Molecular recognition: preparation and characterization of two tripodal anion receptors†
Alireza
Shokri
a,
S. H. M.
Deng
bc,
Xue-Bin
Wang
*bc and
Steven R.
Kass
*a
aDepartment of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: kass@umn.edu
bPhysical Sciences Division, Pacific Northwest National Laboratory, P. O. Box 999, MS K8-88, Richland, Washington 99352, USA. E-mail: xuebin.wang@pnnl.gov
cDepartment of Physics, Washington State University, 2710 University Drive, Richland, Washington 99354, USA
First published on 19th December 2013
Abstract
Two new tripodal hydroxyl-based anion receptors (1 and 2) are reported and their 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molecular complexes with Cl−, H2PO4−, and OAc− along with the (M − 1)− ion of 1 were characterized by negative ion photoelectron spectroscopy in the gas phase and by binding constant determinations in four solvents (i.e., CDCl3, CD2Cl2, CD3COCD3, and CD3CN). An intramolecular hydrogen bond network (HBN) in hexaol 1 was found to diminish its binding whereas the triol 2 is the strongest aliphatic hydroxyl-based receptor to date.
:1 molecular complexes with Cl−, H2PO4−, and OAc− along with the (M − 1)− ion of 1 were characterized by negative ion photoelectron spectroscopy in the gas phase and by binding constant determinations in four solvents (i.e., CDCl3, CD2Cl2, CD3COCD3, and CD3CN). An intramolecular hydrogen bond network (HBN) in hexaol 1 was found to diminish its binding whereas the triol 2 is the strongest aliphatic hydroxyl-based receptor to date.
Introduction
Molecular recognition of negative ions has attracted considerable attention in recent years because of the biological significance of the field, potential applications in sensors and the development of phase transfer reagents.1–4 For example, negative ion gradients across lipid bilayer membranes brought about by transport proteins and anion channels are critical physiological processes.5 The breakdown of this function leads to a wide range of diseases such a cystic fibrosis, nephrolithiasis, osteopetrosis, Angelman syndrome and Bartter's syndrome type III.6–8 There are no cures for most of these afflictions even though the structures of many charge carriers and anion channels are well established. Synthetic receptors can serve as models for the behavior of natural anion channels and provide a platform for probing the fundamental interactions in these species.9 Given that the selectivity of peptide hosts arises in large part due to the displacement of water by hydrogen bond donors,5a functional groups such as carbazoles, ureas, amides, sulfonamides, and alcohols have been incorporated into the structural frameworks