Oxidovanadium(IV), oxidomolybdenum(VI) and cobalt(III) complexes of o-phenylenediamine derivatives: oxidative dehydrogenation and photoluminescence†
Satyabrata
Chaudhuri
a,
Sachinath
Bera
a,
Manas Kumar
Biswas
a,
Amit Saha
Roy
a,
Thomas
Weyhermüller
b and
Prasanta
Ghosh
*a
aDepartment of Chemistry, R. K. Mission Residential College, Narendrapur, Kolkata-700103, India. E-mail: ghosh@pghosh.in; Fax: +91 33 2477 3597; Tel: +91 33 2428 7347
bMax-Planck Institute for Chemical Energy Conversion, Stifstr. 34-36, 45470, Mülheim an der Ruhr, Germany
First published on 7th March 2014
Abstract
Reactions of o-phenylenediamine derivatives (L3H2) incorporating a (Ph)(Py)(H)C–N(H)– function with the oxidovanadium(IV) and oxidomolybdenum(VI) ions afford amide complexes of types [VIVO(L32−)] (3), [VIVO(L3t-Bu 2−)] (4) and cis-[MoVIO2(L32−)] (5) (L3H2 = ((E)-2-(((2-((phenyl(pyridin-2-yl)methyl)amino)phenyl)imino)methyl)phenol); L3t-BuH = ((E)-2,4-di-tert-butyl-6-(((2-((phenyl(pyridin-2-yl)methyl)amino)phenyl)imino)methyl)phenol)), while the similar reaction of L3H2 with the anhydrous CoCl2 in air results in oxidative dehydrogenation (OD) of the (Ph)(Py)(H)C–N(H)– function, affording a cobalt(III) diimine complex, trans-[CoIII(L4−)Cl2] (6) (L4H = 2-((E)-(2-((E)-phenyl(pyridin-2-yl)methyleneamino)phenylimino) methyl)phenol), contradicting the participation of the higher oxidation states of the metal ions in OD reaction of amines. 3–6 are characterized by elemental analyses and mass, IR, 1H NMR and EPR spectra. The molecular geometries of 4·CH3OH, 5 and 6 were confirmed by single crystal X-ray structure determinations. The VIV–Ophenolatocis to the V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and the VIVO lengths in 4·CH3OH are 1.925(2) and 1.612(2) Å. Two cis MoO lengths are 1.710(2) Å and 1.720(2) Å in 5. The aliphatic –C–N– lengths in 4·CH3OH and 5 are 1.448(3) and 1.479(2) Å, while the same is 1.285(4) Å in 6. DFT calculations on 3 and 6 inferred a significant mixing among dM and NN-ligand backbone favoring a t26 state of the metal ion for the OD of the amine fragment to have stronger dM → πketimine* back-bonding. The πNHPh → πaldimine* transition of L3H2 is red shifted in 3 and 4 quenching the emissive πPhenolato → πaldimine* transitions, elucidated by the TD DFT calculations on 3 (and 3+). The πNPh → πaldimine* transitions are blue shifted in the oxidovanadium(V) analogues, [VVO(L32−)]+ (3+) and [VVO(L3t-Bu 2−)]+ (4+), which are fluorescent (3+, λex = 331, λem = 444 nm; 4+, λex = 339, λem = 490 nm) recorded by the fluorescence-spectroelectrochemical measurements in CH2Cl2. 5 and 6 emit weakly at 466 and 473 nm (5, λex = 336 nm, ϕ = 0.003; 6, λex = 324 nm, ϕ = 0.027).
O bond and the VIVO lengths in 4·CH3OH are 1.925(2) and 1.612(2) Å. Two cis MoO lengths are 1.710(2) Å and 1.720(2) Å in 5. The aliphatic –C–N– lengths in 4·CH3OH and 5 are 1.448(3) and 1.479(2) Å, while the same is 1.285(4) Å in 6. DFT calculations on 3 and 6 inferred a significant mixing among dM and NN-ligand backbone favoring a t26 state of the metal ion for the OD of the amine fragment to have stronger dM → πketimine* back-bonding. The πNHPh → πaldimine* transition of L3H2 is red shifted in 3 and 4 quenching the emissive πPhenolato → πaldimine* transitions, elucidated by the TD DFT calculations on 3 (and 3+). The πNPh → πaldimine* transitions are blue shifted in the oxidovanadium(V) analogues, [VVO(L32−)]+ (3+) and [VVO(L3t-Bu 2−)]+ (4+), which are fluorescent (3+, λex = 331, λem = 444 nm; 4+, λex = 339, λem = 490 nm) recorded by the fluorescence-spectroelectrochemical measurements in CH2Cl2. 5 and 6 emit weakly at 466 and 473 nm (5, λex = 336 nm, ϕ = 0.003; 6, λex = 324 nm, ϕ = 0.027).
Introduction
Oxidation of amine is a significant reaction in biology.1 In the laboratory, the transition metal promoted oxidative dehydrogenation (OD) reaction of amines has been an area of research since 1960 and the reaction was first reported by Curtis et al.2 To date, several OD reactions mediated by transition metal ions with different mechanistic aspects have been reported.3 In many cases, participation of the transition metal ions to the −(2e + 2H+) transfer reaction has been proposed. The accepted mechanism is that the metal ion is oxidized first to a higher oxidation state that will either stepwise oxidize the amine by 1e− transfer via a ligand radical intermediate or directly oxidize by 2e− transfer, eliminating protons.3 It has been reported that the OD reactions of [Ru(bpy)2(ampy)]2+ (ampy = 2-(aminomethyl)pyridine),4a [Ru(tame)2]2+ (tame = 1,1,1-tris(aminomethyl)ethane),4b [Ru(en)3]3+ (en = ethylenediamine)4b [Ru(sar)]2+ (sar = sarcophagine, 3,6,10,13,16,19-hexaazabicyclo(6,6,6)eicosane),4c and [Ru(bpy)2(NC5H4CH(CH3)OH)]2+ (NC5H4CH(CH3)OH) = 2-(hydroxymethyl)pyridine)4d proceed via a ruthenium(IV) intermediate. Similarly, intermediacy of M(IV) ions was proposed in the OD reactions of [Os(bpy)2(ampy)]2+,4e [Os(en)3]3+ (ref. 4f) and [Fe(sar)]3+ (ref. 3a) complexes. A Ni(III) intermediate also was proposed to participate in the OD reaction of a tetraazamacrocyclic complex of nickel(II) ion.4g However, the proposals of the participation of Fe(IV), Ni(III), Os(IV) and Ru(IV) ions in OD reactions are futile.The reduction of the metal ion during OD reactions has been established only in the cases of copper(II), iron(III) and ruthenium(III) ions by isolating their reduced analogues. Reduction of copper(II) to copper(I) has been established in OD reactions of [CuII2(H4L)]4+ (L = octaazamacrocyclic dinucleating ligand),5a CuII(boradiazaindacene (BODIPY) derivative)5b and [CuII(L)]2+ (L = N,N-bis-quinolin-2-ylmethyl-cyclohexane-trans-1,2-diamine)5c complexes. Reduction of iron(III) to iron(II) in the OD reactions of [FeIIIH2L]3+ (H2L = 1,9-bis(2′-pyridyl)-5-[(ethoxy-2′′-pyridyl)methyl]-2,5,8-triazanonane),5d tetracyano(1,2-diamine derivative)ferrate(III)5e complexes and formation of a ruthenium(II) analogue in the OD reaction of [RuIII(O–N)(bpy)2]2+ (O–N = unsymmetrical bidentate phenolate type ligand, bpy = 2,2′-bipyridine)5f were authenticated. However, the reports of air and base promoted OD reactions of the amine complexes of nickel(II), cobalt(III) and rare earth metal ions are significant.5g–i
o-Phenylene diamine derivatives are strong chelating agents and furnished several bioactive transition metal complexes.6 Thus, the coordination chemistry of o-phenylenediamine derivatives is the subject of investigation here. Recently, we reported the OD reaction of a tetradentate o-phenylenediamine derivative (L3H2) (L3H2 = (E)-2-(((2-((phenyl(pyridin-2-yl)methyl)amino)phenyl)imino)methyl)phenol). It was disclosed that the reaction of L3H2 with tris(triphenylphosphine)ruthenium(II) precursor results in the OD reaction converting L3H− to L4−, affording the trans-[Ru(L4−)(PPh3)2]+ (2+) cation (L4H = 2-((E)-(2-((E)-phenyl (pyridinyl)methyleneamino)phenylimino)methyl)phenol.7 However, in the presence of an easily reducible iron(III) ion, no OD reaction occurs and the reaction ends up with the formation of an amine complex, cis-[Fe(L3H−)Cl2] (1), as shown in Scheme 1.
