Experimental and theoretical investigation of the magnetic and photoconductive nature of a novel two-dimensional, mixed-valence bis(2-thioxo-1,3-dithiole-4,5-dithiolato)nickelate molecular solid†
Hao
Yang
a,
Jian-Lan
Liu
a,
Lan-Cheng
Zhou
a and
Xiao-Ming
Ren
*abc
aState Key Laboratory of Materials-Oriented Chemical Engineering and College of Science, Nanjing University of Technology, Nanjing 210009, People's Republic of China
bCollege of Materials Science and Engineering, Nanjing University of Technology, Nanjing 210009, People's Republic of China
cState Key Laboratory & Coordination Chemistry Institute, Nanjing University, Nanjing 210093, People's Republic of China. E-mail: xmren@njtech.edu.cn; Fax: +86 25 58139481; Tel: +86 25 58139476
First published on 1st May 2014
Abstract
A two-dimensional, mixed-valence [Ni(dmit)2] compound, [C8-Apy]2[Ni(dmit)2]3 (1) (where dmit2− = 2-thioxo-1,3-dithiole-4,5-dithiolate and Apy+ = 4-amino-1-alkylpyridinium), was prepared and its structure was fully characterized. The molecular solid displays a layered structure along the crystallographic c-axis with a [Ni(dmit)2] layer constructed from alternating straight chains of [Ni(dmit)2]0 and two-legged ladder chains of [Ni(dmit)2]1−. Compound 1 shows an antiferromagnetic, spin-dimer with pairwise interaction magnetic behavior and a semiconducting nature in addition to a rapid, clear and stable response of photoconductivity under UV irradiation. The magnetic coupling nature of 1 was analyzed using the ‘broken-symmetry’ density functional theory (DFT) approach. The electronic band structure was calculated using the CASTEP program and was used to investigate the theoretical transport and photoconductive behavior of 1. This work suggests that mixed-valence [Ni(dmit)2] compounds have great potential as the building blocks for new photoconductor based materials.
Introduction
The metal salt [M(dmit)2] (dmit2− = 2-thioxo-1,3-dithiole-4,5-dithiolate) displays an interesting molecular architecture, with its ten peripheral S atoms enabling a multidimensional electrical network via intermolecular S⋯S bonding interactions in the solid state.1 The electronic structure and physical properties of these metal salts depend on the molecular arrangement of [M(dmit)2] in the solid state, which is also related to the shape and size of the metal counter ions present. Therefore, considerable research effort towards the development of [M(dmit)2] compounds with unique electronic structures and physical properties by rational design of the counter ion structure has been reported.1–5 Nakamura and co-workers2 reported the incorporation of supramolecular M–crown ether cation (where M = alkaline metal ion, alkaline earth metal ion or organic ammonium ion) into the [Ni(dmit)2] system to give a molecular metal compound with Li+ ion conducting channels and a supramolecular rotator that achieved ferroelectricity and polarity control via a switching flip-flop motion of the rotator unit using an external electric field. Kusamoto and co-workers3 reported the novel bilayer Mott salt, (ethyl-4-bromothiazolium)[Ni(dmit)2]2, in which the coexistence of antiferromagnetic (AFM) and ferromagnetic (FM) anionic layers displayed an unusual ferromagnetic anomaly and large negative magnetoresistance. Fourmigué and co-workers4 prepared the first chiral, mixed-valence conducting [Ni(dmit)2] salt incorporating a chiral counter ion with an unusual 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 stoichiometry between the cationic counter ion and [Ni(dmit)2] units.
:5 stoichiometry between the cationic counter ion and [Ni(dmit)2] units.
The conductivity of the [Ni(dmit)2] metal salts has been extensively studied and shows a diverse range of behaviors from the use as Mott insulators,6 semi-conductors7 to metal-like conductors.8 However, investigation into the photoconductivity of [Ni(dmit)2] metal salts is limited.9 There has been an ever-growing use of photodetectors in a wide range of everyday and high-end applications, for example, the use of UV–visible light photoconductors in environmentally-based detectors, space research and optical communications.10
Our research has involved the incorporation of flexible cations, such as 4-amino-1-alkylpyridinium (Cn-Apy+), into the [Ni(dmit)2] metal salt system in an effort to control the stacking pattern and physical properties. We now report the synthesis of a novel, two-dimensional mixed-valence [Ni(dmit)2] metal salt, [C8-Apy]2[Ni(dmit)2]3 (1), which displays an unusual 2:3 stoichiometry between C8-Apy+ and [Ni(dmit)2], in addition to a rapid, clear and stable response of photoconductivity under UV irradiation. The crystal structure, magnetism, conductivity and photoconductivity of 1 were also investigated.
Experimental
Materials
All chemicals and reagents were purchased from commercial sources and used with no further purification. 4,5-Di(thiobenzoyl)-1,3-dithiole-2-thione was prepared according to the procedure reported by Steimecke et al.11 [C8-Apy]Br was prepared using the procedure reported for the synthesis of [C6-Apy]Br.12Synthesis of [C8-Apy]2[Ni(dmit)2]3 (1)
A suspension of 4,5-di(thiobenzoyl)-1,3-dithiole-2-thione (812 mg, 2.0 mmol) in methanol (10 mL) was added to a solution of sodium methoxide [prepared from sodium (184 mg, 8.0 mmol) in methanol (10 mL)] at room temperature under an atmosphere of argon. The reaction mixture was stirred at room temperature for 30 min, resulting in a dark red solution. NiCl2·6H2O (238 mg, 1.0 mmol), a solution of [C8-Apy]Br (287 mg, 1.0 mmol) in methanol (20 mL) followed by a solution of I2 (127 mg, 0.5 mmol) and NaI (150 mg, 1.0 mmol) in methanol (20 mL) were added to the reaction mixture, and the resulting mixture was stirred at room temperature for 2 h and then allowed to stand overnight (∼16 h). The microcrystalline crude product was collected by filtration, washed with MeOH (20 mL) and dried in vacuo. The crude product was recrystallized from acetone to give the title compound 1 as dark green crystals with ca. 80% yield. Anal. Calc. for C44H46N4S30Ni3: C, 29.88; H, 2.62; N, 3.17%. Found: C, 29.85; H, 2.77; N, 3.01%. IR (KBr disc,) ν = 1367, 1323 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1068, 1054 (νCS), 514, 499 (δS–C–S) cm−1. It was noted that the oxidation of ostensibly 1 mmol of [Ni(dmit)2]2− into [Ni(dmit)2]2/3− requires theoretically 1.33 “electron-equivalents” of the oxidant. Even accounting for the “only” 80% yield, this would have required >0.5 mmol of I2; however, only 0.5 mmol of I2 (i.e., 1 mmol of oxidant “electron-equivalents”) was used in the preparation procedure. Such a stoichiometric discrepancy is due to the overoxidation by air.