of supramolecular hosts.10,11 Two strategies often employed to increase selectivity and the binding ability of the receptor are to make use of rigid structures and to enhance the acidities of the hydrogen bond donating groups. Aromatic rings, particularly benzenoid structures, are most commonly used for the former purpose.12 Intramolecular hydrogen bonds recently were also exploited to further freeze receptors into preorganized conformations. Gale et al. incorporated two ortho-phenols into an isophthalamide-based receptor (A) to lock in the syn–syn conformer of the amide groups (Fig. 1).13 This resulted in enhanced binding of I− by an order of magnitude. Similarly, Flood et al. showed that intramolecular hydrogen bonds in an aryl-1,2,3-triazole pentad (B) arising from the incorporation of two aromatic hydroxyl groups provides a more rigid backbone that amplifies the binding to Cl− by a factor of more than 47.14 This strategy also has been employed with a variety of different foldamers such as aromatic oligo-amides, -ureas, and -hydrazides.15–18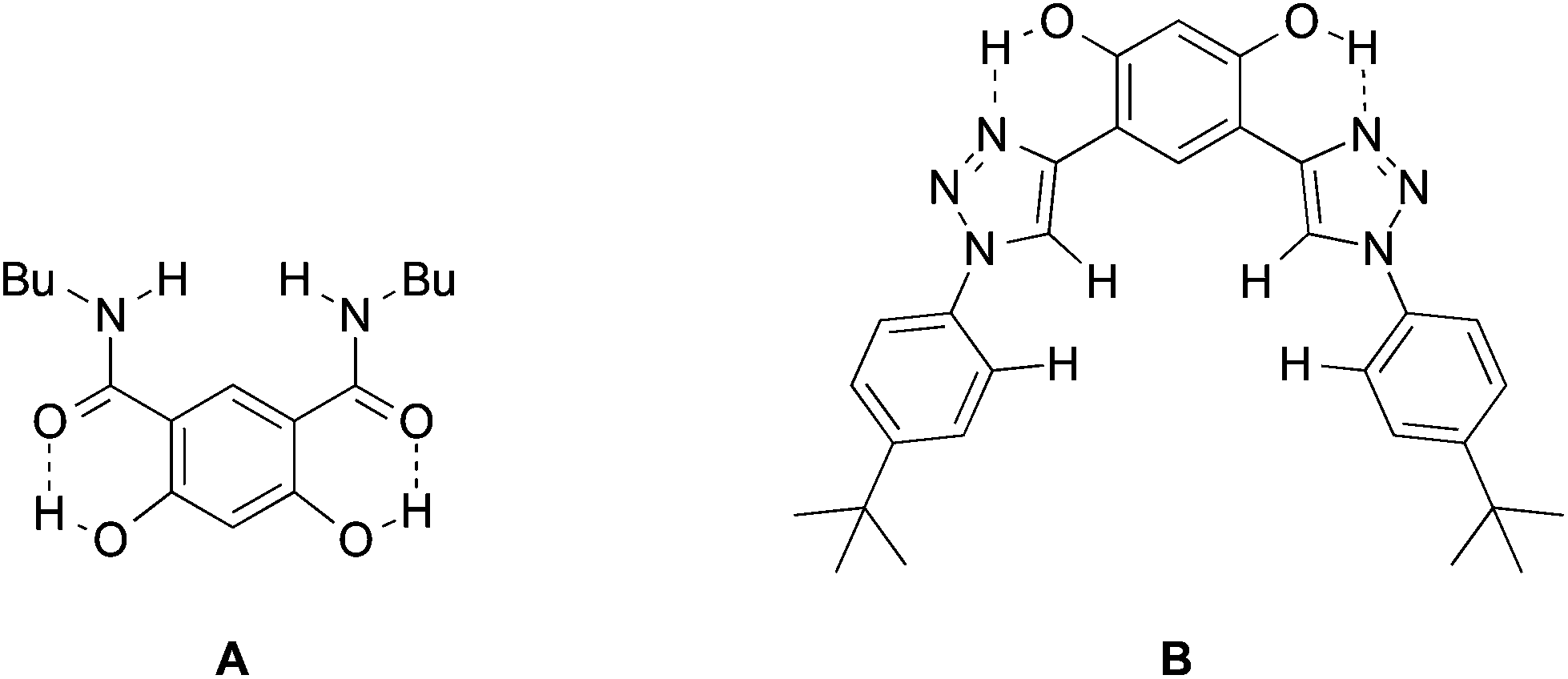 | ||
| Fig. 1 Hydrogen bond conformationally fixed receptors. | ||
In general, the anion affinity of a host can be increased by enhancing its acidity,19 but some exceptions such as ureas vs. thioureas have been reported.20 Electron withdrawing groups tend to further polarize O–H and N–H bonds leading to more favorable hydrogen bonded interactions. For example, several orders of magnitude improvement were observed when nitro groups were used to replace hydrogens on the aryl rings of N,N′-diaryl-(thio)ureas.21 We recently reported that the incorporation of CF3 substituents into a few polyols enhance their Cl− association constants by a factor of 22 for each CF3 group.22 Hydrogen bond networks were also found to stabilize negatively charged centers, and the DMSO acidities of a series of polyols revealed that this stabilization can go beyond O−⋯HO interactions (i.e., the primary hydrogen bonds).23–25 This suggests that secondary hydrogen bonds (i.e., 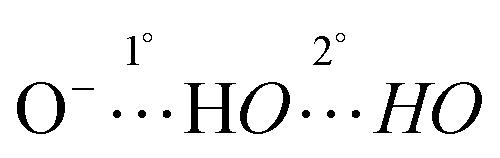 ) can be employed to enhance anion binding since they were shown to increase acidity. Herein, two new hydroxyl-based anion receptors, 1 and 2 (Fig. 2) are reported, their molecular complexes with Cl−, H2PO4−, and OAc− and the (M − 1)− anion of 1 (1a) were probed by photoelectron spectroscopy in the gas phase, binding constants with three of these anions and NO3− were measured in four different solvents, and the resulting experimental findings are supplemented with extensive computations. Taken together, these results provide new perspectives in the field of supramolecular chemistry.