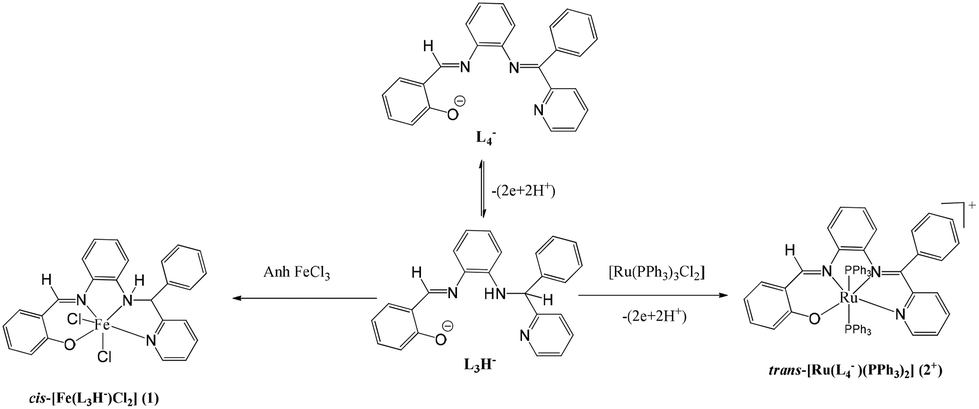 | ||
| Scheme 1 | ||
In this work the role of the metal ions in the OD reaction of the (Ph)(Py)(H)C–N(H)– function of L3H2 was further investigated. The question is whether the reaction requires the higher oxidation state of the metal ion to promote the OD reaction acclaimed so far. To explore it, the chemistry of L3H2 towards the oxidovanadium(IV) and oxidomolybdenum(VI) ions, which are redox active and participate in electron transfer reaction at lower potential and at neutral pH in several redox-enzymes, has been investigated.8,9 Further, oxidovanadium(IV) and dioxidomolybdenum(VI) complexes have been reported as effective catalysts for oxo transfer,10a epoxidation of olefins,10b hydrosilylation of carbonyls10c and oxidative bromination10d reactions. The reactions of oxidovanadium(IV), oxidomolybdenum(VI) and cobaltous ions with L3H2 and L3t-BuH2 in air were performed. Surprisingly, the OD reaction does not occur, with oxidizing oxidovanadium(IV) and oxidomolybdenum(VI) ions yielding only the amide products, [VIVO(L32−)] (3), [VIVO(L3t-Bu 2−)] (4) and cis-[MoVIO2(L32−)] (5) (L3t-BuH2 = (E)-2,4-di-tert-butyl-6-(((2-((phenyl(pyridin-2-yl)methyl)amino)phenyl)imino)methyl) phenol), while the cobaltous ion promotes the OD reaction, affording a cobalt(III) complex of the ketimine derivative, trans-[CoIII(L4−)Cl2] (6).
The metal ion dependent fluorescence features of the organic chromophore is a significant investigation.11 It is observed that L3H2 is fluorescent due to the internal charge transfer (ICT) from the πphenolato to the πaldimine* orbital (λex = 330; λem = 470 nm).7 Lifetimes measurements and time resolved emission spectra (TRES) have confirmed that the lower energy excited state at 390 nm has a higher non-radiative rate constant (knr). It was noted that due to molecular aggregation at higher concentration, the fluid solution fluorescence spectra of L3H2 depend on concentration, which has been investigated by 1H NMR and temperature dependent fluorescence spectra.7 An interesting observation is that the molecular aggregation of L3H2 depends reversibly on temperature. At higher concentration, in addition to the emission band at 470 nm, L3H2 displays a lower energy emission band at 525 nm, which disappears upon dilution. It was recorded that 1 has eighty fold stronger emission than L3H2 itself, while the ketimine analogue 2+ ion is non-emissive.7 The fluid solution fluorescence features of 3, 4, 5 and 6 are also recorded at 298 K. It is found that 3 and 4 in fluid solutions at 298 K are non-emissive while the electrogenerated one-electron oxidized analogues, [VVO(L32−)]+ (3+) and [VVO(L3t-Bu 2−)]+ (4+) are emissive. The complex 5 is weakly emissive. The fluorescence of L3H2 ligand is completely quenched in presence of the reducing cobalt(II) ion, while 6 is brightly emissive.
In this article, to substantiate the role of the metal ions in the OD reaction of L3H2, syntheses, spectra and X-ray structures including the diverse fluorescence spectra and the redox series of 3, 4, 5 and 6 are reported. Density functional theory (DFT) and time dependent (TD) DFT calculations were performed to elucidate the fluorescent as well as the quenched electronic states of the complexes.
Experimental section
Materials and physical measurements
Reagents or analytical grade materials were obtained from commercial suppliers and used without further purification. VO(acac)2 (acac = acetylacetonate) was prepared by the reported procedure.10e Spectroscopic grade solvents were used for spectroscopic and electrochemical measurements. After evaporating MeOH solvents of the sample under high vacuum, elemental analyses and spectral measurements were performed. The C, H and N content of the compounds were obtained using a Perkin-Elmer 2400 series II elemental analyzer. Infrared spectra of the samples were measured from 4000 to 400 cm−1 with KBr pellets at room temperature on a Perkin-Elmer Spectrum RX 1 FT-IR spectrophotometer. 1H NMR spectra in CDCl3 were obtained on a Bruker DPX-300 MHz spectrometer with tetramethylsilane (TMS) as an internal reference. ESI mass spectra were recorded on a micromass Q-TOF mass spectrometer. Electronic absorption spectra in solutions at 298 K were recorded on a Perkin-Elmer Lambda 750 spectrophotometer in the range of 3000–200 nm. Magnetic susceptibility at 298 K was measured on a Sherwood Magnetic Susceptibility Balance. The electroanalytical instrument, BASi Epsilon-EC, for cyclic voltammetric experiments in CH2Cl2 solutions containing 0.2 M tetrabutylammoniumhexafluorophosphate as supporting electrolyte was used. A BASi platinum working electrode, platinum auxiliary electrode and Ag/AgCl reference electrode were used for the measurements. The redox potential data are referenced vs. the ferrocenium/ferrocene, Fc+/Fc, couple. In all cases, the experiments were performed with multiple scan rates to analyze the reversibility of the electron transfer waves. BASi Epsilon-EC was used for spectroelectrochemistry measurements. The X-band electron paramagnetic resonance (EPR) spectra were measured on a Magnettech GmbH MiniScope MS400 spectrometer (equipped with temperature controller TC H03), where the microwave frequency was measured with a frequency counter FC400.The EPR spectra of CH2Cl2 solutions of the paramagnetic complexes 3 and 4 were recorded at 298 K. The EPR spectrum of the CH2Cl2 frozen glass of 3 at 25 K was also recorded. The fluorescence spectra of the complexes were recorded in CH2Cl2 at 298 K. The spectral features of 3+ and 4+ ions were recorded by fluorescence spectro-electrochemical measurements in CH2Cl2 solvent at 298 K.
Excitation and emission spectra were recorded using quartz sample tubes in a Perkin Elmer LS 55 luminescence spectrophotometer. The fluorescence quantum yield (ϕD) was determined in each case by comparing the corrected emission spectrum of the samples with that of anthracene in MeOH (ϕD = 0.20) and CH2Cl2 (ϕD = 0.30) using the following equation12 considering the total area under the emission curve.
