELXS 97, PLATON v1.15,29 ORTEP-3v2,30 WinGX system Ver-1.8031 and TOPOS.32 Crystallographic and structure refinement data of all compounds are given in Table 1. Selected bond lengths, bond angles are displayed in Tables 2 and 3.
Table 1 Crystallographic and structural refinement parameters for 1 and 2
| |
1
|
2
|
|
R
1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = [∑ (w(Fo2 − Fc2)2)/∑w(Fo2)2]1/2.
|
| Formula |
C54H60Co2N12O17 |
C54H60Ni2N12O17 |
| Formula weight |
1266.99 |
1266.52 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
C2/c |
C2/c |
|
a/Å |
19.652(5) |
19.6754(3) |
|
b/Å |
11.540(5) |
11.3812(3) |
|
c/Å |
27.611(5) |
27.5363(5) |
|
α/° |
90 |
90 |
|
β/° |
107.031(5) |
107.342(1) |
|
γ/° |
90 |
90 |
|
V/Å3 |
5987(3) |
5885.9(2) |
|
Z
|
4 |
4 |
|
D
c/g cm−3 |
1.403 |
1.427 |
|
μ/mm−1 |
0.632 |
0.719 |
|
F(000) |
2624 |
2632 |
|
θ range/° |
1.5–27.5 |
1.5–27.6 |
| Reflections collected |
47915 |
47291 |
| Unique reflections |
6852 |
6795 |
| Reflections I > 2σ(I) |
6057 |
5990 |
|
R
int
|
0.023 |
0.027 |
| Goodness-of-fit (F2) |
1.05 |
1.12 |
|
R
1 (I > 2σ(I))a |
0.0386 |
0.0418 |
| wR2 (I > 2σ(I))a |
0.1367 |
0.1280 |
| Δρmax/min/e Å3 |
−0.62, 1.05 |
−0.51, 0.71 |
Table 2 Selected bond lengths (Å) and bond angles (°) for 1
|
Symmetry code: a = 1/2 + x, 1/2 + y, z.
|
| Co1–O1 |
2.0546(17) |
Co1–O1W |
2.1467(19) |
| Co1–O2W |
2.1710(19) |
Co1–N1 |
2.152(2) |
| Co1–N5 |
2.182(2) |
Co1–O4a |
2.0682(17) |
| O1–Co1–O1W |
89.90(6) |
O1–Co1–O2W |
91.26(6) |
| O1–Co1–N1 |
88.46(7) |
O1–Co1–N5 |
90.91(7) |
| O1–Co1–O4a |
176.64(6) |
O1W–Co1–O2W |
178.20(6) |
| O1W–Co1–N1 |
91.09(7) |
O1W–Co1–N5 |
87.77(7) |
| O1W–Co1–O4a |
93.46(6) |
O2W–Co1–N1 |
87.57(7) |
| O2W–Co1–N5 |
93.59(7) |
O2W–Co1–O4a |
85.39(6) |
| N1–Co1–N5 |
178.69(7) |
O4a–Co1–N1 |
91.63(7) |
| O4a–Co1–N5 |
89.08(7) |
|
|
Table 3 Selected bond lengths (Å) and bond angles (°) for 2
|
Symmetry code: a = 1/2 + x, 1/2 + y, z.
|
| Ni1–O1 |
2.0452(17) |
Ni1–O1W |
2.1051(16) |
| Ni1–O2W |
2.1203(16) |
Ni1–N1 |
2.096(2) |
| Ni1–N5 |
2.123(2) |
Ni1–O4a |
2.0518(17) |
| O1–Ni1–O1W |
88.51(7) |
O1–Ni1–O2W |
92.17(7) |
| O1–Ni1–N1 |
88.57(7) |
O1–Ni1–N5 |
90.78(7) |
| O1–Ni1–O4a |
177.72(6) |
O1W–Ni1–O2W |
179.31(7) |
| O1W–Ni1–N1 |
91.55(7) |
O1W–Ni1–N5 |
87.42(7) |
| O1W–Ni1–O4a |
93.74(7) |
O2W–Ni1–N1 |
88.32(7) |
| O2W–Ni1–N5 |
92.72(7) |
O2W–Ni1–O4a |
85.58(7) |
| N1–Ni1–N5 |
178.79(8) |
O4a–Ni1–N1 |
91.03(7) |
| O4a–Ni1–N5 |
89.66(7) |
|
|
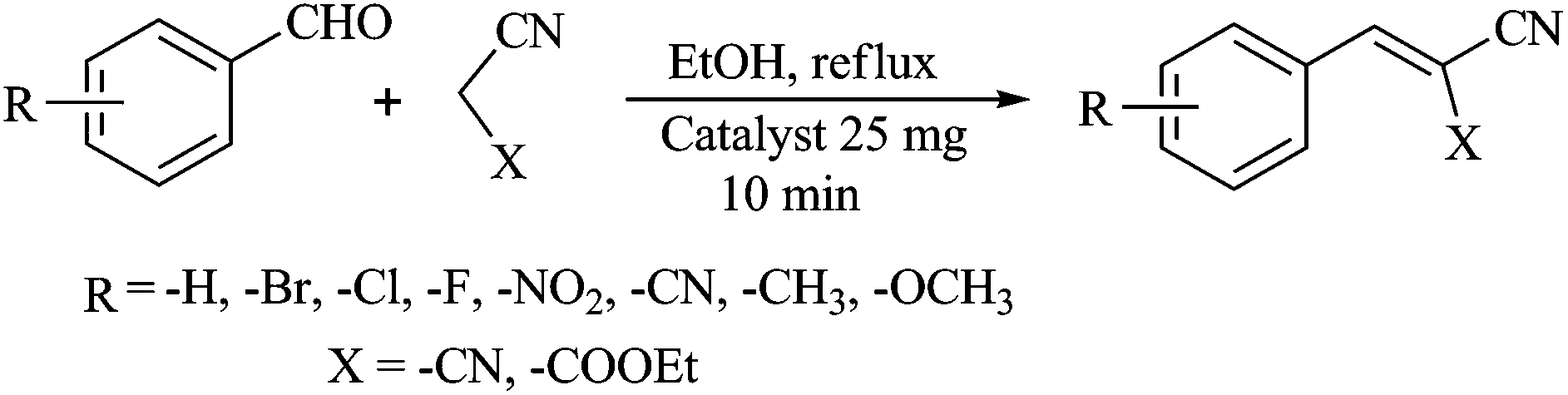
General procedure for the Knoevenagel condensation
The Knoevenagel condensations between substituted benzaldehydes and active methylene compounds were carried out according to the following procedure. A mixture of the substituted benzaldehyde (10 mmol), malononitrile or ethyl cyanoacetate (10 mmol) and 25 mg of catalyst (compound 1 or 2) in ethanol (20 ml) were taken in a round bottom flask and heated to reflux for 10 min (Scheme 1). After completion of the reaction, the catalysts were separated by filtration and washed with ethanol (3 × 10 ml). The combined filtrates were concentrated and the crude reaction products were purified by column chromatography over silica gel (mesh 60–120) using a petroleum ether–ethyl acetate mixture as eluent to give the desired product. The products were then analyzed by 1H NMR, elemental analysis and compared with authentic samples.
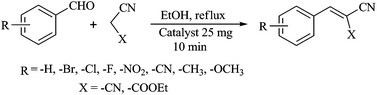 |
| | Scheme 1 Knoevenagel condensation reaction catalyzed by compounds 1 and 2. | |
Results and discussion
Structural description of {[M2(azpy)3(p-bdc)2(H2O)4]·(CH3OH)2(H2O)3}n [M = Co (1) and Ni (2)]