) can be employed to enhance anion binding since they were shown to increase acidity. Herein, two new hydroxyl-based anion receptors, 1 and 2 (Fig. 2) are reported, their molecular complexes with Cl−, H2PO4−, and OAc− and the (M − 1)− anion of 1 (1a) were probed by photoelectron spectroscopy in the gas phase, binding constants with three of these anions and NO3− were measured in four different solvents, and the resulting experimental findings are supplemented with extensive computations. Taken together, these results provide new perspectives in the field of supramolecular chemistry.
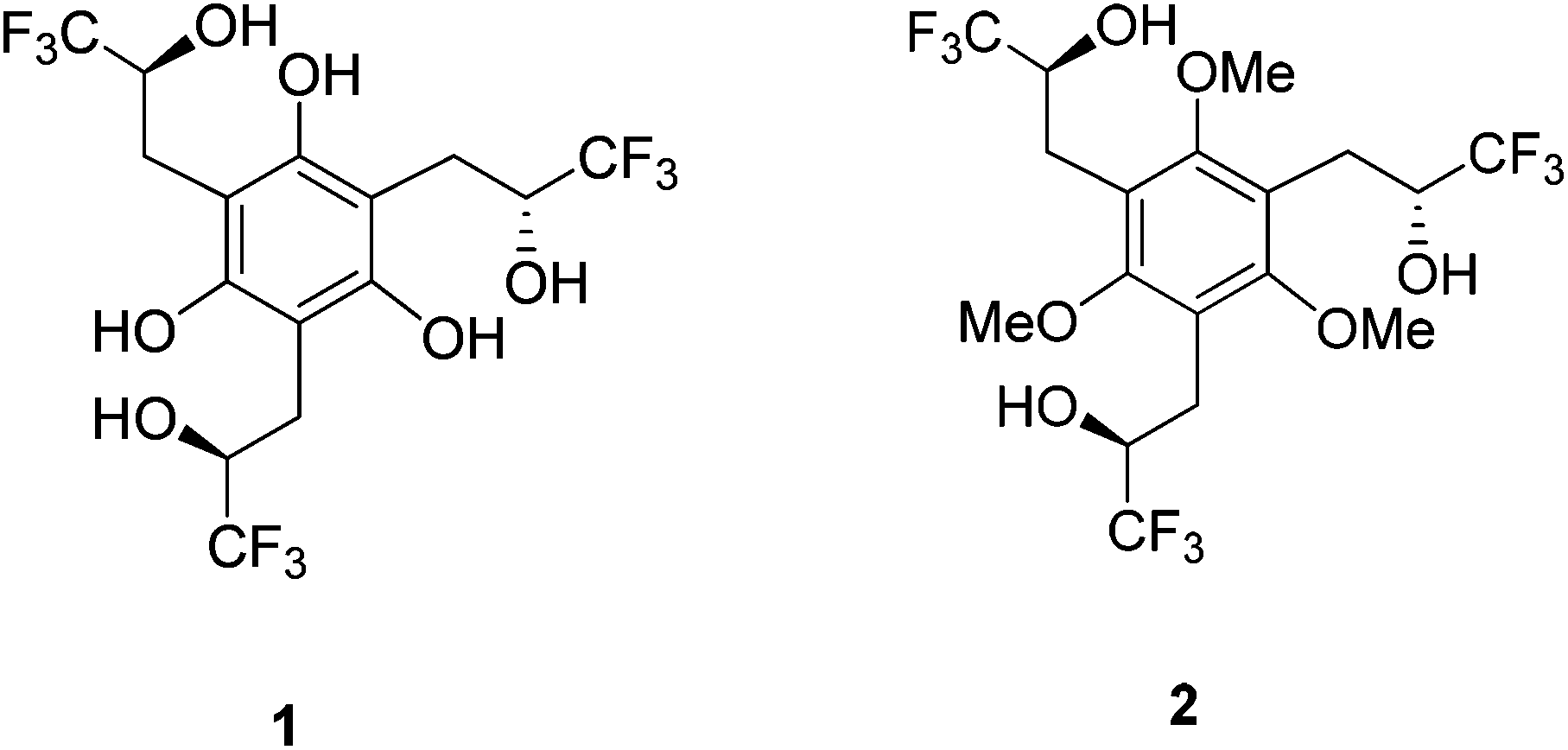 | ||
| Fig. 2 Trifluoromethyl-containing polyol receptors. | ||
Results
Hydroxyl-based receptors 1 and 2 were synthesized as outlined in Scheme 1. Reduction of the previously reported tribenzylic nitrile 426 with diisobutylaluminum hydride (DIBAL-H) afforded the trisaldehyde 3 in modest yield. Nucleophilic addition of three equivalents of (trifluoromethyl)trimethylsilane in the presence of fluoride anion was subsequently carried out to give the desired triol 2 in a 34% yield. Cleavage of all three ethers with boron tribromide resulted in the formation of the desired hexaol receptor 1 in a very efficient (i.e., 90% yield) transformation. | ||
| Scheme 1 Synthesis of anion hosts 1 and 2. a DIBAL-H, CH2Cl2, 3 days; H2SO4, 3 h, 40%. b CF3TMS, CsF, monoglyme, 18 h; TBAF, THF, 3 h, 34%. c BBr3, CH2Cl2, 90%. | ||
Electrospray ionization of an aqueous methanolic solution of 1 afforded its conjugate base while spraying 1 and 2 in the presence of ammonium salts (i.e., NH4X, X = Cl−, OAc−, and H2PO4−) generated their corresponding 1:1 adduct ions. Photoelectron spectra of all seven of these species were recorded at 20 K with a F2 excimer laser at 157 nm (7.867 eV, Fig. 3 and 4). Each spectrum shows two or more broad features resulting from detachment of an electron to afford ground and excited states of the corresponding radical. The adiabatic electron detachment energies (ADEs) were obtained from the rapidly rising onset energies of the lowest energy features whereas the vertical detachment energies (VDEs) were obtained from the maxima of these bands. All of the results are summarized in Table 1.
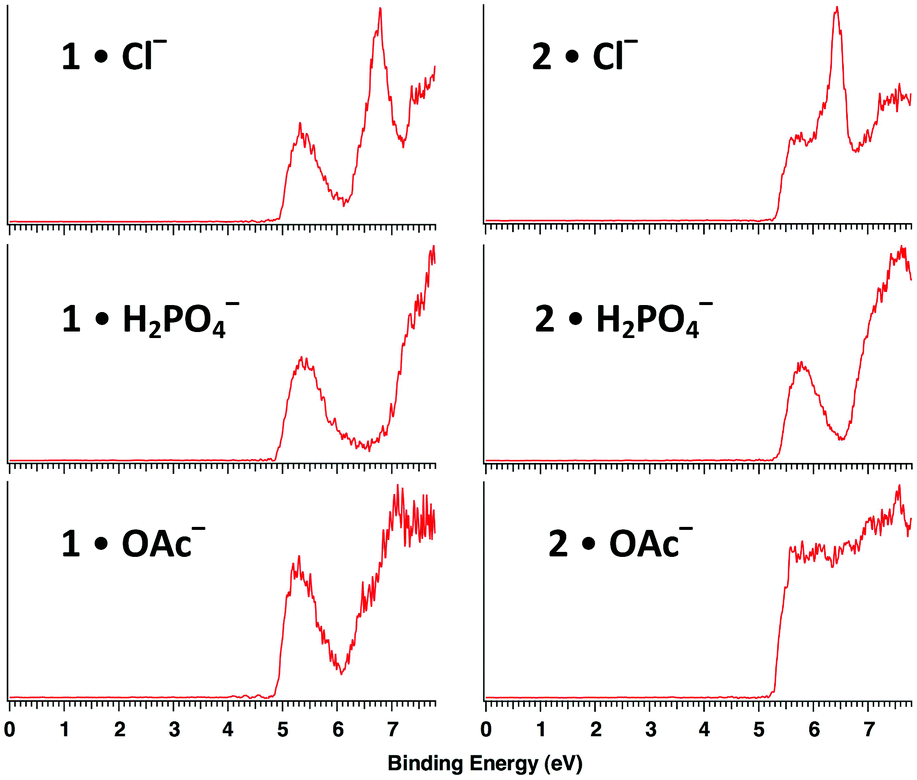 | ||
| Fig. 3 Low temperature (20 K) photoelectron spectra of hexaol 1 and triol 2 complexes with Cl−, H2PO4−, and OAc− at 157 nm (7.867 eV). | ||