C), 1068, 1054 (νCS), 514, 499 (δS–C–S) cm−1. It was noted that the oxidation of ostensibly 1 mmol of [Ni(dmit)2]2− into [Ni(dmit)2]2/3− requires theoretically 1.33 “electron-equivalents” of the oxidant. Even accounting for the “only” 80% yield, this would have required >0.5 mmol of I2; however, only 0.5 mmol of I2 (i.e., 1 mmol of oxidant “electron-equivalents”) was used in the preparation procedure. Such a stoichiometric discrepancy is due to the overoxidation by air.
Physical measurements
Elemental analyses (C, H and N) were performed using a Vario EL III elemental analyzer. Powder X-ray diffraction (PXRD) data were collected using a Bruker D8 Advance powder diffractometer with Cu Kα radiation (λ = 1.5418 Å). FT-IR spectra were recorded using an IF66 V FT-IR (4000–400 cm−1) spectrophotometer on KBr discs. Magnetic susceptibility data were measured for polycrystalline samples using a Quantum Design MPMS-5S superconducting quantum interference device (SQUID) magnetometer over a temperature range of 1.8–400 K. Electrical conductivity measurements were made using a Keithley 2440 5A SourceMeter for the compressed powder pellet using a two-probe method. Photoconductivity was measured for a single crystal using a Zahner Zennium IM6 Electrochemical Workstation. The photocurrent I response to UV irradiation (λ = 375 nm; 3.3 eV) was measured using a needle-shaped single crystal at room temperature and two gold wires with a diameter of 80 μm attached on the two opposite surfaces of a cross section of the needle crystal.X-Ray single crystallography
Single crystal X-ray diffraction data were collected using a Bruker Smart Apex II CCD detector with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 296 K using the ω-scan technique. Data were integrated using the SAINT program and subsequently used for the intensity corrections of the Lorentz and polarization effects. An empirical absorption correction was applied using the SADABS program.13 The structure was solved by direct methods and all non-hydrogen atoms were anisotropically refined on F2 by the full-matrix least-squares technique using the SHELXL-97 crystallographic software package.14 All hydrogen atoms were placed at the calculated positions and refined as riding on the parent atoms. The experimental details for data collection, structure refinement and crystallography are summarized in Table 1.| a R 1 = ∑||Fo| − |Fc||/|Fo| and wR2 = [∑w(∑Fo2 − Fc2)2/∑w(Fo2)2]1/2. | |
|---|---|
| Temp. (K) | 296(2) |
| Wavelength (Å) | 0.71073 |
| Formula | C44H46N4S30Ni3 |
| Formula weight | 1769.02 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| CCDC no. | 956073 |
| Crystal system | Triclinic |
| a (Å) | 11.6986(12) |
| b (Å) | 12.1197(12) |
| c (Å) | 14.0622(14) |
| α (°) | 109.910(3) |
| β (°) | 105.249(3) |
| γ (°) | 103.361(3) |
| V (Å3, Z) | 1691.9(3)/1 |
| ρ (g cm−1) | 1.736 |
| μ (mm−1) | 1.790 |
| F(000) | 902 |
| θ Range for data collection (°) | 1.92–27.55 |
| Index ranges | −15 ≤ h ≤ 15 |
| −15 ≤ k ≤ 15 | |
| −18 ≤ l ≤ 18 | |
| R int | 0.0452 |
| Independent reflect./restraints/parameters | 7788/0/369 |
| Refinement method | The least-squares refinement on F2 |
| Goodness-of-fit on F2 | 1.052 |
| R 1, wR2 [I > 2σ(I)]a | 0.0423, 0.0760 |
| R 1, wR2 [all data]a | 0.0827, 0.0894 |
| Residual (e Å−3) | 0.337/−0.424 |
Density functional theory (DFT) calculations for magnetic coupling constants and electronic band structures
All DFT calculations for magnetic coupling constants were carried out using the Gaussian09 program.15 The single point energies of the high-spin triplet and the low-spin broken-symmetric states for the [Ni(dmit)2]22− spin dimer or the [Ni(dmit)2]32− sandwich trimer present in compound 1 were calculated using the DFT approach on the non-modelized molecular geometry obtained from our X-ray single crystal structure analysis (the spin coupling interaction is sensitive to any changes in molecular structure; therefore no simplification or optimization of the molecular structure was made in our calculations). The popular hybrid exchange–correlation functional B3LYP was composed of Becke's three-parameter hybrid exchange functional,16 LYP correlation functional17 and the lanl2dz relativistic effective core potential basis set18 for the Ni element and the 6-311+g(d,p) basis set for C, S elements used in our calculations. The SCF convergence criterion was 10−8.Electronic band structure calculations were performed using the CASTEP program19 for compound 1 on the non-modelized crystal structure obtained from our X-ray single crystal structure analysis. The cations in the crystal structure were removed in the calculations since the conductivity of this molecular solid is related to the anionic [Ni(dmit)2]− and neutral [Ni(dmit)2]0 species. The generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE)20 potentials has been incorporated into our calculations. The k-points sampling in the Brillouin zone were set at 4 × 4 × 3 according to the Monkhorst–Pack scheme.21 For all relevant calculations the plane wave basis set cut-off was fixed at 240 eV and the convergence criterion fixed at 1.0 × 10−5 eV per atom. The other calculation parameters were set at the default values in the CASTEP code.