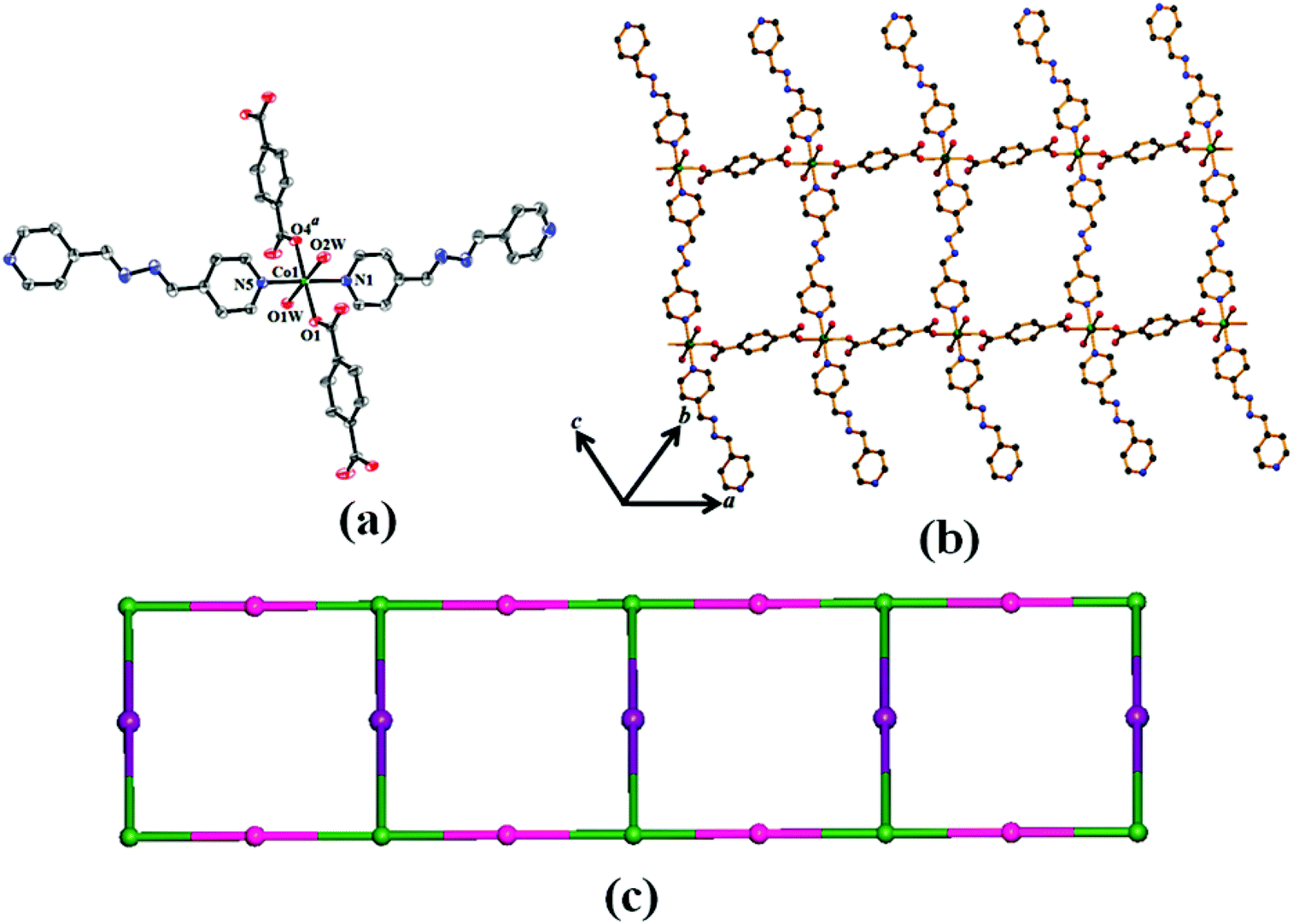
Compounds 1 and 2 are isostructural and both of them crystallize in monoclinic space group C2/c. Single crystal structure analysis reveals that both the compounds have a one-dimensional (1D) ladder structure connected by the 1,4-benzenedicarboxylate (p-bdc) and organic linkers (azpy). The asymmetric unit of 1 and 2 comprises of one Co(II)/Ni(II) center, one p-bdc, one and half an azpy ligand and two coordinated water molecules. There is one methanol and one and half water molecules present as guest molecules in the lattice. Here, each hexa-coordinated metal atom shows a distorted octahedral geometry with the MO4N2 core [Fig. 1(a) for 1 and Fig. S1(a)† for 2] which is reflected by their cisoid and transoid angles along the metal center (Table 2 for 1 and Table 3 for 2). The coordination around each M(II) is furnished by two carboxylate oxygen atoms (O1 and O4a where a = 1/2 + x, 1/2 + y, z for 1 and a = 1/2 + x, 1/2 + y, z for 2) of two different bridging 1,4-benzenedicarboxylates, two nitrogen atoms (N1 and N5) of one bridging and one pendent azpy ligand and two coordinated water molecules (O1W and O2W) [Fig. 1(a) for 1 and Fig. S1(a)† for 2]. In compound 1, the Co(II)–O bond lengths are in the range of 2.0546(17)–2.1710(19) Å and the Co(II)–N bond length varies from 2.152(2)–2.182(2) Å (Table 2); whereas for 2, the Ni(II)–O and Ni(II)–N bond lengths are 2.0452(17)–2.1203(16) Å and 2.096(2)–2.123(2) Å, respectively (Table 3). Here, each 1,4-benzenedicarboxylate binds in a bridging monodentate fashion and each 1,4-benzenedicarboxylate connects two different M(II) centres to create a chain with metal to metal separation of 11.395 Å and 11. 365 Å in 1 and 2, respectively. Two such chains are connected by azpy to form the 1D ladder like structure [Fig. 1(b) for 1 and Fig. S1(b)† for 2] where the pendent azpy ligands are like antennae of the structure on both sides of the ladder.
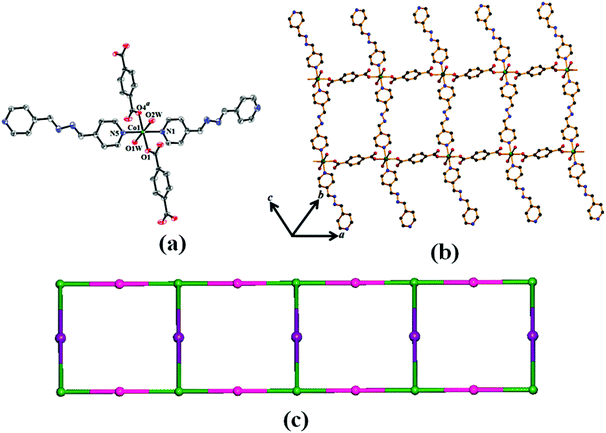 |
| | Fig. 1 (a) ORTEP drawing (40% probability ellipsoid) of 1 showing atom labeling scheme. (b) View of extended 1D ladder structure of compound 1 constructed by 1,4-benzenedicarboxylate, azpy ligand and Co(II). (c) Simplified topological representation of 1D ladder in 2. | |
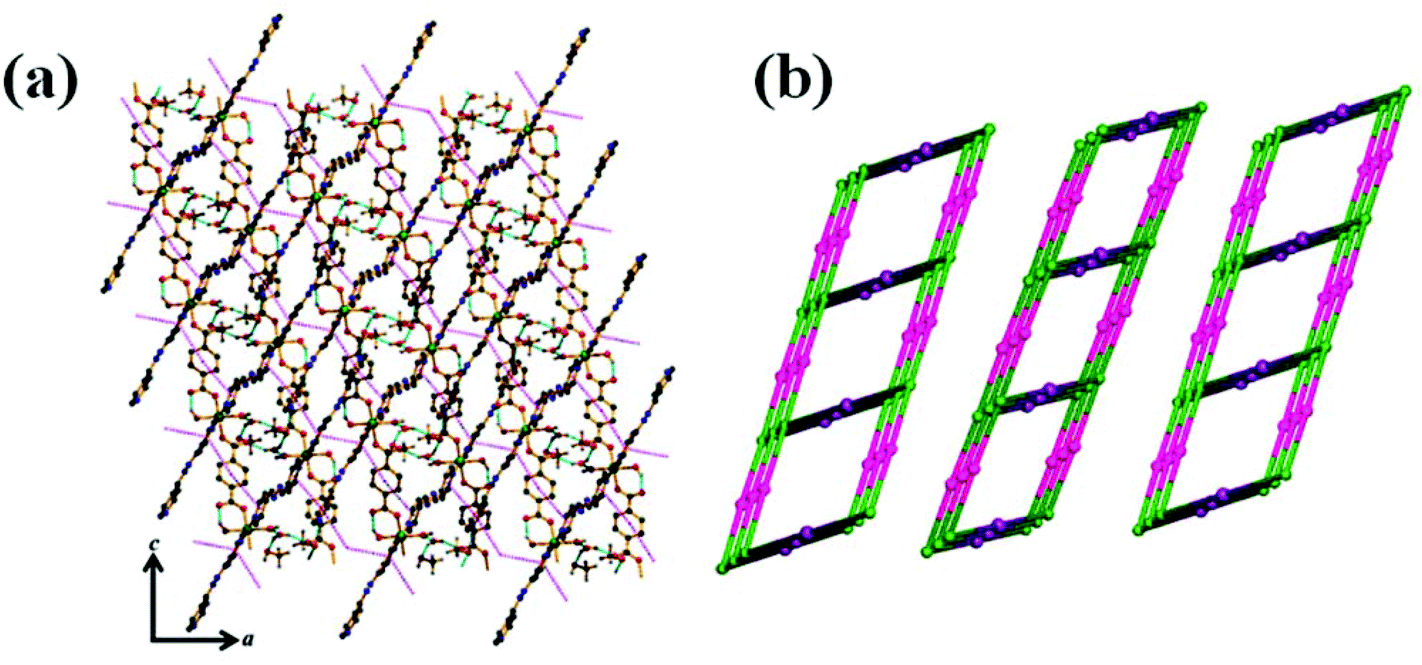
Topological analysis of the 1D ladder by TOPOS32 software suggests the formation of a 3-connected uninodal net with Schläfli symbol {42·6} [Fig. 1(c) for 1 and Fig. S1(c)† for 2]. In the crystal packing, these 1D ladders are stitched by H-bonding constructed by the interaction between the coordinated and lattice water molecules. The lattice methanol molecules are also H-bonded with lattice water molecules and the nitrogen atom (N4) of the pendent azpy ligand (Table S1† for 1 and Table S2† for 2). In addition to that there are intermolecular π–π interactions within the pyridyl rings of inter-sheet adjacent bridging azpy and pendent azpy ligands (centroid–centroid distances are in the range of 3.825(2)–3.914(2) Å and 3.8406(14)–3.9006(17) Å for 1 and 2, respectively) [Table S3† for 1 and Table S4† for 2]. These cooperative supramolecular interactions (H-bonds and π–π interactions) convert the ladder structure into a three-dimensional (3D) supramolecular arrangement [Fig. 2(a) for 1 and Fig. S2(a)† for 2]. The potential solvent-accessible void volume, calculated using PLATON,29 suggests 16.80% and 21.10% void volume of the total crystal volume after of 1 and 2, respectively, after the removal of the guest and coordinated solvent molecules.