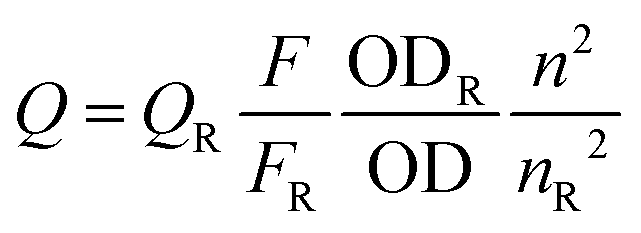 | (1) |
Syntheses
X-Ray crystallographic data collection and refinement of the structures (CCDC 842402 (6), 972492 (4·CH3OH) and 972493 (5))
Single crystals of 4·CH3OH, 5 and 6 were picked up with nylon loops and were mounted on a Bruker AXS Enraf-Nonius Kappa CCD diffractometer equipped with a Mo-target rotating-anode X-ray source and a graphite monochromator (Mo-Kα, λ = 0.71073 Å). 4·CH3OH and 5 were measured at 100 K while 6 was measured at 296 K. Final cell constants were obtained from least squares fits of all measured reflections. The intensity data was corrected for absorptions using intensities of redundant reflections. The structures were readily solved by direct methods and subsequent different Fourier techniques. The crystallographic data of 4·CH3OH, 5 and 6 are listed in Table 1.| 4·CH3OH | 5 | 6 | |
|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b GOF = {∑[w(Fo2 − Fc2)2]/(n − p)}1/2. c wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2 where w = 1/[σ2(Fo2) + (aP)2 + bP], P = (Fo2 + 2Fc2)/3. | |||
| Formula | C33H35N3O2V | C25H19MoN3O3 | C25H18Cl2CoN3O |
| FW | 572.60 | 505.37 | 506.21 |
| Cryst. color | Red | Orange | Green |
| Cryst. syst. | Triclinic | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c |
| a (Å) | 9.340(3) | 7.9292(2) | 9.3416(5) |
| b (Å) | 11.616(7) | 11.4808(6) | 12.8676(7) |
| c (Å) | 13.836(4) | 11.7269(9) | 19.8225(11) |
| α (°) | 76.22(5) | 76.524(4) | 90.00 |
| β (°) | 83.19(5) | 83.276(5) | 103.333(3) |
| γ (°) | 88.86(5) | 81.837(3) | 90.00 |
| V (Å3) | 1447.6(11) | 1023.71(10) | 2318.5(2) |
| Z | 2 | 2 | 4 |
| T (K) | 100(2) | 100(2) | 296(2) |
| 2θ | 60.00 | 62.00 | 48 |
| Calcd (g cm−3) | 1.314 | 1.640 | 1.450 |
| Reflns collected | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 275 275 |
15702 |
9921 |
| Unique reflns | 8372 | 6499 | 3489 |
| Refection [I > 2σ(I)] | 5603 | 6061 | 2567 |
| λ (Å)/μ (mm−1) | 0.71073/0.380 | 0.71073/0.675 | 0.71073/0.993 |
| F(000) | 604 | 512 | 1032 |
| R 1 [I > 2σ(I)]/GOFb | 0.0682/1.036 | 0.0263/1.059 | 0.0432/1.051 |
| R 1 (all data) | 0.1118 | 0.0293 | 0.0609 |
| wR2c [I > 2σ(I)] | 0.1469 | 0.0675 | 0.1145 |
| No. of param./restr. | 378/0 | 289/0 | 289/0 |
| Residual density (e Å−3) | 0.852 | 0.729 | 0.432 |
The Siemens SHELXS-9713a and SHELXL-9713b software packages were used for the solution and the refinement. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed at the calculated positions and refined as riding atoms with isotropic displacement parameters.
Density functional theory (DFT) calculations
All the calculations reported in this article were done with the Gaussian 03W14 program package supported by GaussView 4.1.The DFT15 and TD DFT16 calculations were performed at the level of the Becke three parameter hybrid functional with the non-local correlation functional of Lee–Yang–Parr (B3LYP).17 The gas-phase geometries of 3 with doublet spin state, 3+ and 6 with singlet spin state were optimized using Pulay's Direct Inversion18 in the iterative Subspace (DIIS), ‘tight’ convergent SCF procedure19 ignoring symmetry. The optimized coordinates are listed in Tables S8–S11 (ESI†). In all the calculations, a LANL2DZ basis set,20 along with the corresponding effective core potential (ECP) was used for the metal atom. Valence double zeta with polarization and diffuse functional basis set, 6-31++G**21 were used for the C, N, O and Cl atoms in all the calculations. For the H atoms, the 6-31G basis set was used.22 The percentage contributions of the metal, chloride and ligand to the frontier orbital of the optimized geometries were calculated using the GaussSum program package.23 The sixty excitation energies on the optimized geometries of 3, 3+ and 6 were calculated by TD DFT24 calculations.
Results and discussion
The coordination complexes of the amide and imine derivatives of L3H2 isolated in this work are depicted in Scheme 2. Details of the syntheses of 3–6 are given in the Experimental section. o-Phenylene derivatives are synthesized using the reported procedures.7 L3t-BuH2 and 3–6 are characterized by the elemental analyses and IR, mass, EPR and 1H NMR spectra. The VO stretching vibrations of 3 and 4 are at 966 and 959 cm−1, while the symmetric and asymmetric stretching vibrations of two cis MoO25 resonate at 901 and 915 cm−1. UV/vis absorption spectral data are summarized in Table 2. UV/vis spectra are shown in Fig. S1.† The lower energy absorption bands of 3 and 4 disappear in 3+ and 4+ ions. Complexes 5 and 6 do not display any lower energy absorption bands.
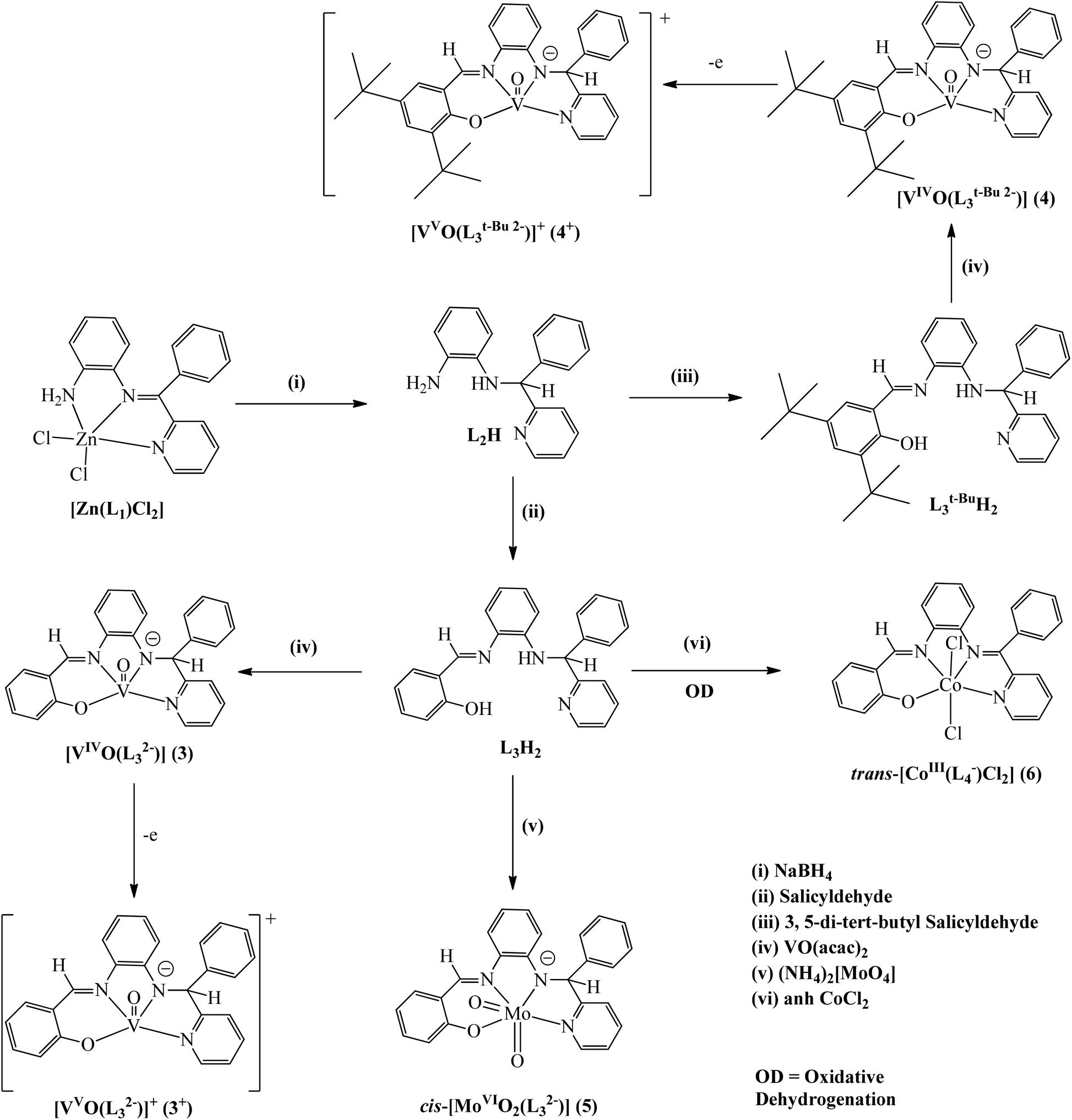 | ||
| Scheme 2 | ||
| Compound | λ max (ε, 104 M−1 cm−1) (nm) |
|---|---|
| 3 | 496 (0.28), 386 (0.44), 327 (0.51)sh, 313 (0.80)sh, 300 (1.24), 266 (1.67) |
| 3 + | 491 (0.13)sh, 407 (0.33)sh, 376 (0.49), 331 (0.64)sh |
| 4 | 492 (0.43), 393 (0.63), 333 (1.72)sh, 316 (2.30)sh, 298 (2.78), 264 (3.20) |
| 4 + | 512 (0.13)sh, 421 (0.55), 357 (0.74)sh |
| 5 | 450 (0.31), 351 (1.01), 326 (1.71)sh 310 (2.23), 256 (2.67)sh |
| 6 | 423 (0.24), 352 (0.41)sh, 331 (0.53)sh, 303 (0.66), 250 (1.10), 206 (1.74) |
The paramagnetic 3 and 4 complexes are redox active. The redox series of 3 and 4 were investigated by cyclic voltammetry in CH2Cl2 containing 0.2 M tetrabutylammoniumhexafluorophosphate as supporting electrolyte. The cyclic voltammogram of 3 is shown in Fig. S2.† The anodic redox waves of 3 and 4 at 0.35 and 0.33 V are assigned to the VO3+/VO2+ redox couple.