Results and discussion
Crystal structure
[C8-Apy]2[Ni(dmit)2]3 (1) crystallizes in the triclinic space group P. An asymmetric unit was comprised of one C8-APy+ cation together with one and a half planar [Ni(dmit)2] units (Fig. 1a). The bond lengths and angles observed in the C8-APy+ cation are within that expected. All atoms are located in the general positions of the [Ni(dmit)2] moiety containing Ni(1), whilst the Ni(2) atoms coincide with an inversion center to give a corresponding [Ni(dmit)2] moiety with Ci symmetry. The typical bond lengths observed in the two different [Ni(dmit)2] moieties are summarized in Table S1.† The total charge of one and a half [Ni(dmit)2] moieties is −1 because the C8-APy+ cation has a +1 charge. Therefore, the average oxidation state of the [Ni(dmit)2] moieties in the asymmetric unit is −2/3. The theoretical analysis based on DFT calculations at the b3lyp/lanl2dz level of theory predicted that the lowest unoccupied molecular orbital (LUMO) of the neutral complex [Ni(dmit)2]0 consists of the 3d orbitals of Ni and the π(CC) orbital of the ligand (Fig. 1b). In the anionic complex [Ni(dmit)2]1−, the LUMO is bonding with respect to the π(CC) bond and antibonding with respect to the σ(C–S) and σ(Ni–S) bonds. The addition of one electron to the LUMO was expected to strengthen the π(CC) bond and weaken the σ(C–S) and σ(Ni–S) bonds. As a result, the bond distance of the π(CC) bond is reduced and the bond distances of both σ(C–S) and σ(Ni–S) bonds are increased with increasing the n value in the series [Ni(dmit)2]n− (n = 0, 1 and 2). Upon careful analysis of the π(CC), σ(C–S) and σ(Ni–S) bond distances in compound 1, we can conclude that the [Ni(dmit)2] moiety containing Ni(2) is a neutral species and the [Ni(dmit)2] moiety containing Ni(1) has a −1 charge (Table S1†). The assignment for Ni(1) and Ni(2) in compound 1 was further confirmed by (1) IR spectra analysis (Table S2 and Fig. S2† where the vibration bands of νCC, νCS and νC–S contributed from both [Ni(dmit)2]1− and [Ni(dmit)2]0 species are observed, respectively) and (2) the energy band structure calculation (see Fig. S4,† where two band structures with different situations, namely, presence/absence of a neutral [Ni(dmit)2] molecule (with Ni(2)), are shown, and the comparison of the two band structures provided information about the inhomogeneity of the valence state between the three molecules in a trimer).
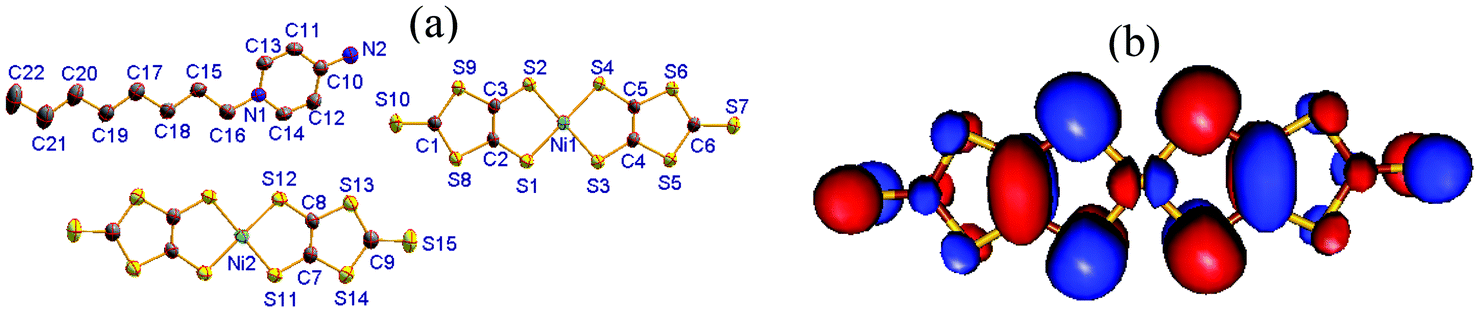 | ||
| Fig. 1 (a) Molecular structure of compound 1 with all non-hydrogen atoms labeled. (b) LUMO of neutral [Ni(dmit)2]0 obtained from DFT calculations at the B3LYP/lanl2dz level of theory. | ||
The C8-APy+ cation and the mixed-valence [Ni(dmit)2] species form alternating layers along the crystallographic c-axis direction as shown in Fig. 2a. The neighboring cations adopt a parallel arrangement along the b-axis direction and an anti-parallel alignment along the a-axis direction (Fig. 2b) within the cationic layer. The anionic layer is constructed from the π-type anion trimers adopting an alignment in the [Ni(dmit)2]1−/[Ni(dmit)2]0/[Ni(dmit)2]1− sandwich in which two anions are related to each other through an inversion center (Fig. 2c). The [Ni(dmit)2]− anions located between neighboring trimers are arranged in a two-legged ladder chain via shorter lateral-to-lateral S⋯S bonding interactions along the crystallographic a- and b-axes (Fig. 2d). The neutral [Ni(dmit)2]0 species between adjacent trimers form a one-dimensional straight chain through head-to-tail S⋯S bonding interactions along the crystallographic b-axis direction within the mixed-valence [Ni(dmit)2] molecule layer (Fig. 2e). The short interatomic distances are less than the sum of van der Waals radii of two S atoms (3.7 Å) and are listed below: dS(4)⋯S(9)#1 = 3.619(1), dS(2)⋯S(2)#1 = 3.480(1), dS(6)⋯S(8)#2 = 3.461(1) and dS(6)⋯S(10)#2 = 3.450(1) Å within a ladder chain; dS(13)⋯S(13)#3 = 3.469(1) Å within a [Ni(dmit)2]0 chain and dNi(2)⋯S(1)#4 = 3.561(1) and dNi(1)#4⋯S(12) = 3.588(1) Å between the longitudinal offset [Ni(dmit)2]0 and [Ni(dmit)2]1− species (symmetric codes #1 = 1 − x, −y, 1 − z; #2 = x, −1 + y, z; #3 = −x, 1 − y, 1 − z and #4 = x, 1 + y, z).