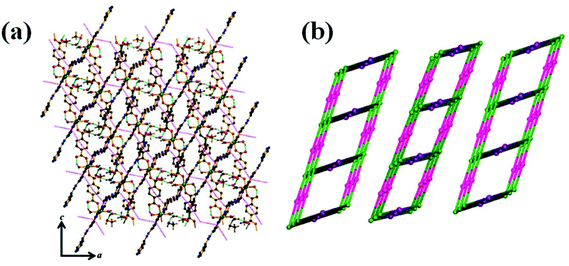 |
| | Fig. 2 (a) Supramolecular 3D network in 1 (π–π interaction: pink dotted lines & H-bonding: cyan dotted lines). (b) Simplified topological disposition view of adjacent ladders in 1. | |
Framework stability: thermogravimetric (TG) and powder X-ray diffraction (PXRD) analysis
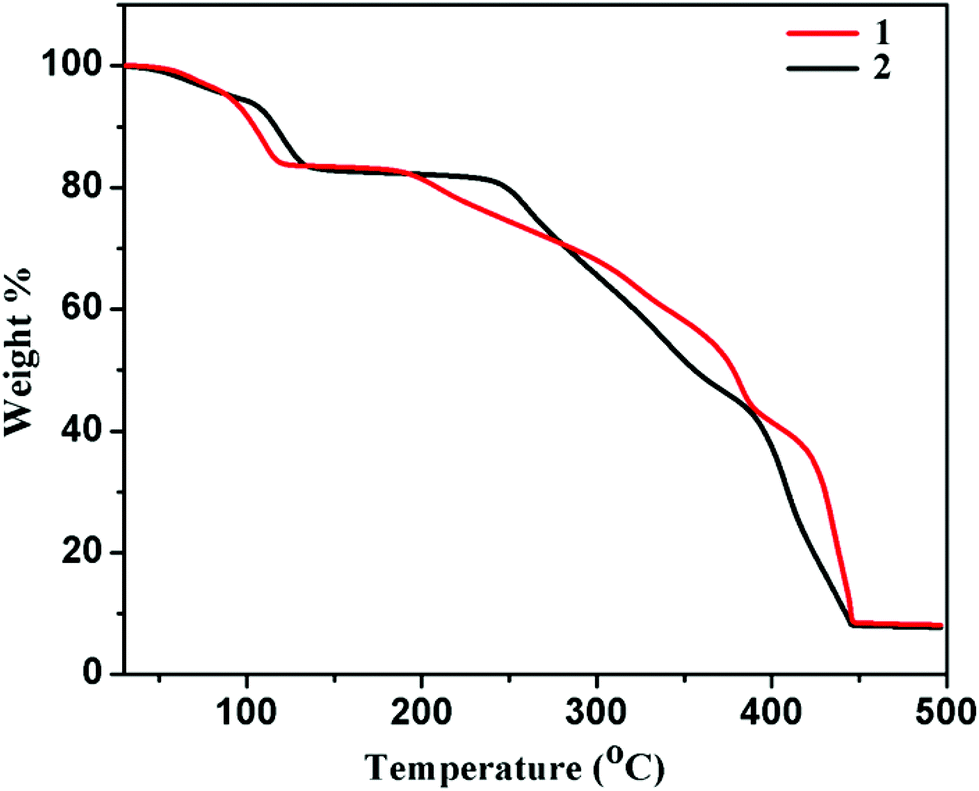
Thermogravimetric analyses (TGA) and powder X-ray diffraction (PXRD) measurements were carried out to study the stability of the framework compounds. TGA of compounds 1 and 2 was performed in the temperature range of 30–500 °C under a nitrogen atmosphere. The TGA profile of compound 1 (Fig. 3) indicates a weight loss of ∼9.4% at 100 °C, which corroborates the removal of all guest solvent (three water and two methanol) molecules (calc. 9.3%). The second step was observed at 115 °C with a weight loss of ∼15.9%, indicating the removal of coordinated water molecules (calc. 15%). Then the desolvated framework is stable up to 180 °C. After that, it gradually decomposes into unidentified products. Similarly compound 2 (Fig. 3), shows a stepwise release of methanol and water molecules. The first step indicates the release of three lattice water and two lattice methanol molecules (calc. wt loss = 9.32%; exp. wt loss ∼9.41%) at 115 °C, and the second one corresponds to the release of the other four coordinated water molecules at 130 °C (calcd wt loss = 15%, total exp. wt loss ∼15.54%). The desolvated framework of 2 is stable up to 240 °C without any further weight loss.
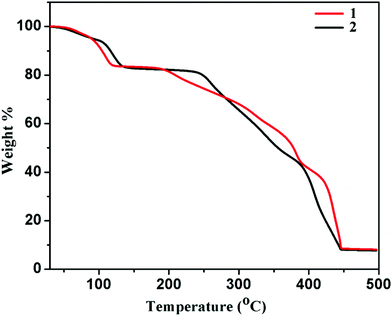 |
| | Fig. 3 TGA curves of compounds 1 and 2 in the temperature range 30–500 °C under a nitrogen atmosphere (heating rate 2 °C min−1). | |
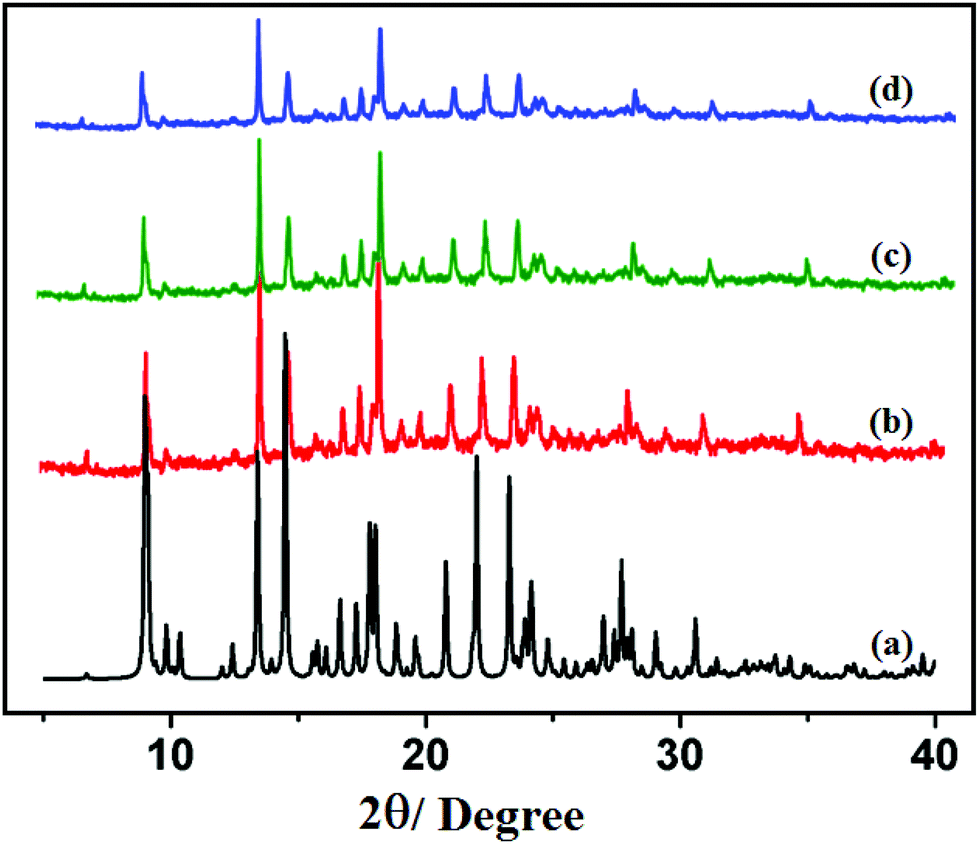
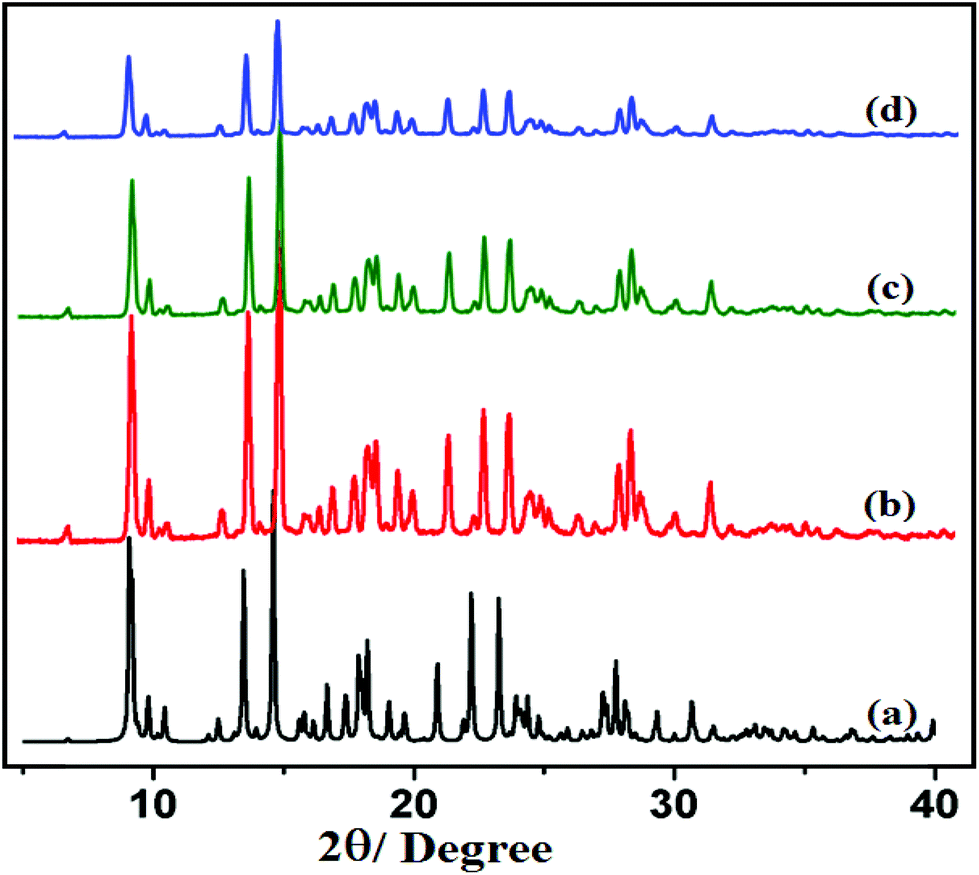
The PXRD patterns of compounds 1 and 2 are shown in Fig. 4 and 5, respectively. For both compounds, good correspondence between the different peaks in the simulated and as-synthesized patterns is found which indicates the phase purity of the as-synthesized compounds. In cases of desolvated framework, the PXRD patterns are almost the same as the simulated as well as the as-synthesized pattern only with some loss of intensities. This indicates that for the heated samples, there are no structural changes in the frameworks even upon removal of the lattice solvent molecule.