 | ||
| Fig. 4 Low temperature (20 K) photoelectron spectrum of deprotonated hexaol 1a at 157 nm (7.867 eV). | ||
:1 anion complexes of 1 and 2
| Anion | Exptla | Calc.b | VDE | ||
|---|---|---|---|---|---|
| ADE | VDE | ADE | |||
| B3LYP | M06-2X | B3LYP | |||
| a Uncertainties are ±0.10 eV. b Computed values correspond to B3LYP/ aug-cc-pVDZ and M06-2X/maug-cc-pVT(+d)Z energies at 0 K. Thermal corrections to 20 K are negligible (<0.01 eV) and were omitted. | |||||
| 1a | 4.00 | 4.32 | 3.72 | 3.91 | 4.03 |
| 1·Cl− | 5.00 | 5.32 | 4.58 | 5.02 | 4.76 |
| 1·OAc− | 4.95 | 5.30 | 4.68 | 4.87 | 5.11 |
| 1·H2PO4− | 4.95 | 5.35 | 4.73 | 4.92 | 5.18 |
| 2·Cl− | 5.38 | 5.65 | 4.83 | 5.11 | 5.36 |
| 2·OAc− | 5.32 | 5.62 | 4.76 | 5.01 | 5.28 |
| 2·H2PO4− | 5.40 | 5.72 | 4.78 | 4.97 | 5.48 |
B3LYP geometry optimizations were carried out with the aug-cc-pVDZ basis set on all of the cluster ions,27,28 the conjugate base of 1 (1a), and their corresponding radicals. The structures of the chloride adducts are illustrated in Fig. 5 and complete details for all of the structures (i.e., geometries and energies) are given in the ESI.† M06-2X29 single point energies with the maug-cc-pVT(+d)Z basis set30 were also computed and the resulting ADEs are provided in Table 1. As previously reported, B3LYP does reasonably well in reproducing the experimental values but the M06-2X functional is superior.29 In this case, the errors span from 0.22–0.62 eV (B3LYP) and 0.02–0.43 eV (M06-2X) with average errors of 0.42 (B3LYP) and 0.18 (M06-2X) eV.
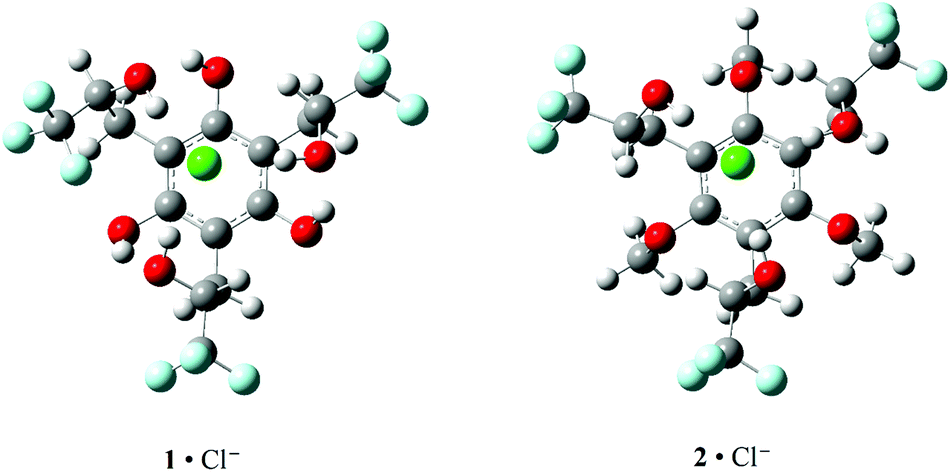 | ||
| Fig. 5 Computed B3LYP/aug-cc-pVDZ structures for 1·Cl− and 2·Cl−. The OH⋯Cl− distances are 2.132, 2.191, and 2.245 Å in 1·Cl− and 2.133, 2.157, and 2.167 Å in 2·Cl−. | ||
Binding affinities of 1 and 2 with Cl−, H2PO4−, NO3− and OAc− were investigated in three or four different solvents (i.e., CD3CN, CD3COCD3, CD2Cl2, and CDCl3) by 1H NMR spectroscopy, and the results are summarized in Table 2. Host concentrations were kept low over a range from 0.1–2.5 mM where there was no evidence for self aggregation while 25–100 mM guest solutions were used in carrying out the titrations. Anion-induced downfield shifts of the 1H NMR signals of the aliphatic hydroxyl and methine groups were followed to obtain the binding isotherms and non-linear fits