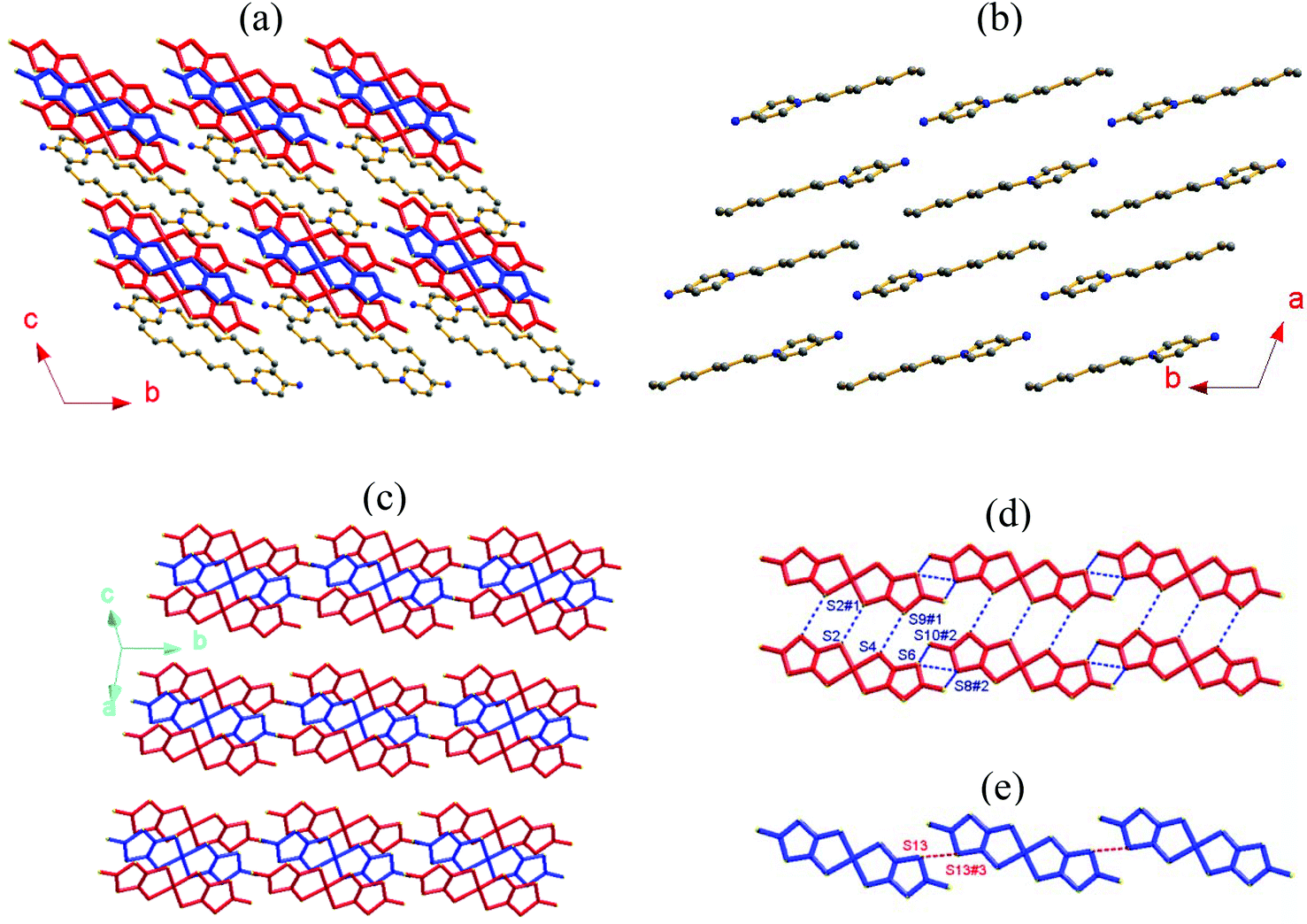 | ||
| Fig. 2 (a) Alternating layers of C8-APy+ cations (H atoms are omitted for clarity) and mixed-valence [Ni(dmit)2] species (where blue = neutral species and red = anionic species) viewed along the crystallographic a-axis in compound 1; (b) the cationic layer; (c) the [Ni(dmit)2] species layer; (d) the two-legged [Ni(dmit)2]1− ladder chain and (e) the one-dimensional [Ni(dmit)2]0 chain (where the dotted lines represent S⋯S bonding interactions <3.7 Å). | ||
Relationship of magnetic properties and structure
The magnetic susceptibility (χm) of compound 1 as a function of temperature (in a range of 1.8–400 K) is shown in Fig. 3. A broad maximum centered at ∼120 K, followed by an exponential drop, is observed in the χm − T curve upon cooling, indicating the existence of a gap in the spin excitation spectrum, and is characteristic of a low-dimensional AFM exchange system. In addition, the Curie–Weiss type magnetic behavior dominates in the low temperature region (<20 K).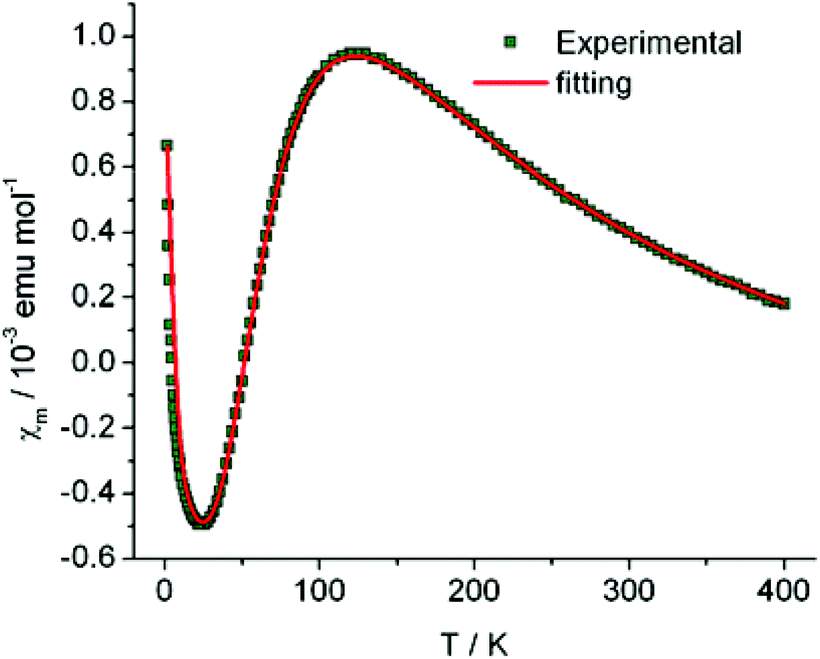 | ||
| Fig. 3 Temperature dependence of the molar magnetic susceptibility (χm) with one [Ni(dmit)2]1− species per formula unit in compound 1 (where solid line = fit plot and open circles = experimental data). | ||
To understand the magnetic coupling between neighboring [Ni(dmit)2]− anions in compound 1, a broken symmetry DFT approach22 was employed to evaluate the magnetic coupling constants.
The magnetic coupling between neighboring [Ni(dmit)2]− anions can be transmitted through non-bonding interactions, especially through S atoms, due to the delocalization of the unpaired electron over the whole skeleton of the [Ni(dmit)2]− anion. Using the X-ray crystal structure analysis (Fig. 2c and 2d), three types of magnetic couplings were identified, one within the trimer with the magnetic coupling constant J1 and the other two within the two-legged ladder chain, with the magnetic coupling constant J2 corresponding to the ladder rung and J3 the ladder rail direction.