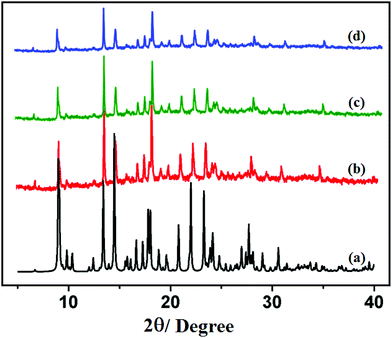 |
| | Fig. 4 PXRD patterns in different states: (a) simulated from X-ray single crystal data; (b) bulk as-synthesized compound; (c) at 150 °C; (d) recovered compound after catalytic use of 1. | |
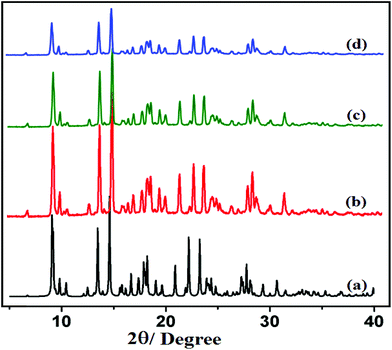 |
| | Fig. 5 PXRD patterns in different states: (a) simulated from X-ray single crystal data; (b) bulk as-synthesized compound; (c) at 150 °C; (d) recovered compound after catalytic use of 2. | |
Adsorption properties
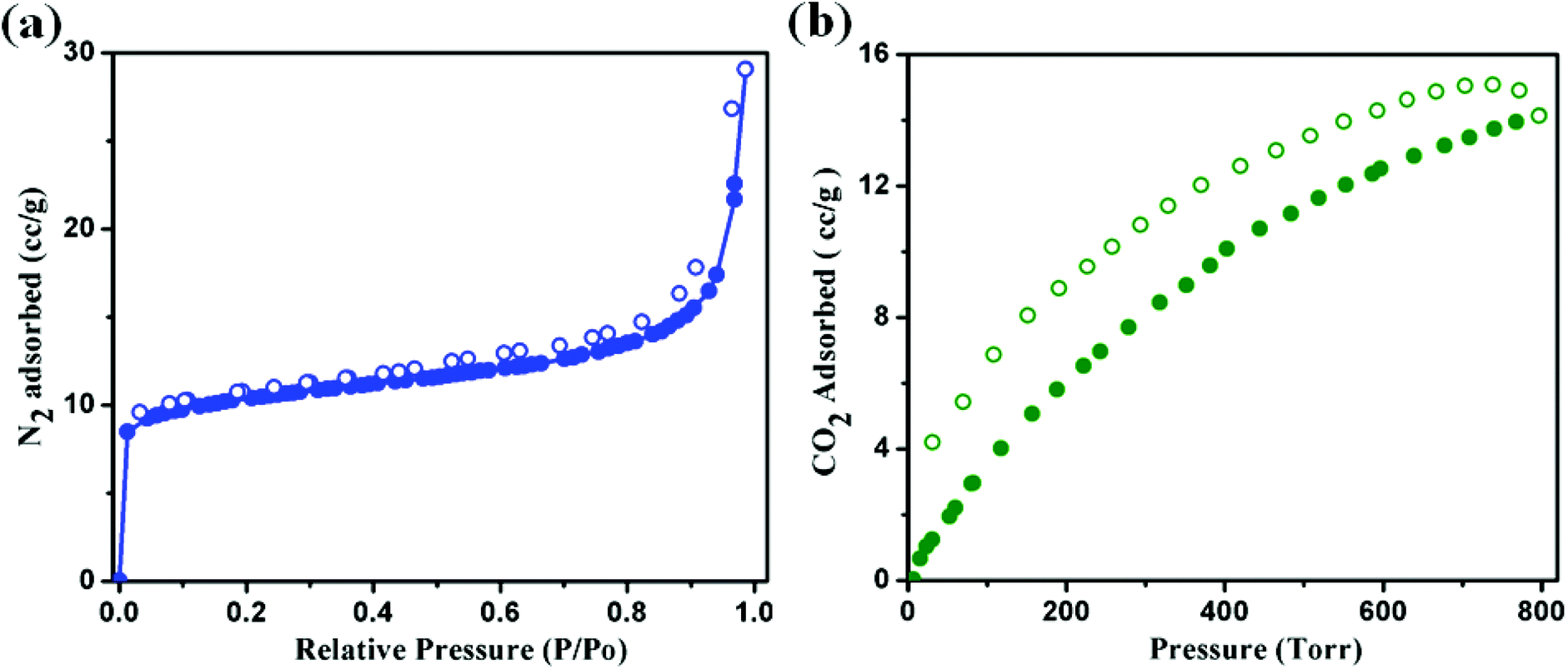
From the structural analysis it is observed that both the structures have measurable voids. Considering their identical structure we took the most stable Ni(II) analogue (2) as a representative to study the permanent porosity and the nature thereof. The dehydrated framework of compound 2 was subjected to sorption studies with N2 (kinetic diameter 3.64 Å) and CO2 (kinetic diameter 3.3 Å) at 77 K and 273 K, respectively (Fig. 6a and b). The sample of 2 was prepared by a thorough washing of the crystals with CHCl3 followed by evacuation at 150 °C for 3 h. The N2 sorption isotherm (Fig. 6a) shows a type II profile, with the final uptake of 29 cc g−1, suggesting only surface adsorption. While for the same compound, the CO2 adsorption isotherm shows the final uptake is 2.78 wt% (14.14 cc g−1) as the pressure approaches to 800 torr and the desorption profile does not follow the adsorption curve (Fig. 6b). The sorption profile shows a hysteresis retaining 4 cc g−1 of CO2 even at very low P/P0 (0.001). This is probably due to the strong interaction of CO2 molecules with the pendent pyridyl nitrogen atom and –CHN– moiety of the azpy ligand. These interactions may facilitate the selectivity in adsorption as well as the kinetic trapping of gas molecules, retaining some of the CO2 molecules.
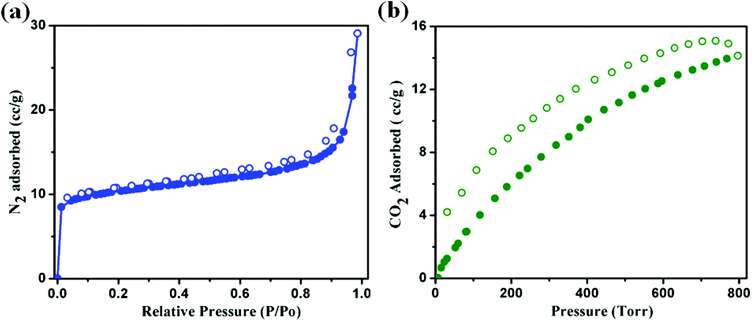 |
| | Fig. 6 (a) N2 adsorption isotherms below 1.0 bar pressure of 2 at 77 K. (b) CO2 adsorption isotherms below 800 Torr pressure of 2 at 273 K temperature. Filled and open circles represent adsorption and desorption respectively. | |
Catalytic Knoevenagel condensation reactions
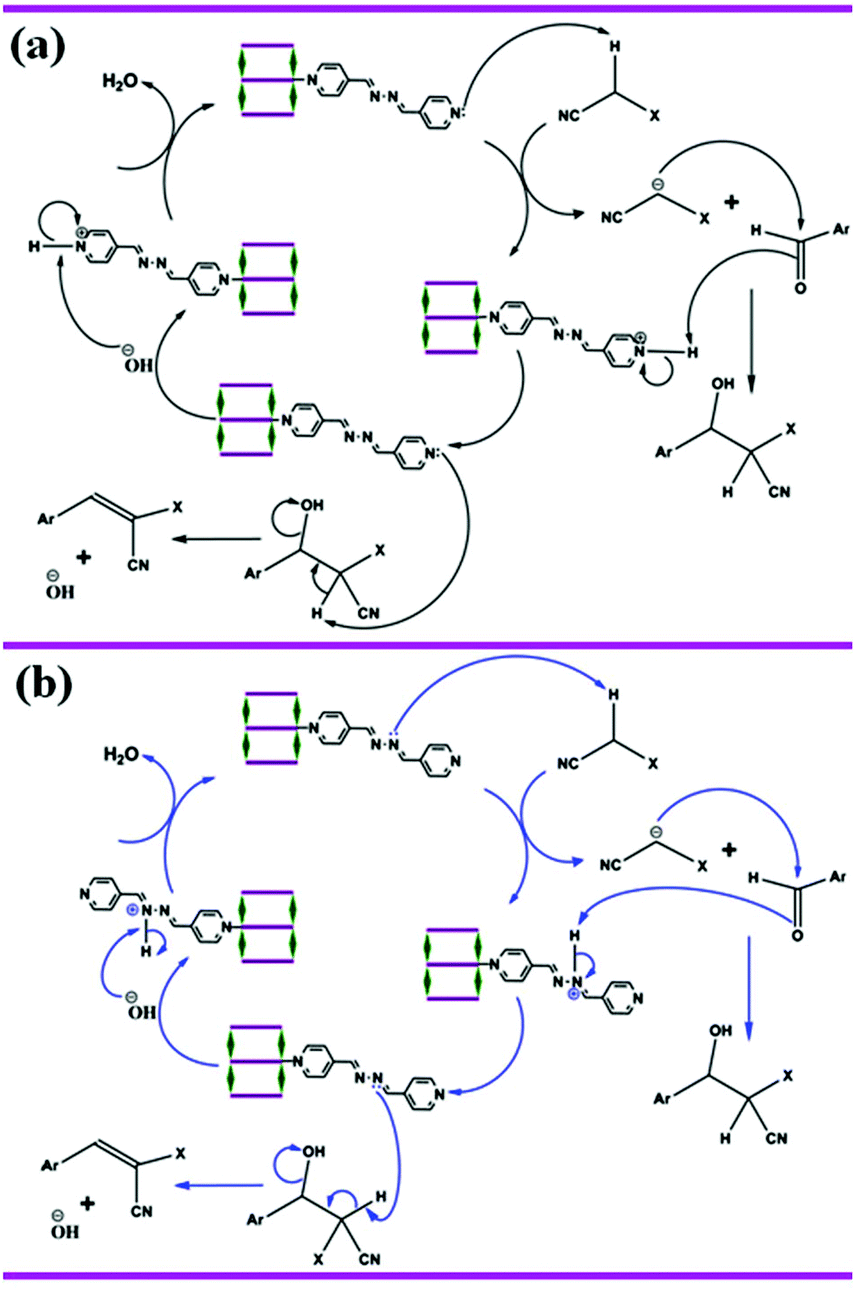
The Knoevenagel reaction is well known and is a very important in reaction methodology, not only as a weak base-catalyzed model reaction but also a reaction that generates a C–C bond. To probe the catalytic activity of 1 and 2, we have chosen the Knoevenagel condensation reaction of substituted benzaldehydes with methylene compounds (malononitrile and ethyl cyanoacetate). The results of the catalytic Knoevenagel condensation reaction for different substrates are summarized in Table 4. The yields of the respective adducts of different benzaldehydes with malononitrile and ethyl cyanoacetate clearly indicate efficient catalytic activity. Both of the compounds do not have effective pore and thus the catalytic reactions mainly occurred on the surface of 1 and 2. A plausible mechanism for the MOF (1 or 2) catalyzed Knoevenagel condensation of different substituted aromatic aldehydes with active methylene compounds is depicted in Scheme 2. Both the compounds contain –CHN– groups of bridging/pendent azpy ligands and also free pyridyl moieties of pendent azpy. These can function as highly reactive base catalysts under such mild conditions. Here the pyridyl/imine nitrogen acts as a Brønsted proton scavenger from the active methylene group of malononitrile or ethyl cyanoacetate, which is acidic in nature due to the presence of two strong electron withdrawing groups.33 The conjugate base generated here is stable due to conjugation with the cyano or acetate groups. Now, this anion undergoes nucleophilic attack on the carbonyl carbon atom of the substituted aromatic aldehydes.33 After two cycles of protonation and deprotonation, the MOF catalysts return back to the original structure and the product is released from the surface of the MOF. A probable mechanism has been depicted in Scheme 2.