of the data provided the 1:1 association constants.31 Hexaol 1 was not soluble enough in CDCl3 to measure its binding constants, it did not associate with H2PO4− and OAc− in CD3CN, its titration profiles could not be fit in CD2Cl2, and it was not studied in acetone-d6. The association constants of triol 2 were determined in all four solvents but the values were too large to measure by 1H NMR (>105 M−1) for Cl− and OAc− in (CD3)2CO. Chloride affinities of 1 and 2 were also determined at three temperatures (i.e., −24.2, 22.0, and 51.3 or 59.1 °C) spanning up to an 83 °C temperature range in order to obtain the thermodynamic binding parameters (ΔG, ΔH, TΔS, and ΔCp), which are given in Table 3.
:1 complexes of 1 and 2 with tetrabutylammonium salts (Bu4N+X−, X− = Cl−, H2PO4−, NO3−, and OAc−) in four different solvents at 22 °C
| Cmpd | T (°C) | K (M−1) | ΔH | TΔS | ΔCp | ΔG |
|---|---|---|---|---|---|---|
| a Enthalpies, TΔS, and ΔG are in kcal mol−1 whereas ΔCp is in kcal mol−1 K−1. | ||||||
| 1 | −24.2 | 4740 | ||||
| 22.0 | 667 | 10.8 | 14.8 | 0.6 | −4.0 | |
| 59.1 | 30700 |
|||||
| 2 | −24.2 | 97900 |
||||
| 22.0 | 8240 | −2.1 | 3.0 | 0.2 | −5.1 | |
| 51.3 | 6310 | |||||
Discussion
To design molecular receptors, hydrogen bond donors are commonly preorganized in different orientations that are controlled by the covalent architecture of the host. Macrocycles and polyfused rings such as calixarenes, steroids, and cis,cis-1,3,5-substituted cyclohexanes are widely employed subunits in molecular recognition studies.32–34 Additional preorganization brought about by intramolecular hydrogen bonds has been used to provide conformational control. It is also well know that increasing the acidity of a hydrogen bond donor in a receptor typically leads to enhanced binding of anions. As a result, computations were carried out on hexaol 1 and triol 2 since it was expected that they would benefit from several key features including a rigid central benzene moiety, three electron withdrawing trifluoromethyl groups to enhance the acidity of the aliphatic alcohols, and three phenolic substituents in the former compound to provide additional hydrogen bond stabilization of its anion–bound complexes. The B3LYP/aug-cc-pVDZ structures of the chloride clusters with the anti diastereomers of 1 and 2 (i.e., the RRS/SSR isomers) form three hydrogen bonds between the secondary