The <S2>HS, <S2>BS and J values for compound 1 were calculated using eqn (S2)–(S4) (see the ESI†) and are summarized in Table 2. These results show that: (1) a strong AFM interaction exists within a trimer, a weak AFM interaction along the rail direction and negligible magnetic coupling along the rung direction within the two-legged ladder chains is observed. This indicates that the magnetic behavior of compound 1 is dominated by the magnetic coupling within the trimer. (2) The calculated <S2>HS values in the three types of magnetic couplings are close to the angular momentum eigenvalue found in the high-spin triplet state, ST(ST + 1) = 1(1 + 1) = 2, whilst the calculated <S2>BS values for J1 (1.1294), J2 (1.0096) and J3 (1.0091) show relatively high spin contamination. (3) The calculated magnetic coupling constants, using eqn (S2) and (S4) (see the ESI†), are similar to each other, demonstrating the existence of ‘weak bonding’ within the spin dimer. The frontier molecular orbitals of the BS states of J1 and J3 indicate the existence of a partial overlap between the two SOMOs within the sandwich trimer and spin localized in an individual [Ni(dmit)2]− anion for the magnetic exchange pathway of J3 (Fig. 4). In combination with the spin density distribution analysis, we can conclude that the magnetic couplings between the neighboring [Ni(dmit)2]− anions are achieved through orbital overlap together with a weak spin polarization mechanism within the sandwich trimer. In addition, only a spin polarization mechanism is observed for the ladder chain along the rail direction in compound 1.
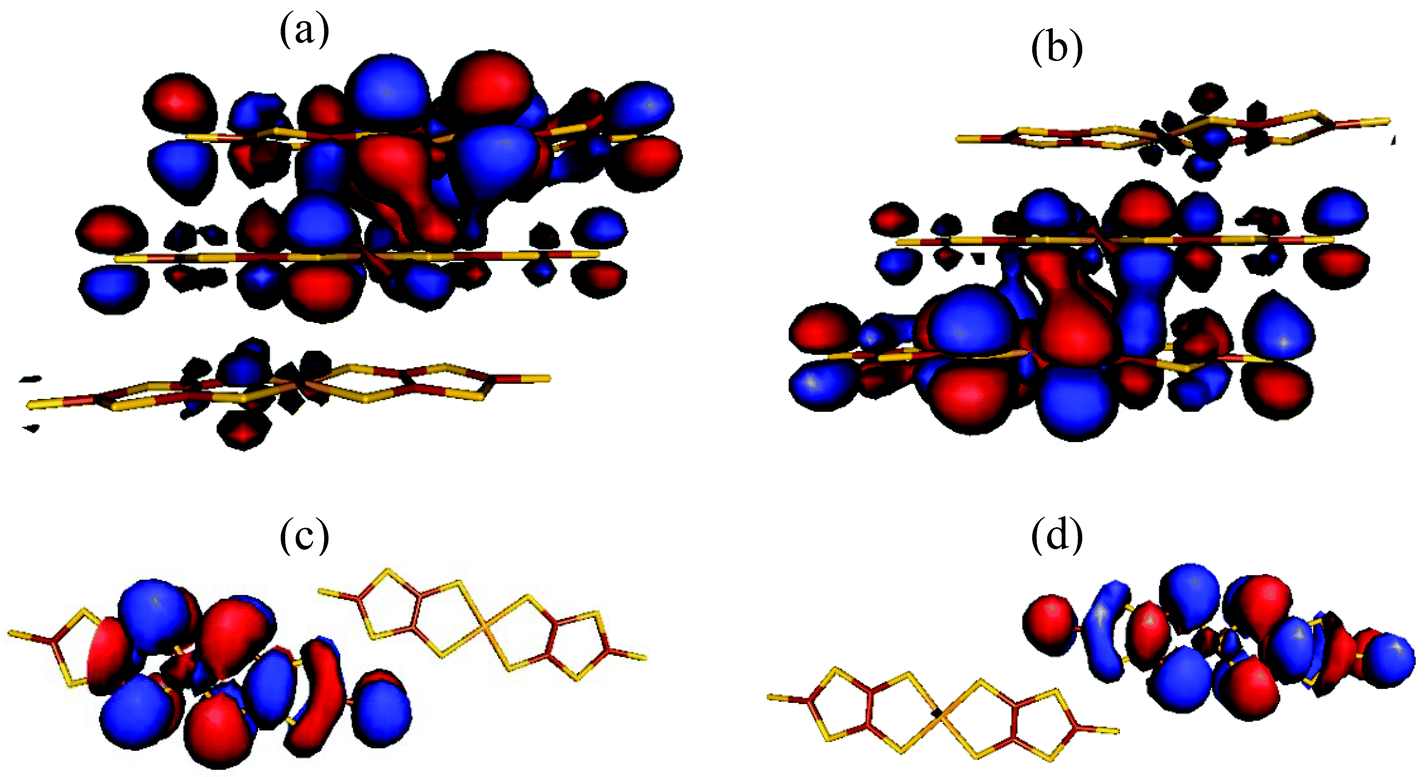 | ||
| Fig. 4 SOMOs in the BS states for J1 (a, b) and J3 (c, d), respectively. | ||
| Coupling pathway | J 1/kB (K) | J 2/kB (K) | J 3/kB (K) | <S2>HS | <S2>BS |
|---|---|---|---|---|---|
| a J 1/kB, J2/kB and J3/kB were obtained using eqn (S2)–(S4) (see the ESI). | |||||
| J 1 (within a trimer) | −96.8 | −48.4 | −109.5 | 2.0134 | 1.1294 |
| J 2 (along ladder rung) | 0.098 | 0.049 | 0.098 | 2.0097 | 1.0096 |
| J 3 (along ladder rail) | −1.8 | −0.9 | −1.8 | 2.0095 | 1.0091 |
On the basis of the analysis described beforehand, a simple S = 1/2 spin dimer model was chosen to fit the magnetic susceptibility over the whole temperature range. In addition, to take into account the Curie–Weiss and temperature independent magnetic susceptibilities, the experimental molar magnetic susceptibility is expressed in eqn (1).