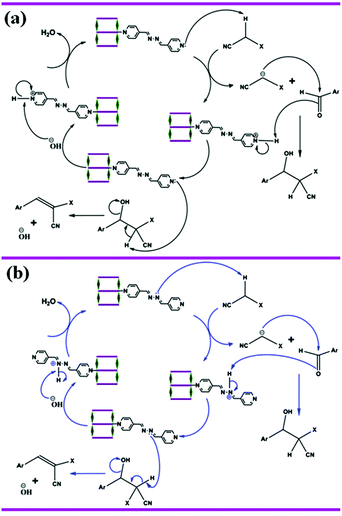 |
| | Scheme 2 Probable mechanism of the Knoevenagel condensation reaction catalyzed by compounds 1 and 2. (a) Proton is scavenged by pyridyl nitrogen (black arrow) of pendent azpy ligand. (b) Proton is scavenged by imine nitrogen (blue arrow) of bridging and pendent azpy ligand. | |
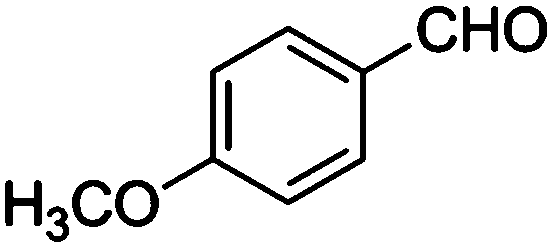
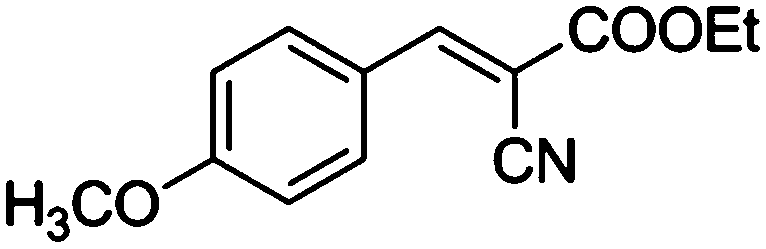
Table 4 Knoevenagel condensation of various aromatic aldehydes with active methylene compounds catalyzed by compounds 1 and 2a
| Entry |
Aldehyde |
X |
Product |
Time (min) |
Isolated yield (wt%) |
| Compound 1 |
Compound 2 |
|
All reactions were performed with 10 mmol of substrates in 10 ml of absolute ethanol at 80 °C using 25 mg of the as-synthesized compounds 1 or 2.
The fifth run.
|
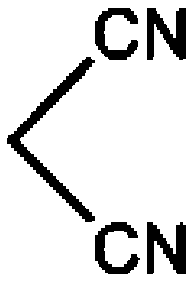
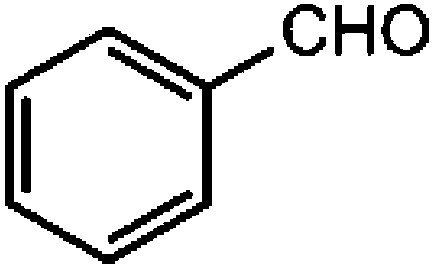
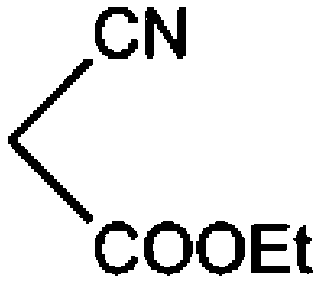
| 1 |
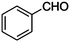
|
CN |
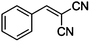
|
10 |
96, 94b |
95, 92b |
| 2 |
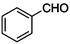
|
COOEt |
|
60 |
92 |
92 |
| 3 |
|
CN |
|
10 |
93 |
93 |
| 4 |
|
COOEt |
|
60 |
91 |
91 |
| 5 |
|
CN |
|
10 |
92 |
93 |
| 6 |
|
COOEt |
|
60 |
92 |
92 |
| 7 |
|
CN |
|
10 |
95 |
96 |
| 8 |
|
CN |
|
10 |
96 |
97 |
| 9 |
|
CN |
|
10 |
94 |
94 |
| 10 |
|
COOEt |
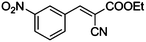
|
60 |
91 |
89 |
| 11 |
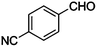
|
CN |
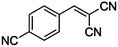
|
10 |
97 |
97 |
| 12 |
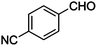
|
COOEt |
|
60 |
93 |
94 |
| 13 |
|
CN |
|
10 |
98 |
97 |
| 14 |
|
COOEt |
|
10 |
95 |
94 |
| 15 |
|
CN |
|
10 |
85 |
83 |
| 16 |
|
CN |
|
10 |
82 |
84 |
| 17 |
|
COOEt |
|
60 |
79 |
81 |
The influence of solvent on the Knoevenagel condensation catalyzed by 1 or 2 was investigated using the model reaction between benzaldehyde and malononitrile and the results are shown in Table 5. It is evident that only in CH3OH and C2H5OH, are the reactions complete within 10 min under refluxing conditions with high yields. However, at 50 °C in C2H5OH–CH3OH, the reaction did not complete, as the yield of the reaction is much higher under refluxing conditions. Thus we may infer that for a certain time, with an increase of temperature the yield of the condensation reaction increases. For solvents like acetonitrile, benzene, CHCl3 and THF, which are less polar than ethanol–methanol, the Knoevenagel condensation did not occur at all within 10 min time either at 50 °C or under refluxing conditions (Table 5, entries 1–7).
Table 5 The influence of solvent on Knoevenagel condensation of benzaldehyde and malononitrilea
| Entry |
Solvent |
Temp. (°C) |
Time (min) |
Yieldb (%) |
| Compound 1 |
Compound 2 |
|
Reaction conditions: benzaldehyde (10 mmol), malononitrile (10 mmol), compound 1 or 2 (25 mg) in each solvent (10 ml).
Isolated yield by column chromatography.
|
| 1 |
C6H6 |
50/80 |
10 |
nr |
nr |
| 2 |
CHC13 |
50/65 |
10 |
nr |
nr |
| 3 |
THF |
50/65 |
10 |
nr |
nr |
| 4 |
CH3CN |
50/80 |
10 |
nr |
nr |
| 5 |
CH3OH |
50 |
10 |
82 |
83 |
| 6 |
CH3OH |
65 |
10 |
94 |
93 |
| 7 |
C2H5OH |
50 |
10 |
54 |
54 |
| 8 |
C2H5OH |
60 |
10 |
72 |
70 |
| 9 |
C2H5OH |
70 |
10 |
83 |
86 |
| 10 |
C2H5OH |
80 |
10 |
96 |
96 |
Considering the moderate reaction temperature we used the more environmentally friendly ethanol as the solvent for studying our model Knoevenagel condensation catalyzed by 1 or 2. At first, a blank reaction of benzaldehyde with malononitrile in refluxing ethanol was performed to confirm that the reaction does not proceed in the absence of a MOF catalyst. Then the catalysis was performed with different substrates. In the cases where the benzaldehyde was substituted with electron withdrawing groups (–NO2, –CN, etc.), the yield increases, whereas electron donating substituents (–CH3, –OCH3, etc.) on the aromatic ring considerably decreases the yield (Table 4). In cases with electron withdrawing groups the possibility of attack of the carbanion (generated from the active methylene group) on the carbonyl carbon is much higher compared to that of electron donating groups. Condensation of substituted benzaldehydes with various substrates possessing active methylene groups is shown in Table 4. Substituents with electron withdrawing nature stabilize the carbanion (via conjugation) compared to those with electron donating nature. So malononitrile displays the maximum conversion compared to ethyl cyanoacetate. When substituents are present in the ortho position of the aromatic ring the yield of the product is minimized. This could be attributed to steric hindrance at the reaction site.