hydroxyl groups and the chloride anion (Fig. 5). As a result, the B3LYP/aug-cc-pVDZ and M06-2X/maug-cc-pVT(+d)Z clustering enthalpies (i.e., ΔH for Cl− + 1 or 2 → 1·Cl− or 2·Cl−) are remarkably favorable. That is, ΔH = −41.2 (B3LYP) and −47.9 (M06-2X) kcal mol−1 (2) and −41.3 (B3LYP) and −51.2 (M06-2X) kcal mol−1 (1) whereas the binding of methanol to chloride anion has been measured to be exothermic by −17.5 ± 0.3 kcal mol−1.35 The association of three molecules of methanol to Cl−, however, has been determined to be −43.1 ± 0.535 kcal mol−1 which is similar to the computed values for 2. The M06-2X binding affinity for the hexaol is larger than for the triol by 3.3 kcal mol−1, and can be attributed to the presence of the three intramolecular hydrogen bonds between the aryl OH donors and the oxygen acceptors of the aliphatic hydroxyl groups in the chloride anion complex of 1.The experimentally determined ADEs for 1 and 2 clustered to Cl−, OAc− and H2PO4− (i.e., 1·X− and 2·X−) are well reproduced computationally, but the values for the triol are larger than those for the hexaol. This unexpected result is due to the fact that ADE(2·X−) – ADE(1·X−) corresponds to the difference in the clustering energies of 1 and 2 with X− and X˙. For example, Cl− binds more favorably to the hexaol than the triol by 0.1 (B3LYP) and 3.3 kcal mol−1 (M06-2X) and this leads to a larger ADE for the former complex. This difference is offset and overcome, however, by the greater preference for the association of 1 to Cl˙ by 5.8 (B3LYP) and 5.4 kcal mol−1 (M06-2X). In retrospect maybe this is not surprising because the abstraction of a hydrogen atom from one of the secondary hydroxyl groups leads to an alkoxy radical that appears to rearrange to a stabilized phenoxyl radical with little or no barrier.‡
In making clusters of 1, but not 2, its conjugate base (1a) was observed. The photoelectron spectrum of the (M − 1)− ion consequently was recorded at 20 K (Fig. 4). Computations indicate that the most acidic site in the hexaol is one of the phenolic hydroxyl groups and the resulting anion is stabilized by a total of five hydrogen bonds – two to the charged site, two O–H⋯F interactions, and one O–H⋯OH hydrogen bond. The ADE of 1a is 4.00 ± 0.10 eV which is well reproduced by both the B3LYP (3.72 eV) and M06-2X (3.91 eV) predictions. It is also 1.75 eV (40.3 kcal mol−1) larger than for phenoxide anion36 as a result of the hydrogen bond network and electron withdrawing CF3 groups in this ion.