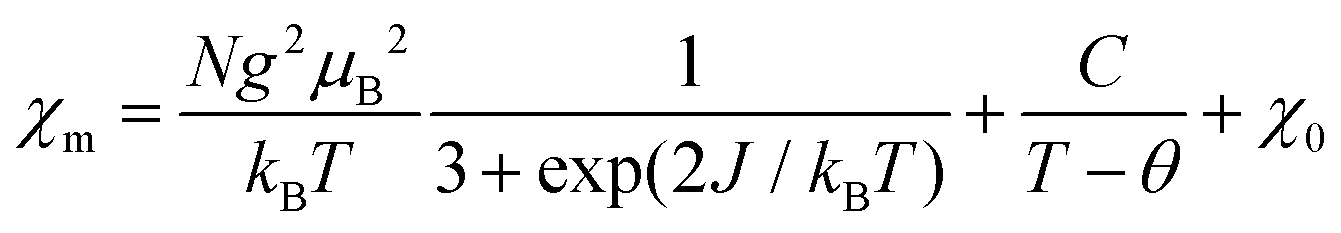 | (1) |
In eqn (1), the first term represents the magnetic susceptibility contribution from a spin dimer with two S = 1/2 monomers, deduced from spin Hamiltonian H = −2JS1S2, and J is the intradimer magnetic coupling constant. The C/(T − θ) term arises from the magnetic impurity and χ0 is the sum of diamagnetism and possible van Vleck paramagnetism related to the coupling of ground and excited states via a magnetic field.23 The χm – T plot in the temperature range 1.8–400 K (Fig. 3) was reproduced using eqn (1) with the parameters: J/kB = −99.7(0) K (corresponding to Δ/kB = 199.4 K), χ0 = −6.8(2) × 10−4 emu mol−1, C = 4.03(3) × 10−3 emu K mol−1 and θ = −1.19(3) K with a g-factor = 2.045(1). The fitted J value is close to the calculated J1 value. In addition, on the basis of the fitted Curie constant, the amount of S = 1/2 magnetic impurities was estimated to be ∼1.1%.
Conductivity and photoconductivity
Compound 1 displays semi-conductor character, the σ = 1.5 × 10−6 S cm−1 at 303 K (30 °C) as shown in Fig. 5a. The activation energy was estimated to be 0.33 eV by fitting the variable temperature conductivity in the temperature range of 300–420 K using the Arrhenius equation (eqn (2)).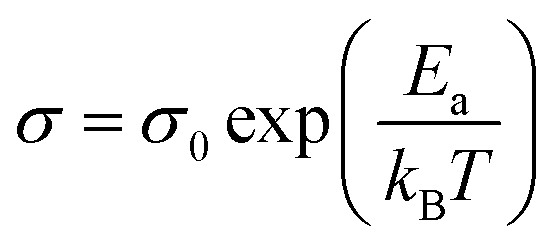 | (2) |
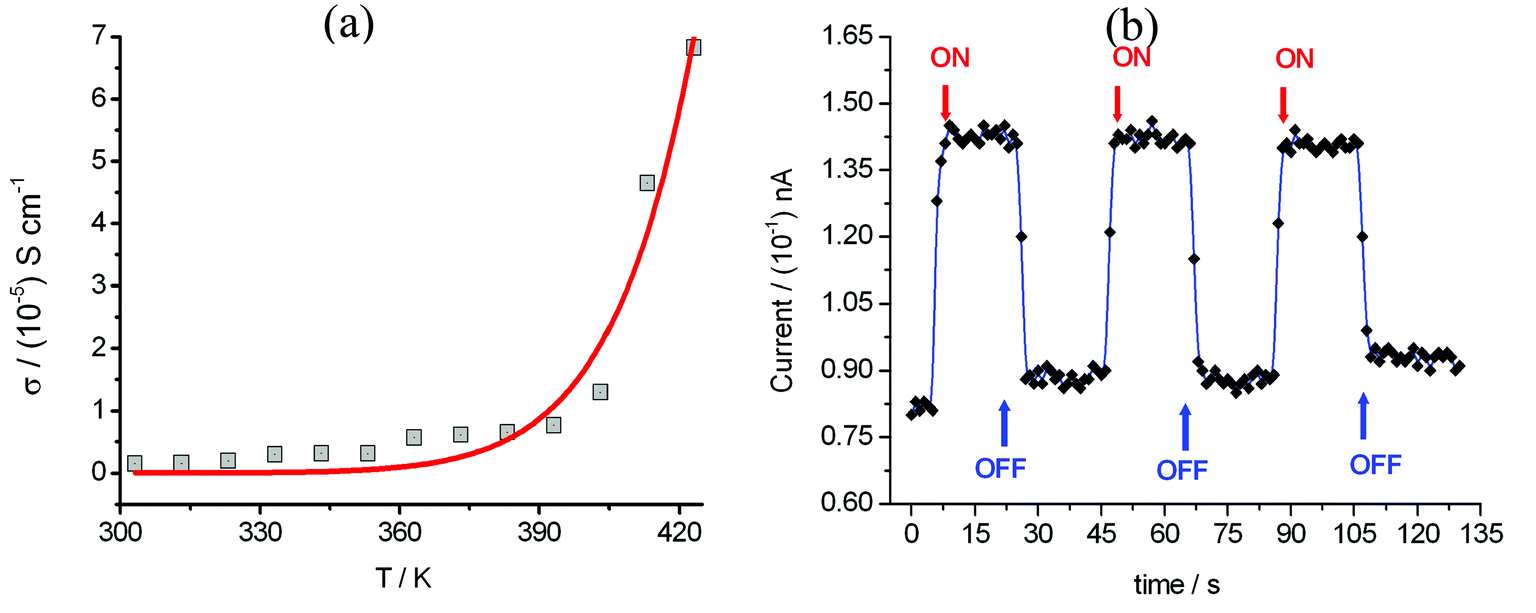 | ||
| Fig. 5 (a) Plot of electrical conductivity (σ) of compound 1versus temperature from 303–423 K (red line = theoretically produced plot using eqn (2)). (b) The photocurrent I response to UV irradiation (λUV = 375 nm; 3.3 eV) was measured using a needle-shaped single crystal at 300 K with the applied voltage = 0.1 V and light intensity = 45 mW cm−2, and two gold wires with a diameter of 80 μm were attached on the two opposite surfaces of a cross section of the needle crystal. | ||
The photoconductivity for compound 1 was investigated and the photocurrent was monitored as a function of time with an alternating ON and OFF UV irradiation (Fig. 5b). After a few seconds of irradiation, the current increases sharply, to reach a saturation value. The relaxation back to the original dark current was observed after switching off the illumination.