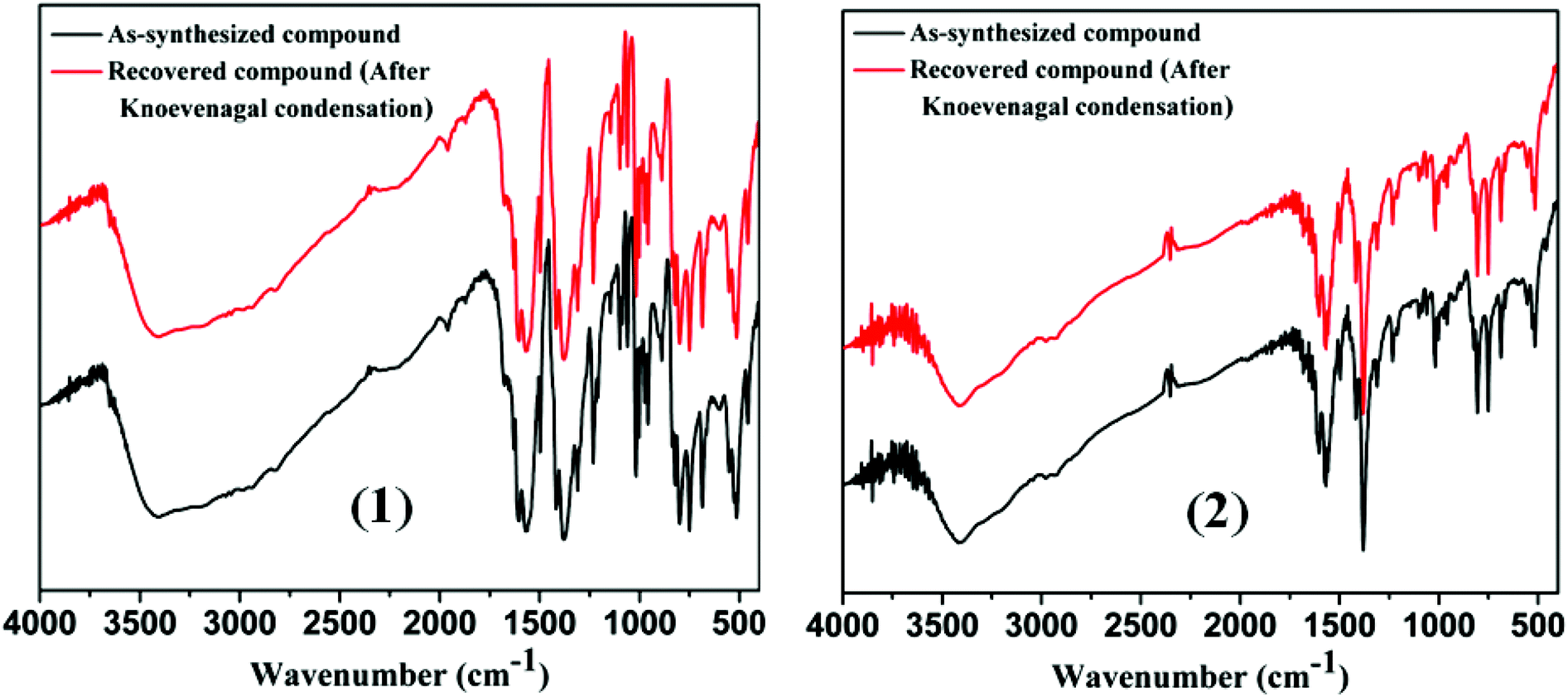
The key advantages of the use of heterogeneous catalysts are easy recovery of the catalyst from the reaction mixtures and its reuse which has been shown in all instances in the present study. In order to check the stability of the MOF catalysts, we have also characterized the recovered MOFs. After the catalytic reactions were over, the MOF was recovered by centrifugation, washed thoroughly with ethanol and dried. The recovered MOF catalyst was then subjected to X-ray powder diffraction and IR spectral analysis. Comparison of IR spectra (Fig. 7) and X-ray diffraction patterns (Fig. 4 and 5) of the as-synthesized and the recovered MOF convincingly demonstrated that the structural integrity of the compound remained unaltered after the Knoevenagel condensation reactions. For the recycling study, we performed Knoevenagel condensation reactions using the same model reaction between benzaldehyde and malononitrile with recovered MOFs 1 and 2 in ethanol. After recycling five times, and to our surprise, the condensation yield was as high as that of the first cycle (Table 6). With our results, we made a comparative study to see how well our present protocol holds up with respect to existing solid catalysts.34 The results of our findings are summarized in Table 7, which shows that the reported protocol offers considerable improvement over existing reports not only in terms of catalyst loading but also in terms of reaction time and yield.
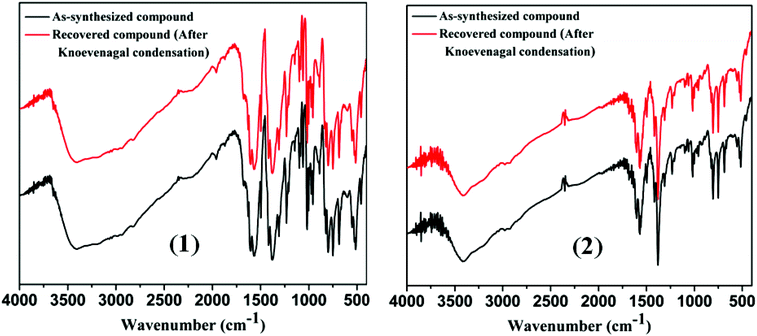 |
| | Fig. 7 Comparison of IR spectra of pure and recovered compound after Knoevenagel condensation reaction of 1 and 2, respectively. | |
Table 6 Reusability of the coordination polymer catalysts 1 and 2a
| Entry |
Recycling run |
Time (min) |
Yieldb (%) |
| Compound 1 |
Compound 2 |
|
Reaction conditions: benzaldehyde (10 mmol), malononitrile (10 mmol), compound 1 or 2 (25 mg) in each solvent (10 ml).
Isolated yield by column chromatography.
|
| 1 |
First |
10 |
96 |
95 |
| 2 |
Second |
10 |
96 |
95 |
| 3 |
Third |
10 |
95 |
94 |
| 4 |
Forth |
10 |
95 |
93 |
| 5 |
Fifth |
10 |
94 |
92 |
Table 7 List of catalytic activities of some of the MOFs and common solid catalysts for the Knoevenagel condensation of benzaldehyde with malononitrile and ethyl cyanoacetate
| Aldehyde |
Active methylene compound |
Name of catalyst |
Amount of catalyst (mg) |
T (°C) |
Time |
Yield (%) |
Reference |
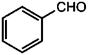
|
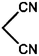
|
BEA |
200 |
130 |
4 h |
100 |
34a
|
| MFI |
200 |
130 |
6 h |
96 |
34a
|
| CuBTC |
200 |
130 |
1 h |
100 |
34a
|
| Al-MIL-101-NH2 |
30 |
80 |
30 min |
61 |
34b
|
| Fe-MIL-101-NH2 |
30 |
80 |
30 min |
78 |
34b
|
| AlPNO (amorphous) |
200 |
80 |
30 min |
33 |
34c
|
| {[Co(azpy)3(p-bdc)2(H2O)4]·(CH3OH)2(H2O)3}n |
25 |
80 |
10 min |
96 |
This work |
| {[Ni(azpy)3(p-bdc)2H2O)4]·(CH3OH)2(H2O)3}n |
25 |
80 |
10 min |
94 |
This work |
| |
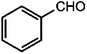
|
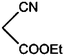
|
BEA |
200 |
130 |
24 h |
66 |
34a
|
| MFI |
200 |
130 |
24 h |
39 |
34a
|
| CuBTC |
200 |
130 |
24 h |
67 |
34a
|
| ED-MIL-101-NH2 |
30 |
60 |
15 h |
89 |
34d
|
| BD-MIL-101-NH2 |
30 |
60 |
15 h |
98 |
34d
|
| DD-MIL-101-NH2 |
30 |
60 |
15 h |
82 |
34d
|
| {[Co(azpy)3(p-bdc)2(H2O)4]·(CH3OH)2(H2O)3}n |
25 |
80 |
1 h |
92 |
This work |
| {[Ni(azpy)3(p-bdc)2H2O)4]·(CH3OH)2(H2O)3}n |
25 |
80 |
1 h |
92 |
This work |
Magnetic properties of 1 and 2
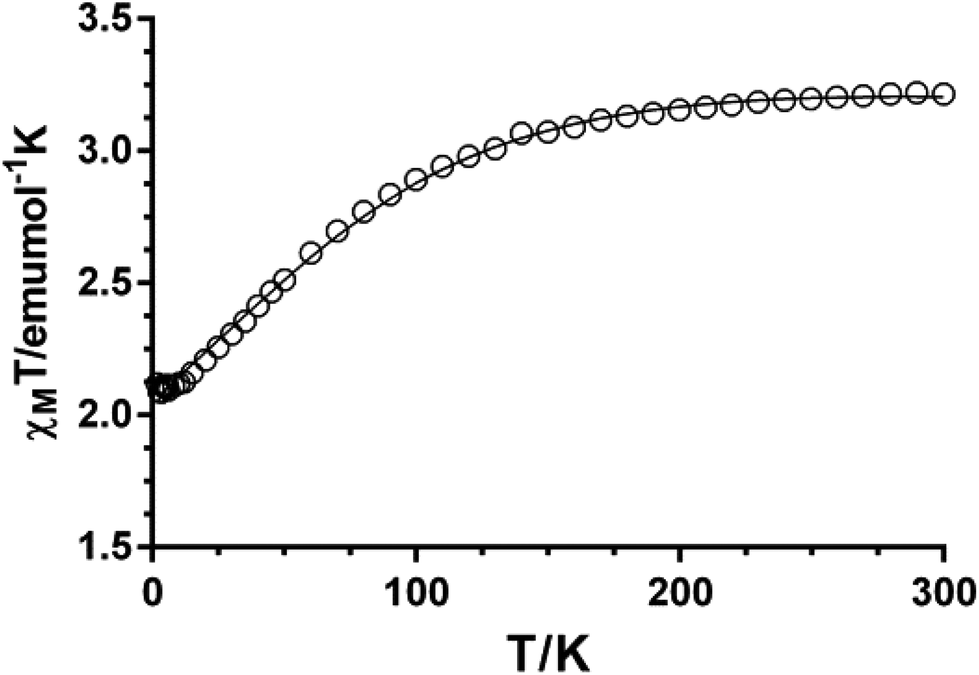
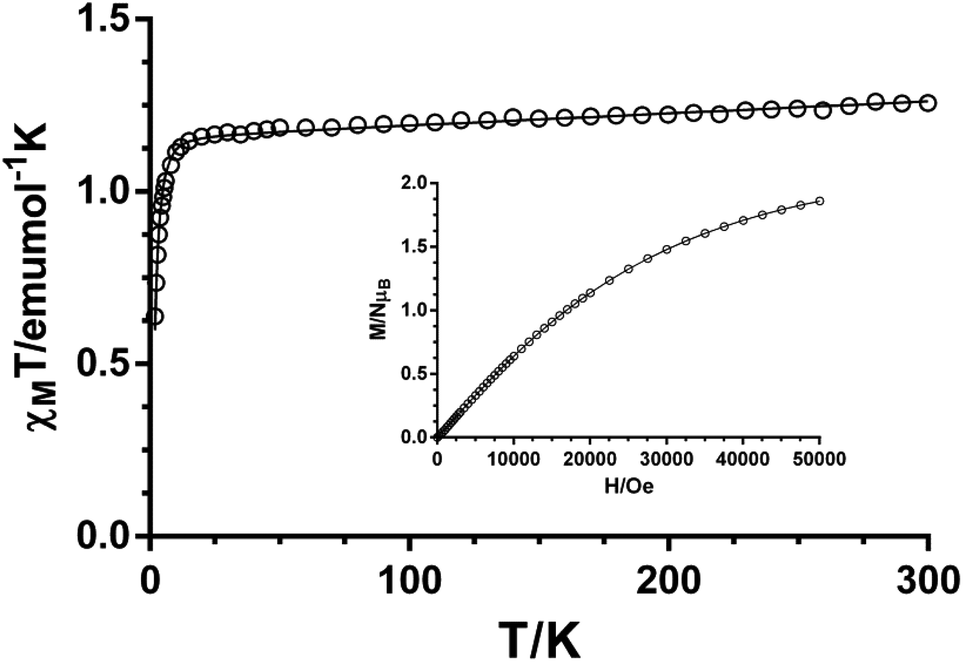
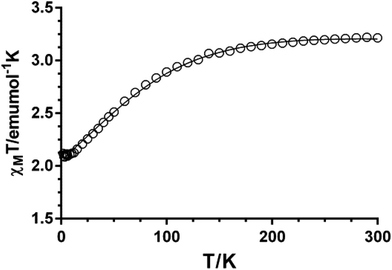
The χMT value for compound 1 at room temperature (3.21 cm3 mol−1 K) is significantly larger than the spin-only value for a high-spin Co(II) ion (S = 3/2, 1.875 cm3 mol−1 K with g = 2), which is indicative of the unquenched orbital contribution of the Co(II) ion in distorted octahedral geometry (Fig. 8). The χMT product remains almost constant from room temperature to 100 K and then slightly decreases reaching a value of 2.11 cm3 mol−1 K at 2 K. Because the shortest Co–Co distance through the ligand is 11.395 Å, the exchange interaction between the Co(II) ions can be considered as negligible. Therefore, this decrease in χMT is most likely due to spin–orbit coupling (SOC) effects rather than intra- and intermolecular antiferromagnetic interactions. The magnetic susceptibility data for 1 were analyzed by introducing SOC effects through the Hamiltonian: Ĥ = αλ![[L with combining circumflex]](https://www.rsc.org/images/entities/b_i_char_004c_0302.gif) ·Ŝ + Δ[2z − ( + 1)/3] + βH(−α + geŜ) where λ is the spin–orbit coupling parameter, α is the orbital reduction factor, and Δ the axial and orbital splitting of the T1 term (Fig. 8). Using the MAGSAKI package,35 the best-fit was found with the values λ = −126 cm−1, α = 1.395, Δ = −691 cm−1 and TIP = 428 × 10−6 cm3 mol−1, with an agreement factor R = 2.9 × 10−5. The negative value of Δ agrees well with the tetragonally compressed octahedral geometry of the CoO4N2 coordination environment in 1.