No electron transfer occurs in the reactions of L3H2 with VO(acac)2 and molybdate ion producing amide complexes 3, 4 and 5. However, the reaction of CoCl2 with L3H2 affords ketimine complex, 6. In the reaction with CoCl2, both the metal ion and the ligand undergo oxidation. Overall it is a −(3e + 2H+) transfer reaction involving an external dioxygen molecule as an oxidizing agent. Scheme 3 illustrates the probable intermediates of this −(3e + 2H+) transfer redox reaction, which involves: the coordination of the M(II) ion to the monoanionic L3H− ligand affording A, deprotonation of the monoanionic L3H− ligand to the dianionic L32− affording B, and the oxidation of the dianionic L32− to L4− by an external oxygen molecule affording C. The intermediate A has been isolated as a iron(III) complex as 1 (Scheme 1).7 The intermediate B has been isolated as oxidovanadium(IV) and cis-dioxidomolybdenum(VI) complexes as 3, 4 and 5. In case of ruthenium, C is the final product furnishing the 2+ ion (Scheme 1). However, in the case of the cobalt(II) ion, the eg1 electron is delocalized over the low-lying πdiimine* orbital and reacts easily with air, affording the cobalt(III) complex, D.
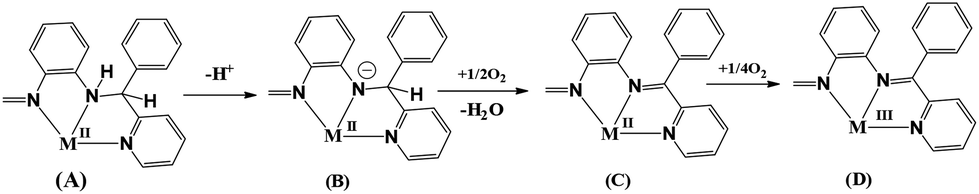 | ||
| Scheme 3 | ||
The OD reaction with the cobalt(II) ion is informative. The higher valent cobalt(IV) ion will never be achieved in air. The conversion of cobalt(II) to cobalt(III) ion in presence of a chelating ligand is easier, however, cobalt(III) ion is not an oxidizing agent. It completely defies the participation of the higher oxidation state of the metal ion in an OD reaction of the amine. One of the important roles of the metal ions is to de-protonate the NH functionality upon coordination, which is achieved in the cases of oxidovanadium and oxidomolybdenum ions. The oxidation occurs via the external O2 molecule facilitating the dM → πimine* back-bonding, which is favored with a t26 state, e.g. Ru(II) and Co(III) ions. For effective back-bonding increasing the M–N bond order, it needs a lower oxidation state of the metal ions. Oxidovanadium(IV) and dioxidomolybdenum(VI) ions in 3–5 are, respectively, d1 and d0 ions and lack the ability to back donate, significantly disfavoring the OD of the amide ligand.
Molecular geometries
Molecular bond parameters and cis or trans geometries of 3–6 were confirmed by the single crystal X-ray structure determinations of 4·CH3OH, 5 and 6. 4·CH3OH crystallizes in the P space group. The molecular geometry of 4·CH3OH in the crystals along with the atom labeling scheme is illustrated in Fig. 1. The significant bond parameters are summarized in Table 3. The tetra-dentate L3t-Bu 2− dianionic ligand spans the sites of the square (with a mean deviation of 0.09 Å) of the distorted square pyramid coordination sphere around the vanadium ion. The vanadium ion is displaced towards the oxido group by 0.65 Å. The oxidovanadium, V(1)–O(40) and the V(1)–Ophenolatoi.e. V(1)–O(1) bond lengths, 1.612(2) and 1.925(2) Å, respectively, correlate well with the presence of the oxidovanadium(IV) ion in 4·CH3OH.26 The C(8)–N(9) and C(17)–N(16) lengths, 1.303(3) and 1.448(3) Å, respectively, are consistent with the existence of the aldimine, –CHN– and (Ph)(Py)(H)C–N(H)– functionalities in 4·CH3OH.7
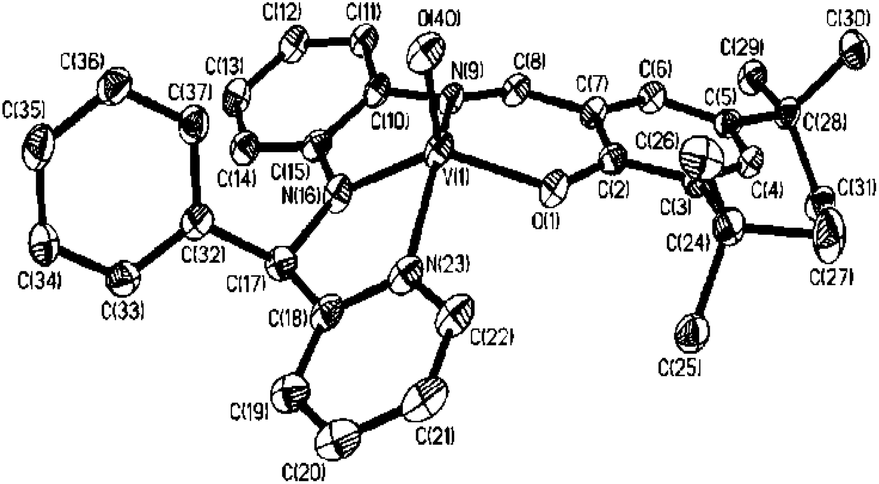 | ||
| Fig. 1 Molecular geometry of 4·CH3OH in crystals (50% ellipsoids; CH3OH and H atoms are omitted for clarity). | ||
| Exp. | Cal. | |
|---|---|---|
| 4·CH3OH | 3 | |
| V(1)–O(1) | 1.925(2) | 1.923 |
| V(1)–N(9) | 2.052(2) | 2.066 |
| V(1)–N(16) | 1.950(2) | 1.968 |
| V(1)–N(23) | 2.099(3) | 2.110 |
| V(1)–O(40) | 1.612(2) | 1.599 |
| C(8)–N(9) | 1.303(3) | 1.302 |
| N(16)–C(17) | 1.448(3) | 1.448 |
| O(1)–V(1)–N(16) | 135.08(9) | 133.75 |
| N(9)–V(1)–N(23) | 146.09(10) | 147.79 |
5 crystallizes in the P space group. An ORTEP plot of the molecule and the atom labeling scheme are illustrated in Fig. 2. Significant bond parameters are listed in Table 4. The orientation of the L32− ligand in 5 is different from that in 4·CH3OH. The Mo–Npy (N(23)) bond is perpendicular to the MoO(1)N(9)N(16) plane, making the two oxido groups cis to each other. Two MoO bond lengths, 1.7096(11) and 1.7198(11) Å, are similar to those reported in cis-dioxidomolybdenum(VI) complexes.25 The C(8)–N(9) length, 1.294(2) Å, authenticates the aldimine (–CHN–) functional group, while the C(17)–N(16) length of 1.479(2) correlates well with a C–N single bond.