For photoconductivity, several different mechanisms have been reported to occur in [M(dmit)2] (M = Ni or Pd) molecular solids. Ishikawa and co-workers24 reported the compounds Et2Me2Sb[Pd(dmit)2]2 and Cs[Pd(dmit)2]2 that display an ultrafast photoconductive response. These molecular solids possess two different crystalline phases with distinct conductivity, and their photoinduced phase transition leads to the ultrafast photoconductive response. Naito and co-workers9 reported a novel charge-transfer (CT) photoconductor, BPY[Ni(dmit)2]2 (where: BPY = N,N′-ethylene-2,2′-bipyridinium), in which the molecular orbitals of BPY2+ and [Ni(dmit)2]− mix to form energy bands. The interband transitions occur in the UV spectroscopic region corresponding to the CT bands between BPY2+ and [Ni(dmit)2]−, generating new carriers of holes and electrons and is responsive to the photoconductivity of BPY[Ni(dmit)2]2. On the basis of the variable temperature magnetic susceptibility measurements (Fig. 3) and the DSC analysis over the temperature range of 293–473 K, no phase transition was observed for compound 1. Therefore, a photoinduced phase transition mechanism for photoconductivity can be excluded for compound 1.
The energy band structures for the two-dimensional, mixed valence layer in compound 1 were analyzed and the calculated bands along the high symmetry directions of the Brillouin zone are shown in Fig. 6. The valence band (VB) was observed near the Fermi level and a small dispersion along the c-axis (approximately from G to Z in the k-space) indicative of weak bonding interactions between the mixed valence layers and the two-dimensional electronic nature of this molecular solid. These results are in agreement with the X-ray crystal structure analysis in which neighboring mixed valence layers are separated by a cationic layer. The VB is relatively sharp along the direction from G to Y in the k-space, indicating the existence of stronger bonding interactions in the mixed valence layer. The calculated energy gap between the highest occupied and the lowest unoccupied bands is 0.519 eV, a value almost double that obtained from the conductivity measurements and attributed to the limitations of DFT methods.25 The orbitals of the [Ni(dmit)2]− and [Ni(dmit)2]0 species mix to form energy bands in compound 1 and the photoconductivity of 1 arising due to interband transitions in the UV spectroscopic region corresponds to the CT bands between the [Ni(dmit)2]− and [Ni(dmit)2]0 species. However, further investigation is needed to understand the novel photoconductivity of compound 1 (Fig. 6a).
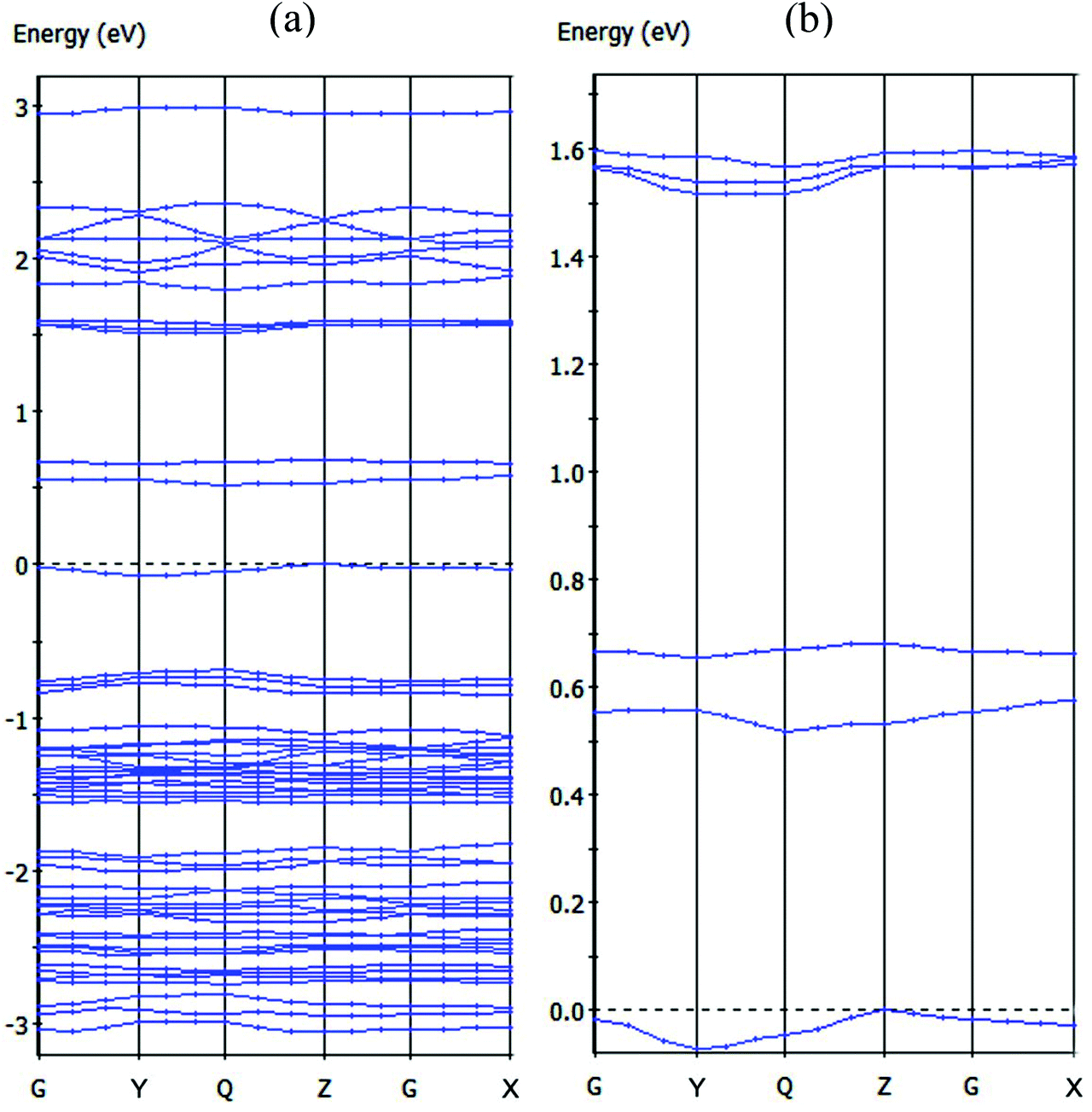 | ||
| Fig. 6 (a) Dispersion relations of the mixed valence layer in compound 1 [where Fermi levels are shown by dashed lines and the k-points: G = (0, 0, 0), X = (0.5, 0, 0), Y = (0, 0.5, 0), Z = (0, 0, 0.5) and Q = (0, 0.5, 0.5)]. (b) Magnified view of the highest occupied band and several lowest unoccupied bands. | ||
Conclusion
In summary, a novel, two-dimensional, mixed-valence [Ni(dmit)2] compound, [C8-Apy]2[Ni(dmit)2]3 (1), has been prepared possessing an unusual 2:3 stoichiometry between the monocation C8-Apy+ and [Ni(dmit)2] species. The [Ni(dmit)2] layer is constructed from alternating straight chains of [Ni(dmit)2]0 and two-legged ladder chains of [Ni(dmit)2]1−. Compound 1 displays semi-conductor character and AFM dimer magnetic behavior in addition to a rapid, clear and stable response of photoconductivity under UV irradiation. The magnetic coupling nature was analyzed using the ‘broken-symmetry’ DFT approach and the novel photoconductivity attributed to the electronic transitions between the [Ni(dmit)2]1− and [Ni(dmit)2]0 species in 1. This work suggests that mixed-valence [Ni(dmit)2] compounds have great potential as the building blocks for new photoconductor based materials.