·Ŝ + Δ[2z − ( + 1)/3] + βH(−α + geŜ) where λ is the spin–orbit coupling parameter, α is the orbital reduction factor, and Δ the axial and orbital splitting of the T1 term (Fig. 8). Using the MAGSAKI package,35 the best-fit was found with the values λ = −126 cm−1, α = 1.395, Δ = −691 cm−1 and TIP = 428 × 10−6 cm3 mol−1, with an agreement factor R = 2.9 × 10−5. The negative value of Δ agrees well with the tetragonally compressed octahedral geometry of the CoO4N2 coordination environment in 1.
 |
| | Fig. 8 Temperature dependence of χMT for 1. | |
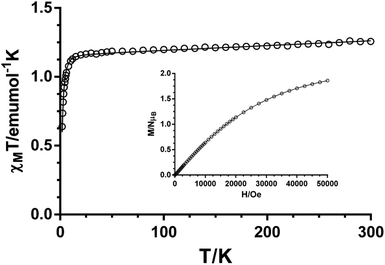
The temperature dependence of the χMT product for compound 2 (χMT being the molar magnetic susceptibility per Ni(II) ion) is displayed in Fig. 9. The χMT value at room temperature of 1.26 cm3 K mol−1 is typical for non-interacting Ni(II) ions (S = 1) with g > 2, as expected for the long Ni–Ni distances through the ligand (15.64 Å) and the space (12.03 Å). When temperature is lowered, χMT remains almost constant until 15 K and then sharply decreases to a value of 0.61 cm3 K mol−1 at 2 K. The decrease at low temperature is due to the zero-field splitting of the 4A2g ground state in Oh geometry, due to a combination of the tetragonal distortion and spin–orbit coupling. We have further analyzed the magnetic properties of 2 by using the Hamiltonian H = D[S2z − S(S + 1)/3] + gμBHS, where S is the spin ground state and D is the axial anisotropy, μB is the Bohr magneton and H the applied magnetic field. Simultaneous fitting of the magnetization and magnetic susceptibility data to the above Hamiltonian using the program PHI36 afforded the following magnetic parameters: |D| = 5.2 cm−1, g = 2.15 and TIP = 340 × 10−6 cm3 mol−1. The DNi value is in agreement with those reported in the literature.37
 |
| | Fig. 9 Temperature dependence of χMT and field-dependence of magnetization at 2 K (inset) for 2. | |
Conclusions
In summary, we have successfully synthesized two mixed ligand one-dimensional (1D) ladder like coordination polymers of Co(II) and Ni(II) containing pendent linear Schiff base linkers. Both the coordination polymers contain an imine group as well as non-coordinated pyridyl groups and efficiently conduct the Knoevenagel condensation of different substituted benzaldehydes with different methylene active compounds, as solid basic catalysts. During the catalytic reaction they display excellent conversions in a short reaction time and there is no need for inert atmosphere to carry out this reaction, as the MOF based catalysts are highly stable in air. It is interesting to note here, that among all the tested solvents, the more economic and environmentally friendly ethanol gives a significantly good yield. Moreover, at the end of each catalytic reaction, the catalysts were easily separated from the reaction system by filtration and then conveniently recycled and reused several times without any loss of activity. The attractive features of these types of coordination polymer catalysts could help to reduce disposal costs and could prove themselves to be significant to the chemical industry for their use in greener and continuous chemical processes. Therefore, compounds 1 and 2 are nice recyclable catalytic systems which can function reliably in an ethanol medium producing promising yields for base catalyzed Knoevenagel condensations. Further investigation towards designing more coordination polymer based catalysts and also investigation of other base catalyzed reactions using coordination polymers are currently in progress.
Acknowledgements
The authors gratefully acknowledge the financial assistance given by CSIR, Govt. of India (grant to DG). BB and DKM acknowledge UGC for the research fellowship. The X-ray diffractometer facility under the DST-FIST program of Department of Chemistry (JU) is also gratefully acknowledged. We gratefully acknowledge Dr Rahul Banerjee at NCL, Pune, India, for the gas adsorption study and some valuable discussions during the preparation of the manuscript.
References
-
(a) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed;
(b) C. Janiak, Dalton Trans., 2003, 2781 RSC;
(c) S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed;
(d) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef PubMed;
(e) A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780 RSC;
(f) K. Biradha, A. Ramanan and J. J. Vittal, Cryst. Growth Des., 2009, 9, 2969 CrossRef CAS;
(g) A. U. Czaja, N. Trukhanb and U. Müller, Chem. Soc. Rev., 2009, 38, 1284 RSC;
(h) N. R. Champness, Dalton Trans., 2011, 40, 10311 RSC.
-
(a) R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230 RSC;
(b) O. K. Farha, A. Ö. Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944 CrossRef CAS PubMed;
(c) K. Jayaramulu, S. K. Reddy, A. Hazra, S. Balasubramanian and T. K. Maji, Inorg. Chem., 2012, 51, 7103 CrossRef CAS PubMed;
(d) K. Sumida, D. Stück, L. Mino, J.-D. Chai, E. D. Bloch, O. Zavorotynska, L. J. Murray, M. Dincă, S. Chavan, S. Bordiga, M. Head-Gordon and J. R. Long, J. Am. Chem. Soc., 2013, 135, 1083 CrossRef CAS PubMed.
-
(a) X. Zhao, B. Xiao, J. A. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012 CrossRef CAS PubMed;
(b) H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523 CrossRef CAS PubMed;
(c) W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Chem. Mater., 2010, 22, 5964 CrossRef CAS;
(d) P. Pachfule and R. Banerjee, Cryst. Growth Des., 2011, 11, 5176 CrossRef CAS;
(e) G. Férey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Llewellyn, G. D. Weireld, A. Vimont, M. Daturif and J.-S. Changg, Chem. Soc. Rev., 2011, 40, 550 RSC.
-
(a) S. Sen, N. N. Nair, T. Yamada, H. Kitagawa and P. K. Bharadwaj, J. Am. Chem. Soc., 2012, 134, 19432 CrossRef CAS PubMed;
(b) S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376 CrossRef CAS PubMed;
(c) X.-Y. Dong, R. Wang, J.-B. Li, S.-Q. Zang, H.-W. Houa and T. C. W. Makac, Chem. Commun., 2013, 49, 10590 RSC;
(d) T. Panda, T. Kundu and R. Banerjee, Chem. Commun., 2013, 49, 6197 RSC;
(e) T. Yamada, K. Otsubo, R. Makiurac and H. Kitagawa, Chem. Soc. Rev., 2013, 42, 6655 RSC.
-
(a) G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762 CrossRef CAS PubMed;
(b) Y. Bai, G. J. He, Y. G. Zhao, C. Y. Duan, D. B. Dang and Q. J. Meng, Chem. Commun., 2006, 1530 RSC;
(c) B. Gole, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 12137 RSC;
(d) S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int. Ed., 2013, 52, 2881 CrossRef CAS PubMed.
-
(a) P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 5974 CrossRef CAS PubMed;
(b) K. E. deKrafft, Z. G. Xie, G. H. Cao, S. Tran, L. Q. Ma, O. Z. Zhou and W. B. Lin, Angew. Chem., Int. Ed., 2009, 48, 9901 CrossRef CAS PubMed;
(c) I. Imaz, M. Rubio-Martinez, L. Garcia-Fernandez, F. Garcia, D. Ruiz-Molina, J. Hernando, V. Puntes and D. Maspoch, Chem. Commun., 2010, 46, 4737 RSC.