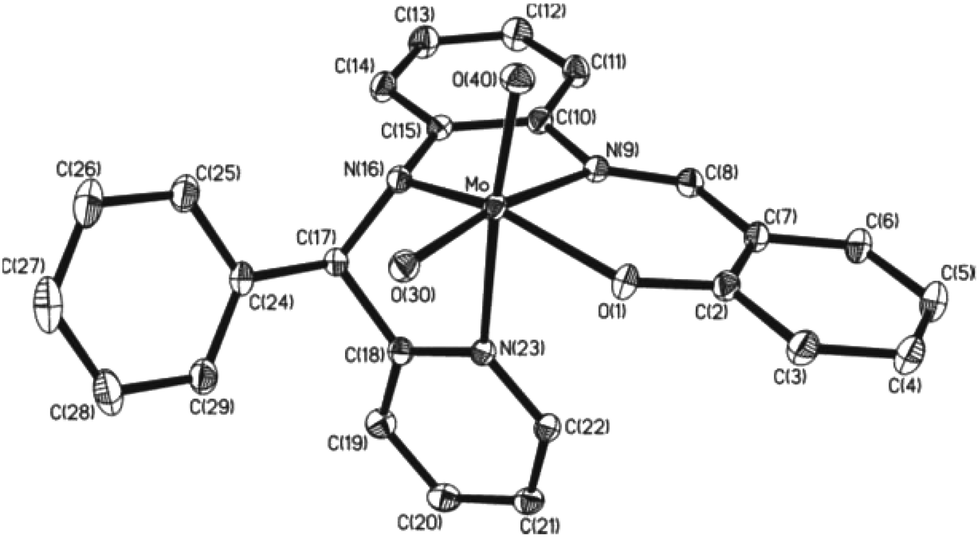 | ||
| Fig. 2 Molecular geometry of 5 in crystals (50% ellipsoids; H atoms are omitted for clarity). | ||
| Mo–O(1) | 1.9692(11) | C(8)–N(9) | 1.2934(19) |
| Mo–N(9) | 2.2958(13) | N(16)–C(17) | 1.4789(18) |
| Mo–N(16) | 2.0219(12) | O(1)–Mo–N(16) | 147.57(5) |
| Mo–N(23) | 2.3637(12) | N(9)–Mo–N(23) | 77.79(4) |
| Mo–O(30) | 1.7198(11) | O(40)–Mo–O(30) | 107.32(5) |
| Mo–O(40) | 1.7096(11) |
6 crystallizes in the P21/c space group. The molecular geometry of 6 in the crystals and the atom labeling scheme is depicted in Fig. 3. Significant bond parameters are summarized in Table 5. The orientation of the tetra-dentate L4− ligand is different from the dianionic L3t-Bu 2− and L32− ligands present in 4·CH3OH and 5. In contrast to the non-planner geometries of L3t-Bu 2− and L32−, the L4− in 6 is completely planar, excluding the pendent phenyl groups, and occupies the square plane of the CoN3OCl2 octahedron enforcing the two chloride ligands trans to each other.
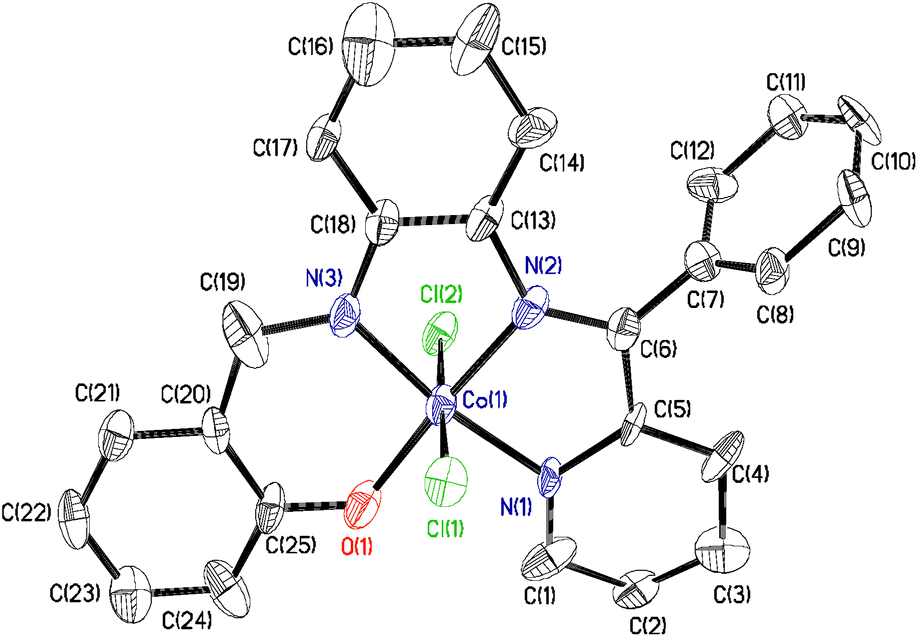 | ||
| Fig. 3 Molecular geometry of 6 in crystals (50% ellipsoids; H atoms are omitted for clarity). | ||
| Exp. | Cal. | |
|---|---|---|
| Co(1)–O(1) | 1.872(2) | 1.888 |
| Co(1)–N(1) | 1.933(3) | 1.9451 |
| Co(1)–N(2) | 1.880(3) | 1.916 |
| Co(1)–N(3) | 1.870(3) | 1.896 |
| Co(1)–Cl(1) | 2.2333(10) | 2.306 |
| Co(1)–Cl(2) | 2.2637(10) | 2.306 |
| N(2)–C(6) | 1.285(4) | 1.299 |
| N(3)–C(19) | 1.297(4) | 1.306 |
| Cl(1)–Co(1)–Cl(2) | 177.55(4) | 177.33 |
| O(1)–Co(1)–N(2) | 176.30(12) | 176.85 |
| N(3)–Co(1)–N(1) | 170.08(13) | 169.75 |
The N(3)–C(19) and N(2)–C(6) lengths, 1.285(4) and 1.297(4) Å, are consistent with the existence of the aldimine (–CHN–) and ketimine ((Ph)(py)CN–) functional groups in 6.7 The bond parameters and the planarity confirm the −(2e + 2H+) oxidation of L32− to L4− in 6. The two trans CoIII–Cl lengths are 2.233(2) and 2.364(2) Å.
The trend of M–Nketimine and M–Namine bond lengths in 1–6 complexes is noteworthy. All three types of bond lengths, M–Namine, M–Namide and M–Nketimine, with 3d and 4d metal ions have successfully been determined. The experimental bond lengths are listed in Table 6. It is observed that the M–Nketimine lengths are significantly shorter than the M–Namine and M–Namide lengths. In 2+, the RuII–Nketimine length, 1.976(6) Å, is intermediate between the reported RuII–Namine and RuIINimide lengths. The average RuII–Namine and RuII–Niminoquinone distances in o-phenylenediamine complexes are 2.132 and 2.080 Å.27 The reported average RuIINimide length is 1.753 Å.28 It claims that the bond order of the RuII–Nketimine in 2+ ion is higher than one. A similar trend has been recorded in the case of Co(III) complex 6 also. The CoIII–Nimine distance, 1.880(3) Å, is shorter than FeIII–Namine and VIV–Namide distances (Table 6). The observed CoIII–Naldimine length in 6 is 1.870(3) Å. In a o-phenylenediamine complex, CoIII–Namine length is 1.982(8)–2.016(3) Å,29 while the CoIIINimide length in a cobalt(III) aryl imido complex is 1.675 Å.30 In 6, the CoIII–Nketimine length being intermediate between the CoIII–N single and double bonds corresponds to a bond order higher than one. The features are explained by the mixing of the dM–π* orbitals (vide infra) that stabilizes the lower oxidation states of the metal ions and increases the M–Nketimine bond order.