Acknowledgements
The authors would like to thank the Priority Academic Program Development of Jiangsu Higher Education Institutions and the National Nature Science Foundation of China (grant no. 91122011, 21071080 and 21271103) for financial support.Notes and references
- R. Kato, Chem. Rev., 2004, 104, 5319 CrossRef CAS PubMed; K. Kanoda and R. Kato, Annu. Rev. Condens. Matter Phys., 2011, 2, 167 CrossRef PubMed; T. Kusamoto, H. M. Yamamoto, N. Tajima, Y. Oshima, S. Yamashita and R. Kato, Inorg. Chem., 2012, 51, 11645 CrossRef PubMed.
- T. Nakamura, T. Akutagawa, K. Honda, A. E. Underhill, A. T. Coomber and R. H. Friend, Nature, 1998, 394, 159 CrossRef CAS PubMed; T. Akutagawa, H. Koshinaka, D. Sato, S. Takeda, S.-I. Noro, H. Takahashi, R. Kumai, Y. Tokura and T. Nakamura, Nat. Mater., 2009, 8, 342 CrossRef PubMed.
- T. Kusamoto, H. M. Yamamoto, N. Tajima, Y. Oshima, S. Yamashita and R. Kato, Inorg. Chem., 2013, 52, 4759 CrossRef CAS PubMed.
- J. Lieffrig, O. Jeannin, P. Auban-Senzier and M. Fourmigué, Inorg. Chem., 2012, 51, 7144 CrossRef CAS PubMed.
- Y. K. Han, D. K. Seo, H. Kang, W. Kang and D. Y. Noh, Inorg. Chem., 2004, 43, 7294 CrossRef CAS PubMed; D. Y. Noh, A. E. Underhill and M. B. Hursthouse, Synth. Met., 2001, 120, 1053 CrossRef; D. Y. Noh, H. J. Lee, H. Kang, W. Kang and U. Lee, Mol. Cryst. Liq. Cryst., 2002, 376, 269 CrossRef.
- P. Cassoux, L. Valade, H. Kobayashi, A. Kobayashi, R. A. Clark and A. E. Underhill, Coord. Chem. Rev., 1991, 110, 115 CrossRef CAS.
- A. E. Pullen and R.-K. Olk, Coord. Chem. Rev., 1999, 188, 211 CrossRef CAS.
- M. Bousseau, L. Valade, J. Legros, P. Cassoux, M. Garbauskas and L. V. Interrante, J. Am. Chem. Soc., 1986, 108, 1908 CrossRef CAS.
- T. Naito, T. Karasudani, S. Mori, K. Ohara, K. Konishi, T. Takano, Y. Takahashi, T. Inabe, S. Nishihara and K. Inoue, J. Am. Chem. Soc., 2012, 134, 18656 CrossRef CAS PubMed.
- Q. H. Li, T. Gao, Y. G. Wang and T. H. Wang, Appl. Phys. Lett., 2005, 86, 123117 CrossRef PubMed; T. Y. Wei, C. T. Huang, B. Hansen, Y. F. Lin, L. J. Chen, S. Y. Lu and Z. L. Wang, Appl. Phys. Lett., 2010, 96, 013508 CrossRef PubMed.
- G. Steimecke, H.-J. Sieler, R. Kirmse and E. Hoyer, Phosphorus, Sulfur Silicon Relat. Elem., 1979, 7, 49 CrossRef CAS.
- H. B. Duan, X. M. Ren, L. J. Shen, W. Q. Jin, Q. J. Meng, Z. F. Tian and S. M. Zhou, Dalton Trans., 2011, 40, 3622 RSC.
- G. M. Sheldrick, SADABS, Program for Bruker Area Detector Absorption Correction, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- W. Y. C. Lee and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef.
- T. H. Dunning Jr. and P. J. Hay, Modern Theoretical Chemistry, Plenum, New York, 1976, pp. 1–28 CrossRef CAS PubMed; P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS PubMed; W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef PubMed; P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef PubMed.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS; V. Milman, B. Winkler, J. A. White, C. J. Pickard, M. C. Payne, E. V. Akhmatskaya and R. H. Nobes, Int. J. Quantum Chem., 2000, 77, 895 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- L. Noodleman and J. G. Norman Jr., J. Chem. Phys., 1979, 70, 4903 CrossRef CAS PubMed.
- J. H. Van Vleck, The theory of electric and magnetic susceptibilities, Oxford University Press, Oxford, U.K., 1932 Search PubMed.
- T. Ishikawa, N. Fukazawa, Y. Matsubara, R. Nakajima, K. Onda, Y. Okimoto, S. Koshihara, M. Lorenc, E. Collet, M. Tamura and R. Kato, Phys. Rev. B: Condens. Matter, 2009, 80, 115108 CrossRef CAS; N. Fukazawa, T. Tanaka, T. Ishikawa, Y. Okimoto, S. Koshihara, T. Yamamoto, M. Tamura, R. Kato and K. Onda, J. Phys. Chem. C, 2013, 117, 13187 Search PubMed.
- R. W. Godby, M. Schluther and L. J. Sham, Phys. Rev. B, 1987, 36, 6497 CrossRef CAS; C. M. I. Okoye, J. Phys.: Condens. Matter, 2003, 15, 5945 CrossRef; R. Terki, G. Bertrand and H. Aourag, Microelectron. Eng., 2005, 81, 514 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the broken-symmetry DFT approach, average bond lengths, the characteristic vibration frequencies (cm−1) for [Ni(dmit)2]n− (n = 0, 1), calculated overlap and transfer integrals for each spin dimer in the two-legged ladder chain of 1. Figures include IR spectrum, TG, DTA and PXRD profiles. CCDC 956073. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qi00106g |
| This journal is © the Partner Organisations 2014 |