-
(a) P. Kanoo, C. Madhu, G. Mostafa, T. K. Maji, A. Sundaresan, S. K. Pati and C. N. R. Rao, Dalton Trans., 2009, 5062 RSC;
(b) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC;
(c) P. Mahata, S. Natarajan, P. Panissod and M. Drillon, J. Am. Chem. Soc., 2009, 131, 10140 CrossRef CAS PubMed;
(d) Z. M. Wang, K. L. Hu, S. Gao and H. Kobayashi, Adv. Mater., 2010, 22, 1526 CrossRef CAS PubMed;
(e) B. K. Tripuramallu, P. Manna, S. N. Reddy and S. K. Das, Cryst. Growth Des., 2012, 12, 777 CrossRef CAS;
(f) S. Goswami, A. Adhikary, H. S. Jena, S. Biswas and S. Konar, Inorg. Chem., 2013, 52, 12064 CrossRef CAS PubMed.
-
(a) T. Uemura, R. Kitaura, Y. Ohta, M. Nagaoka and S. Kitagawa, Angew. Chem., Int. Ed., 2006, 45, 4112 CrossRef CAS PubMed;
(b) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC;
(c) M. C. Das, S. Xiang, Z. Zhang and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 10510 CrossRef CAS PubMed;
(d) R. Sen, D. Saha and S. Koner, Chem. – Eur. J., 2012, 18, 5979 CrossRef CAS PubMed;
(e) B. Gole, A. K. Bar, A. Mallick, R. Banerjee and P. S. Mukherjee, Chem. Commun., 2013, 49, 7439 RSC.
-
(a) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982 CrossRef CAS PubMed;
(b) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607 CrossRef CAS PubMed;
(c) J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75 CrossRef CAS PubMed;
(d) S. Neogi, M. K. Sharma and P. K. Bharadwaj, J. Mol. Catal. A: Chem., 2009, 299, 1 CrossRef CAS PubMed;
(e) M. Savonnet, S. Aguado, U. Ravon, D. Bazer-Bachi, V. Lecocq, N. Bats, C. Pinel and D. Farrusseng, Green Chem., 2009, 11, 1729 RSC;
(f) T. Uemura, N. Yanaia and S. Kitagawa, Chem. Soc. Rev., 2009, 38, 1228 RSC;
(g) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC;
(h) A. Dhakshinamoorthy, M. Opanasenko, J. Čejka and H. Garcia, Adv. Synth. Catal., 2013, 355, 247 CAS.
-
(a) M. L. Kantam, B. M. Choudary, Ch. V. Reddy, K. K. Rao and F. Figueras, Chem. Commun., 1998, 1033 RSC;
(b) S. Abelló, F. Medina, D. Tichit, J. Perez-Ramírez, Y. Cesteros, P. Salagrea and J. E. Sueiras, Chem. Commun., 2005, 1453 RSC;
(c) A. M. Frey, S. K. Karmee, K. P. de Jong, J. H. Bitter and U. Hanefeld, ChemCatChem, 2013, 5, 594 CrossRef CAS.
-
(a) B. Das and N. Chowdhury, J. Mol. Catal. A: Chem., 2007, 263, 212 CrossRef CAS PubMed;
(b) M. Mokhtar, T. S. Saleh and S. N. Basahel, J. Mol. Catal. A: Chem., 2012, 353, 122 CrossRef PubMed.
-
(a) R. Ballini, G. Bosica, D. Livi, A. Palmieri, R. Maggib and G. Sartorib, Tetrahedron Lett., 2003, 44, 2271 CrossRef CAS;
(b) T. Arai, M. Watanabe, A. Fujiwara, N. Yokoyama and A. Yanagisawa, Angew. Chem., Int. Ed., 2006, 45, 5978 CrossRef CAS PubMed.
-
(a) M. Maheswara, V. Siddaiah, G. L. V. Damu, Y. K. Rao and C. V. Rao, J. Mol. Catal. A: Chem., 2006, 255, 49 CrossRef CAS PubMed;
(b) P. Kalita and R. Kumar, Microporous Mesoporous Mater., 2012, 149, 1 CrossRef CAS PubMed.
-
(a) J. S. Yadav, P. P. Rao, D. Sreenu, R. S. Rao, V. N. Kumar, K. Nagaiah and A. R. Prasad, Tetrahedron Lett., 2005, 46, 7249 CrossRef CAS PubMed;
(b) Z. N. Siddiqui and K. Khan, New J. Chem., 2013, 37, 1595 RSC.
-
(a) N. Kan-nari, S. Okamura, S.-i. Fujita, J.-i. Ozaki and M. Arai, Adv. Synth. Catal., 2010, 9, 1476 CrossRef;
(b) G. B. B. Varadwaj, S. Rana and K. M. Parida, Dalton Trans., 2013, 42, 5122 RSC.
-
(a) E. Knoevenengel, Chem. Ber., 1898, 31, 2585 CrossRef;
(b) G. Jones, Org. React., 1967, 15, 204 CAS;
(c) L. F. Tietze and P. Saling, Synlett, 1992, 281 CrossRef CAS PubMed;
(d) I. Kim, S. G. Kim, J. Choi and G. H. Lee, Tetrahedron, 2008, 64, 664 CrossRef CAS PubMed.
-
(a) P. S. Rao and R. V. Venkataratnam, Tetrahedron Lett., 1991, 32, 5821 CrossRef CAS;
(b) F. Texier-Boullet and A. Foucaud, Tetrahedron Lett., 1982, 23, 4927 CrossRef CAS;
(c) G. Dai, D. Shi, L. Zhou and Y. Huaxue, Chin. J. Appl. Chem., 1995, 12, 104 Search PubMed.
- T. I. Reddy and R. S. Varma, Tetrahedron Lett., 1997, 38, 1721 CrossRef CAS.
- S. Wada and H. Suzuki, Tetrahedron Lett., 2003, 44, 399 CrossRef CAS.
-
(a) K. M. Parida and D. Rath, J. Mol. Catal. A: Chem., 2009, 310, 93 CrossRef CAS PubMed;
(b) F. Shang, J. Sun, S. Wua, Y. Yang, Q. Kan and J. Guan, Microporous Mesoporous Mater., 2010, 134, 44 CrossRef CAS PubMed.
- K. M. Parida, S. Mallick, P. C. Sahoo and S. K. Rana, Appl. Catal., A, 2010, 381, 226 CrossRef CAS PubMed.
- N. T. S. Phan and C. W. Jones, J. Mol. Catal. A: Chem., 2006, 253, 123 CrossRef CAS PubMed.
- K. R. Reddy, K. Rajgopal, C. U. Maheswari and M. L. Kantam, New J. Chem., 2006, 30, 1549 RSC.
-
(a) R. Dey, R. Haldar, T. K. Maji and D. Ghoshal, Cryst. Growth Des., 2011, 11, 3905 CrossRef CAS;
(b) B. Bhattacharya, R. Dey, P. Pachfule, R. Banerjee and D. Ghoshal, Cryst. Growth Des., 2013, 13, 731 CrossRef CAS;
(c) B. Bhattacharya, R. Dey, D. K. Maity and D. Ghoshal, CrystEngComm, 2013, 15, 9457 RSC;
(d) B. Bhattacharya, R. Haldar, R. Dey, T. K. Maji and D. Ghoshal, Dalton Trans., 2014, 43, 2272 RSC;
(e) R. Dey, B. Bhattacharya, P. Pachfule, R. Banerjee and D. Ghoshal, CrystEngComm, 2014, 16, 2305 RSC.
- A. R. Kennedy, K. G. Brown, D. Graham, J. B. Kirkhouse, M. Kittner, C. Major, C. J. McHugh, P. Murdoch and W. E. Smith, New J. Chem., 2005, 29, 826 RSC.
-
SMART (V 5.628), SAINT (V 6.45a), XPREP, SHELXTL, Bruker AXS Inc., Madison, WI, 2004 Search PubMed.
-
G. M. Sheldrick, SADABS (Version 2.03), University of Göttingen, Germany, 2002 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. J. Farrugia, WinGX, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
-
(a) V. A. Blatov, A. P. Shevchenko and V. N. J. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS;
(b) V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 378 RSC.
-
(a) R. W. Hein, M. D. Astle and J. R. Shelton, J. Org. Chem., 1961, 26, 4874 CrossRef CAS;
(b) G. Li, J. Xiao and W. Zhang, Green Chem., 2011, 13, 1828 RSC;
(c) J. Mondal, A. Modak and A. Bhaumik, J. Mol. Catal. A: Chem., 2011, 335, 236 CrossRef CAS PubMed.
-
(a) M. Opanasenko, A. Dhakshinamoorthy, M. Shamzhy, P. Nachtigall, M. Horáček, H. Garcia and J. Čejka, Catal. Sci. Technol., 2013, 3, 500 RSC;
(b) M. Hartmann and M. Fischer, Microporous Mesoporous Mater., 2012, 164, 38 CrossRef CAS PubMed;
(c) S. Ernst, M. Hartmann, S. Sauerbeck and T. Bongers, Appl. Catal., A, 2000, 200, 117 CrossRef CAS;
(d) P. Kasinathan, Y.-K. Seo, K.-E. Shim, Y. Kyu Hwang, U.-H. Lee, D. W. Hwang, D.-Y. Hong, S. B. Halligudi and J.-S. Chang, Bull. Korean Chem. Soc., 2011, 32, 2073 CrossRef CAS.
- H. Sakiyama, J. Chem. Software, 2001, 7, 171 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164 CrossRef CAS PubMed.
-
(a) S. K. Dey, M. S. El Fallah, J. Ribas, T. Matsushita, V. Gramlich and S. Mitra, Inorg. Chim. Acta, 2004, 357, 1517 CrossRef PubMed;
(b) K. K. Nanda, R. Das, L. K. Thompson, K. Venkatsubramanian and K. Nag, Inorg. Chem., 1994, 33, 5934 CrossRef CAS;
(c) R. Das, K. K. Nanda, K. Venkatsubramanian, P. Paul and K. Nag, J. Chem. Soc., Dalton Trans., 1992, 1253 RSC;
(d) P. Mukherjee, M. G. B. Drew, C. J. Gómez-García and A. Ghosh, Inorg. Chem., 2009, 48, 5848 CrossRef CAS PubMed;
(e) Z. Q. Pan, K. Ding, H. Zhou, Q. R. Cheng, Y. I. Cheng and Q. M. Huang, Polyhedron, 2011, 30, 2268 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of H-bonding and π–π interactions, structural figures of compounds reported in this paper and 1H NMR of all known products found in the reactions related to catalysis, elemental analysis study of the isolated products. CCDC 981262 and 981263. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00032c |
|
| This journal is © the Partner Organisations 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.