Hexaol 1 adopts a conformation(s) in acetonitrile where intramolecular hydrogen bonds are formed, and the secondary hydroxyl groups act as hydrogen bond donors and the phenolic oxygen atoms serve as hydrogen bond acceptors. This structural assignment is based on the downfield 1H NMR chemical shifts of the aliphatic OH resonances at 5.30 and 5.36 ppm compared to those in the triol at 4.21 ppm. This preorganization might be expected to enhance the anion-binding of 1 by reducing the entropic penalty for association. Small equilibrium constants for 1:1 binding with Cl− and NO3− were determined in CD3CN contrary to expectation, but the entropy is quite favorable as anticipated and this can be viewed as a manifestation of the hydrophobic effect.37,38 That is, binding results in the release of solvent molecules despite the unfavorable enthalpy for this process. In contrast, triol 2 was found to bind to all four anions (i.e., Cl−, H2PO4−, NO3− and OAc−) and while it displays relatively little selectivity presumably because of the flexibility of its scaffold, to the best of our knowledge it is the strongest known aliphatic hydoxyl-based receptor to date. This is due, at least in the case of Cl−, to the favorable binding enthalpy and entropy. One can rationalize the thermochemical differences between 1 and 2 based upon their dipole moments and reorganization energies upon binding. That is, 1 and 2 have computed dipole moments of 1.58 and 3.80 D, respectively, so one would expect the solvent to organize more strongly around the triol. This should lead to a more favorable entropy of binding for 1, which is what is observed (i.e., TΔS = 14.8 (1) vs. 3.0 kcal mol−1 (2)). The reorganization energies are analogous to vertical detachment energies (VDEs) in that they correspond to the difference between the energy for the adopted structure of the host in its bound complex and its free form. These energies were computed for the hexaol and the triol and are 6.4 (B3LYP) and 4.9 kcal mol−1 (M06-2X) larger for the former compound. This is consistent with the binding enthalpies obtained from the Van't Hoff plots as the hexaol pays a larger energetic price than the triol to adopt its anion-bound conformation (i.e., ΔH is less favorable for 1).