| Bond type | Length | Complexes |
|---|---|---|
| FeIII–Namine | 2.192(2) | 1 |
| RuII–Nketimine | 1.976(6) | 2 + |
| VIV–Namide | 1.950(2) | 4 |
| MoVI–Namide | 2.022(2) | 5 |
| CoIII–Nketimine | 1.880(3) | 6 |
EPR spectra, fluorescence and fluorescence-spectroelectrochemistry
The EPR spectra with simulation are shown in the panels (a–c) of Fig. S3.† The spectra with the hyperfine coupling from 51V nuclei corroborate with s = 1/2 spin state and (3, giso = 1.9806, A = 86.9 × 10−4 cm−1; 4, giso = 1.9778, A = 86.7 × 10−4 cm−1) and are consistent with the presence of the oxidovanadium(IV) ion in 3 and 4. The g values of the axial spectrum (panel (b) of Fig. S3†) of the CH2Cl2 frozen glass of 3 at 25 K are: g‖ = 1.9590, A‖ = 156.4 × 10−4 cm−1; g┴ = 1.9828, A┴ = 103.7 × 10−4 cm−1. Analysis of the EPR spectra of 3+ and 4+ ions confirms that the oxidation is metal centered, concluding that 3+ and 4+ cations are the oxidovanadium(V) complexes of types [VVO(L32−)]+ (3+) and [VVO(L3t-Bu 2−)]+ (4+).3 and 4 are non-emissive while the oxidized analogues 3+ and 4+ ions are emissive at 298 K. In CH2Cl2, 5 and 6 are also fluorescent. The fluorescence data are listed in Table 7 and the relevant spectra are shown in Fig. S4.†
In this regard, it is to be noted that the free L3H2 ligand is fluorescent (λex = 330; λem = 470 nm) due to the internal charge transfer from the πphenolato → πaldimine* orbital.73 and 4 absorb strongly at comparatively longer wavelengths (493 and 497 nm) due to πNPh → πaldimine* transition and the complexes are non-emissive. However similar to 1, in 3+ and 4+ ions these lower energy bands are absent and the cations are fluorescent.
It is to be noted that upon oxidation the lower energy absorption bands gradually disappear while fluorescence intensity at λem = 444 and 490 nm, respectively, for 3+ and 4+ cations gradually increases as depicted in the panels (b) and (d) of Fig. 4. The spectral features of 3+ and 4+ cations are illustrated in Fig. 4. The lower energy absorption bands of 5 and 6 at 450 and 423 nm are weaker and both the complexes are weakly fluorescent as illustrated in Fig. S4† and Table 7.
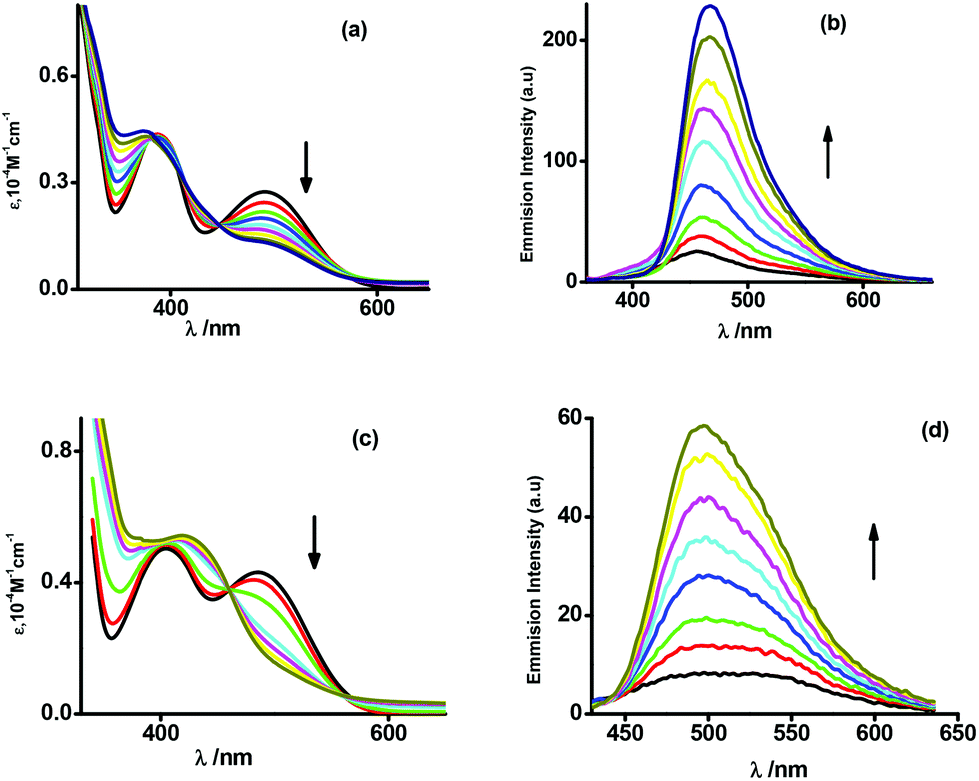 | ||
| Fig. 4 Spectroelectrochemical measurements of the conversion of 3 → 3+ [(a) UV-vis/NIR absorption and (b) fluorescence spectra] and 4 → 4+ [(c) UV-vis/NIR absorption and (d) fluorescence spectra] in CH2Cl2 at 298 K. | ||
The origins of the UV-vis/NIR absorptions of 3–6 were elucidated by the time dependent (TD) density functional theory (DFT) calculations on 3, 3+ and 6. The gas phase geometry of 3 was optimized at the B3LYP/DFT level with the doublet spin state while those of 3+ and 6 were optimized with the singlet spin state. Calculated bond parameters are listed in Tables 3, 5 and S1.† The calculated bond parameters are similar to those obtained from the single crystal X-ray diffraction studies of 4·CH3OH and 6 (Tables 3 and 5). The optimized geometries of 3, 3+ and 6 are shown in Fig. S2.† Excitation energies were calculated by the TD DFT calculations on the optimized geometries. The excitation energies with the oscillator strengths are listed in Table S2.† Fragmentations of the ligand used for the calculations are shown in Fig. S5.† It is reported that L3H2 is emissive due to πPhenolato → πaldimine* transition at λex = 330 nm. 3 and 4 with lower energy absorption bands at λmax = 490 and 500 nm are non-emissive. The TD DFT calculation on 3 has authenticated that the lower energy absorption band of 3 at λmax = 493.57 nm with f = 0.13 is due to πNPh → πaldimine* transitions. The πphenolato → πaldimine* transition of 3 appears at 316.72 nm. However, the non-emissive lower energy absorption band at λmax = 490 nm gradually disappears upon oxidation of 3 to 3+ (panel (a) of Fig. 4) and 3+ becomes emissive. The calculated excitation band of 3+ at 357.8 nm with f = 0.12 is due to πphenolato → πaldimine* transition. Similarly, the calculated πphenolato → πaldimne* emissive excitation wavelength of 6 is 318.06 nm (f = 0.45). The emissive and non-emissive transitions of 3–6 including the L3H2 ligand are illustrated in Scheme S1.†
Molecular orbital analyses
The constituents of the frontier molecular orbitals of 3 and 6 were investigated by DFT calculations using B3LYP functional. Gas phase geometries of 3 and 6 were optimized, respectively, with doublet and singlet spin states. The constituents of the frontier orbitals are analyzed and the data are summarized in Table S3.† The calculations authenticated a significant mixing among the d orbitals and the benzoyl pyridine fragment of the L4− ligand in 6. Analyses have shown that the dxz (HOMO−12) and dyz (HOMO−11) orbitals of the t2 set of 6 exhibit strong interactions with the benzoyl pyridine fragment of the tetra dentate diimine ligand. Similar types of mixing among the d orbitals and the L4− ligand have been attributed in the case of 3 also. However, the d orbitals of the t2 set of the OV(IV) ion interact equally with the phenolato and the benzoyl pyridine fragments. The mixing of the d orbitals results in diverse effects in 3 and 6. In the case of 6, the d6 ion promotes the oxidation of amine to ketimine for π delocalization while the d1 ion stabilizes the hard amide binding in 3. Similarly, the amide binding is stabilized by the hard acid, d0, molybdenum(VI) ion. The result is reversed with the soft d6 ruthenium(II) ion, which converts amine to ketimine for π-delocalization. The OD of the amine to ketimine parallels the chemistry of the conversion of NO → NO+ reducing metal ions in some cases for effective back-bonding with the lower oxidation states of the metal ions. The results correlate well with the reported conversions of copper(II) to copper(I), d10 ion, iron(III) to iron(II) t26 ion and ruthenium(III) to ruthenium(II), t26 ion oxidizing amines to imines.4Conclusion
The role of the oxidation states of the metal ions in oxidative dehydrogenation (OD) reaction of the (Ph)(Py)(H)C–N(H)– functionality of an o-phenylenediamine derivative (L3H2) has been investigated (L3H2 = (E)-2-(((2-((phenyl(pyridin-2-yl)methyl)amino)phenyl)imino)methyl)phenol). Recently, we reported that the reaction of L3H2 with anhydrous FeCl3 affords the amine complex cis-[FeIII(L3H−)Cl2] (1) while the same reaction with [RuII(PPh3)3Cl2] results in an OD reaction affording a ketimine complex, trans-[RuII(L4−)(PPh3)]+ (2+), in good yield (L4H = 2-((E)-(2-((E)-phenyl(pyridin-2-yl)methyleneamino) phenylimino)methyl)phenol)). To summarize the effect of the higher oxidation states of the metal ions to the OD reaction of L3H2, similar reactions of L3H2 with oxidovanadium(IV) and oxidomolybdenum(VI) ions were performed. In each case the reaction produces amide complexes of type [VIVO(L32−)] (3), [VIVO(L3t-Bu 2−)] (4) and cis-[MoVIO2(L32−)] (5). However, the reaction of anhydrous CoCl2 with L3H2 promotes the OD reaction in air yielding a ketimine complex of type trans-[CoIII(L4−)Cl2] (6). The study infers that the OD reaction of L3H2 is not successful with hard metal ions with higher oxidation states, such as FeIII, VIVO and MoVIO4 ions, while the OD reaction occurs with softer, lower valency Ru(II) and Co(II) ions with the filled t26 set that enhances the dM → pπ* back bonding. The work does not justify the previous reports that claim that the metal ion promoted OD reaction of an amine requires the higher oxidation state as an intermediate for oxidation of amines. The work rather concludes that the coordinated amine is de-protonated to an amide that undergoes oxidation to ketimine by an external oxygen molecule to have stronger dM → pπ* back bonding with the lower oxidation states of the metal ions.Fluorescence features of L3H2 and 3–6 are noteworthy. L3H2 is weakly fluorescent (λex = 330; λem = 470 nm) due to a non-emissive lower energy absorption band at 390 nm. 3 and 4 exhibit absorption bands at 493 and 497 nm and are non-emissive, while upon oxidation the lower energy absorption band disappears and [VVO(L32−)]+ (3+) and [VVO(L3t-Bu 2−)]+ (4+) cations are fluorescent, recorded by fluorescence-spectroelectrochemical measurements in CH2Cl2 at 298 K. 5 and 6 display weaker absorption bands at 445 and 423 nm and are weakly fluorescent (5, λex = 336 nm, λem = 466 nm; 6, λex = 324 nm, λem = 473 nm). Moreover, in addition to the oxidation state dependent fluorescence features of 3–4, and oxidovanadium(IV) and dioxidomolybdenum(VI) compounds being effective catalysts for several organic transformations, complexes 3–5 appeared to be significant in coordination chemistry.
Acknowledgements
Financial support received from DST (SR/S1/IC/0026/2012) and CSIR (01/2699/12-EMR-II) New Delhi, India is gratefully acknowledged. SB (CSIR no. 8/531(0006)/2012-EMR-I) and MKB (CSIR no. 8/531(0007)/2012-EMR-I) are thankful to CSIR, New Delhi, India, for fellowships.Notes and references
- (a) C. Anthony, Biochem. J., 1996, 320, 697 CAS; (b) Metal Ions in Biological Systems, ed. P. F. Knowles, D. M. Dooley, H. Sigel and A. Sigel, Marcel Dekker, New York, 1994, vol. 30, p. 361 Search PubMed; (c) M. D. Berry, A. V. Juorio and I. A. Paterson, Prog. Neurobiol., 1994, 42, 375 CrossRef CAS; (d) Principles and Applications of Quinoproteins, ed. W. S. McIntire, C. Hartmann and V. L. Davison, Marcel Dekker, New York, 1993, p. 97 Search PubMed; (e) H. Blaschko, Rev. Physiol., Biochem. Pharmacol., 1974, 70, 83 CrossRef CAS.
- (a) N. F. Curtis, J. Chem. Soc. A, 1971, 2834 RSC; (b) N. F. Curtis, Coord. Chem. Rev., 1968, 3, 3 CrossRef CAS; (c) N. F. Curtis, Y. M. Curtis and H. K. J. Powell, J. Chem. Soc. A, 1966, 1015 RSC.
- (a) F. R. Keene, Coord. Chem. Rev., 1999, 187, 121 CrossRef CAS; (b) S. Minakata, Y. Ohshima, A. Takemiya, I. Ryu, M. Komatsu and Y. Ohshiro, Chem. Lett., 1997, 26, 311 CrossRef; (c) A. J. Bailey and B. R. James, Chem. Commun., 1996, 2343 RSC; (d) P. Muller and D. M. Gilabert, Tetrahedron, 1988, 44, 7171 CrossRef; (e) S. I. Murahashi, T. Naota and H. Taki, J. Chem. Soc., Chem. Commun., 1985, 613 RSC; (f) C. K. Poon and C.-M. Che, J. Chem. Soc., Dalton Trans., 1981, 1019 RSC.
- (a) M. J. Ridd and F. R. Keene, J. Am. Chem. Soc., 1981, 103, 5733 CrossRef CAS; F. R. Keene, M. J. Ridd and M. R. Snow, J. Am. Chem. Soc., 1983, 105, 7075 Search PubMed; (b) P. Bernhard, D. J. Bull, H. B. Burgi, P. Osvath, A. Raselli and A. M. Sargeson, Inorg. Chem., 1997, 36, 2804 CrossRef CAS PubMed; B. C. Lane, J. E. Lester and F. Basolo, Chem. Commun., 1971, 1618 Search PubMed; D. F. Mahoney and J. K. Beattie, Inorg. Chem., 1973, 12, 2561 Search PubMed; (c) P. Bernhard, D. J. Bull, H.-B. Burgi, P. Osvath, A. Raselli and A. M. Sargeson, Inorg. Chem., 1997, 36, 2804 CrossRef CAS PubMed; P. Bernhard and A. M. Sargeson, J. Chem. Soc., Chem. Commun., 1985, 1516 Search PubMed; P. Bernhard, A. M. Sargeson and F. C. Anson, Inorg. Chem., 1988, 27, 2754 Search PubMed; P. Bernhard and A. M. Sargeson, J. Am. Chem. Soc., 1989, 111, 597 Search PubMed; P. Bernhard and F. C. Anson, Inorg. Chem., 1989, 28, 3272 Search PubMed; (d) M. J. Ridd, D. J. Gakowski, G. E. Sneddon and F. R. Keene, J. Chem. Soc., Dalton Trans., 1992, 1949 RSC; (e) F. R. Keene, P. A. Lay, G. E. Sneddon and G. W. Whebell, Aust. J. Chem., 1993, 46, 1763 CrossRef CAS; (f) P. A. Lay, A. M. Sargeson, B. W. Skelton and A. H. White, J. Am. Chem. Soc., 1982, 104, 6161 CrossRef CAS; P. A. Lay and A. M. Sargeson, Inorg. Chim. Acta, 1992, 449, 198 Search PubMed; (g) P. Maruthamuthu, L. K. Patterson and G. Ferraudi, Inorg. Chem., 1978, 17, 3157 CrossRef CAS.
- (a) G. J. Christian, A. Llobet and F. Maseras, Inorg. Chem., 2010, 49, 5977 CrossRef CAS PubMed; G. J. Christian, A. Arbuse, X. Fontrodona, M. A. Martinez, A. Llobet and F. Maseras, Dalton Trans., 2009, 6013 Search PubMed; (b) D. Wang, Y. Shiraishi and T. Hirai, Chem. Commun., 2011, 47, 2673 RSC; (c) V. Amendola, L. Fabbrizzi, E. Mundum and P. Pallavicini, Dalton Trans., 2003, 773 RSC; (d) J. P. Saucedo-Vázquez, V. M. Ugalde-Saldívar, A. R. Toscano, P. M. H. Kroneck and M. E. Sosa-Torres, Inorg. Chem., 2009, 48, 1214 CrossRef PubMed; (e) Y. Kuroda, N. Tanaka, M. Goto and T. Sakai, Inorg. Chem., 1978, 17, 314 CrossRef; (f) R. Mitsuhashi, T. Suzuki and Y. Sunatsuki, Inorg. Chem., 2013, 52, 10183 CrossRef CAS PubMed; (g) C. L. Weeks, P. Turner, R. R. Fenton and P. A. Lay, J. Chem. Soc., Dalton Trans., 2002, 931 RSC; R. K. Wilson and S. Brooker, Dalton Trans., 2013, 42, 12075 Search PubMed; (h) A. Panja and P. Guionneau, Dalton Trans., 2013, 42, 5068 RSC; (i) Q. Li, S. Zhou, S. Wang, X. Zhu, L. Zhang, Z. Feng, L. Guo, F. Wang and Y. Wei, Dalton Trans., 2013, 42, 2861 RSC.
- (a) N. L. Fry, M. J. Rose, D. L. Rogow, C. Nyitray, M. Kaur and P. K. Mascharak, Inorg. Chem., 2010, 49, 1487 CrossRef CAS PubMed; (b) M. J. Rose and P. K. Mascharak, Inorg. Chem., 2009, 48, 6904 CrossRef CAS PubMed; (c) M. J. Rose, N. M. Betterley and P. K. Mascharak, J. Am. Chem. Soc., 2009, 131, 8340 CrossRef CAS PubMed; (d) G. M. Halpenny and P. K. Mascharak, Inorg. Chem., 2009, 48, 1490 CrossRef CAS PubMed; (e) M. J. Rose, C. Nyitray and P. K. Mascharak, Inorg. Chem., 2008, 47, 11604 CrossRef PubMed; (f) M. J. Rose, N. L. Fry, R. Marlow, L. Hinck and P. K. Mascharak, J. Am. Chem. Soc., 2008, 130, 8834 CrossRef CAS PubMed.
- S. Chaudhuri, S. C. Patra, P. Saha, A. Saha Roy, S. Maity, S. Bera, P. S. Sardar, S. Ghosh, T. Weyhermüller and P. Ghosh, Dalton Trans., 2013, 42, 15028 RSC.
- Vanadium references: (a) M. Li, W. Ding, B. Baruah, D. C. Crans and R. J. Wang, Inorg. Biochem., 2008, 102, 1846 CrossRef CAS PubMed; (b) D. Rehder, Bioinorganic Vanadium Chemistry, John Wiley & Sons Ltd., New York, 2008 Search PubMed; (c) C. J. Schneider and V. L. Pecoraro, Vanadium: The Versatile Metal, ACS Symposium Series 974, American Chemical Society, Washington, DC, 2007, p. 148 Search PubMed; (d) D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, Chem. Rev., 2004, 104, 849 CrossRef CAS PubMed.
- Molybdenum references: (a) R. Hille, Dalton Trans., 2013, 42, 3029 RSC and references therein; (b) Y. Zhang, S. Rump and V. N. Gladyshev, Coord. Chem. Rev., 2011, 255, 1206 CrossRef CAS PubMed; (c) R. Hille, Chem. Rev., 1996, 96, 2757 CrossRef CAS PubMed.
- (a) R. Dinda, P. Sengupta, S. Ghosh, H. Mayer-Figge and W. S. Sheldrick, J. Chem. Soc., Dalton Trans., 2002, 4434 RSC; C. J. Whiteoak, G. J. P. Britovsek, V. C. Gibson and A. J. P. White, Dalton Trans., 2009, 2337 Search PubMed; (b) M. A. Katkar, S. N. Rao and H. D. Juneja, RSC Adv., 2012, 2, 8071 RSC; Z. Li, S. Wu, H. Ding, H. Lu, J. Liu, Q. Huo, J. Guan and Q. Kan, New J. Chem., 2013, 37, 4220 Search PubMed; (c) P. M. Reis, C. C. Romão and B. Royo, Dalton Trans., 2006, 1842 RSC; (d) M. R. Maurya, U. Kumarand and P. Manikandan, Dalton Trans., 2006, 3561 RSC; (e) R. A. Row and M. M. Jones, Inorg. Synth., 1957, 5, 113 CrossRef.
- (a) H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC; (b) M. D. Ward, Coord. Chem. Rev., 2010, 254, 2634 CrossRef CAS PubMed; (c) Photofunctional Transition Metal Complexes, ed. V. W. W. Yam, Springer, Series: Structure and Bonding, 2007, vol. 123 Search PubMed; (d) M. D. Ward, Coord. Chem. Rev., 2007, 251, 1663 CrossRef CAS PubMed; (e) M. Hissler, J. E. McGarrah, W. B. Connick, D. K. Geiger, S. D. Cummings and R. Eisenberg, Coord. Chem. Rev., 2000, 208, 115 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed.
- (a) G. M. Sheldrick, SHELXS97, Universität Göttingen, Göttingen, Germany, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL97, Universität Göttingen, Göttingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman Jr., J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, revision E.01, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- (a) The Challenge of d and f Electrons, ed. D. R. Salahub and M. C. Zerner, ACS, Washington, D.C., 1989 Search PubMed; (b) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, U.K., 1989 Search PubMed; (c) W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef; (d) P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- (a) R. E. Stratmann, G. E. Scuseria and M. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS PubMed; (b) M. E. Casida, C. Jamoroski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS PubMed; (c) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- P. J. Pulay, J. Comput. Chem., 1982, 3, 556 CrossRef CAS.
- H. B. Schlegel and J. J. McDouall, Computational Advances in Organic Chemistry, ed. C. Ogretir and I. G. Csizmadia, Kluwer Academic, The Netherlands, 1991, p. 167 Search PubMed.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS PubMed; (b) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS PubMed; (c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS PubMed.
- (a) T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS; (b) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS.
- (a) V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976 CrossRef CAS; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople and M. S. Gordon, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS PubMed; (c) P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209 CrossRef CAS; (d) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS; (e) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839 CrossRef CAS PubMed.
- (a) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed; (b) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- (a) J. A. Schachner, P. Traar, C. Sala, M. Melcher, B. N. Harum, A. F. Sax, M. Volpe, F. Belaj and Z. N. C. Mosch, Inorg. Chem., 2012, 51, 7642 CrossRef CAS PubMed; (b) Y. L. Wong, L. H. Tong, J. R. Dilworth, D. K. P. Ng and H. K. Lee, Dalton Trans., 2010, 39, 4602 RSC; (c) C. Zhang, G. Rheinwald, V. Lozan, B. Wu, P. G. Lassahn, H. Lang and C. Janiak, Z. Anorg. Allg. Chem., 2002, 628, 1259 CrossRef CAS; (d) A. M. Santos, F. E. Kühn, K. B. Jensen, I. Lucas, C. C. Romão and E. Herdtweck, J. Chem. Soc., Dalton Trans., 2001, 1332 RSC.
- (a) S. Kundu, S. Maity, T. Weyhermüller and P. Ghosh, Inorg. Chem., 2013, 52, 7417 CrossRef CAS PubMed; (b) A. Saha Roy, P. Saha, N. Das Adhikary and P. Ghosh, Inorg. Chem., 2011, 50, 2488 CrossRef CAS PubMed and references therein.
- A. Lúci, R. Silva, M. O. Santiago, I. C. N. Diógenes, S. O. Pinheiro, E. E. Castellano, J. Ellena, A. A. Batista, F. B. Do-Nascimento and I. S. Moreira, Inorg. Chem. Commun., 2005, 8, 1154 CrossRef PubMed.
- R. A. Eikey and M. M. Abu-Omar, Coord. Chem. Rev., 2003, 243, 83 CrossRef CAS.
- (a) V. Stavila, A. Gulea, S. Shova, Y. A. Simonov, P. Petrenko, J. Lipkowski, F. Riblet and L. Helm, Inorg. Chim. Acta, 2004, 357, 2060 CrossRef CAS PubMed; (b) Y. Yanase, H. Yoshimura, S. Kinoshita, T. Yamaguchi and H. Wakita, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 36 CrossRef; (c) L. P. Battaglia, A. M. Corradi, C. G. Palmieri, M. Nardelli and M. E. V. Tani, Acta Crystallogr., Sect.B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1114 CrossRef; (d) A. A. Khandar, B. Shaabani, F. Belaj and A. Bakhtiari, Polyhedron, 2006, 25, 1893 CrossRef CAS PubMed.
- X. Hu and K. Meyer, J. Am. Chem. Soc., 2004, 126, 16322 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis/NIR absorption spectra (Fig. S1), cyclic voltammogram of 3 (Fig. S2), X-band EPR spectra of 3 and 4 (Fig. S3), fluorescence spectra of 5 and 6 (Fig. S4), photoactive molecular orbitals (Scheme S1), schematic diagram of the ligand fragmentation considered in MO analyses (Fig. S5), calculated bond lengths of 3, 3+ and 6 (Table S1), TD DFT calculations (Table S2), population analyses of selected molecular orbitals of 6, 3, 3+ (Table S3) and optimized coordinates (Table S4–S6). CCDC 842402, 972492 and 972493. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qi00103b |
| This journal is © the Partner Organisations 2014 |