Solvents are well known to play a critical role in molecular recognition. Neutral receptors bind neutral guests much more effectively in lower dielectric constant media.39 One might assume that the same situation would apply when a host binds an ion, but this need not be the case as charged species are always accompanied by a counterion in condensed media. A free ion would bind more tightly to a receptor in a non-polar solvent but when a counterion is present the oppositely charged species will interact more strongly in this medium. To address this issue, 1:1 host–guest equilibrium constants were measured in four different solvents (i.e., CDCl3, CD2Cl2, CD3COCD3 and CD3CN) between 2 and Cl−, H2PO4−, NO3− and OAc− (Table 2). The results in chloroform (ε = 4.7) and acetonitrile (ε = 35.7) are essentially the same except for nitrate which binds ∼10 times more strongly in the less polar solvent. In contrast, CDCl3 and CD2Cl2 (ε = 8.9) are similar solvents but the equilibrium binding constants vary by factors of ∼2–8. They are larger in the solvent with the bigger dielectric constant and this maybe due to greater ion separation of the tetrabutylammonium salts in dichloromethane. The most dramatic change in the complexation of 2 was observed in acetone-d6. Equilibrium constants that are too big to measure by 1H NMR spectroscopy were obtained with Cl− and OAc−. Acetone does have a larger solvent donor number than acetonitrile, chloroform and dichloromethane (these values are 17, 14.1, 4.0, and 1.0 kcal mol−1, respectively and correspond to the ability to solvate a cation),40 but more work is needed to elucidate the key factors influencing host–guest binding of ions in different solvents and environments.
Experimental
General
All chemicals and solvents were purchased from Sigma-Aldrich and used as received except for the deuterated solvents (i.e., CD3CN, CD3COCD3, CD2Cl2, and CDCl3) which came from Cambridge Isotopes and were dried over activated 3 Å molecular sieves over the course of a few days. Nuclear magnetic resonance spectra were obtained with Bruker HD-500, Varian VI-300 and VI-500 spectrometers, and all of the reported chemical shifts (δ) are referenced to the residual proton signals of the solvents. High resolution mass spectra were obtained with a Bruker BioTof II electrospray ionization–time-of-flight mass spectrometer using ethanolic PEG 300 and 425 solutions as internal standards. TLC analyses were carried out on precoated aluminum-backed 0.25 mm Masherey-Nagel silica gel plates. Medium pressure liquid chromatography (25 − 60 psi) was performed on silica gel using a Biotage Isolera 1. A Thomas Hoover capillary melting point apparatus was used to obtain the uncorrected melting point of 1.:2 hexanes–ethyl acetate) to give 1.2 g (40%) of 3 as a yellow oil. 1H NMR (300 MHz, CDCl3) δ 3.57 (9H, s), 3.67 (6H, s), 9.64 (3H, s). 13C NMR (75 MHz, CDCl3) δ 39.4, 60.8, 116.9, 157.9, 198.8. HRMS-ESI: calc. for C15H18NaO6+ (M + Na)+ 317.0996, found 317.0992.
:1 hexanes–ethyl acetate) of the crude product on silica gel afforded 0.7 g (34%) of 2 as an ∼1:5 syn (RRR/SSS) and anti (RRS/SSR) mixture of diastereomers. Anti-2: 1H NMR (300 MHz, CDCl3) δ 3.04 (m, 6H), 3.31 (m, 3H), 3.82 (s, 6H), 3.84 (s, 3H), 4.21 (bs, 3H). 13C NMR (75 MHz, CDCl3) δ 24.8, 24.9, 61.15, 61.18, 69.1 (q, J = 30.6 Hz), 69.2 (q, J = 30.6 Hz), 119.8, 119.9, 124.7 (q, J = 282.7 Hz), 157.7, 157.8. 19F NMR (282 MHz, CDCl3) −81.15 (d, J = 6.1 Hz), −81.12 (d, J = 8.5 Hz). HRMS-ESI: calc. for C18H22F9O6+ (M + H)+ 505.1267, found 505.1257.
:1 hexanes–ethyl acetate) to afford 0.4 g (90%) of 1 as 1:5 mixture of syn and anti diastereomers. The anti diastereomer was subsequently separated from the syn isomer by flash column chromatography (2:1 hexanes–Et2O) and was obtained as a white solid (m.p. 143–145 °C). 1H NMR (300 MHz, CD3CN) δ 2.61 (m, 3H), 3.05 (m, 3H), 3.96 (m, 3H), 5.30 (d, J = 3.8 Hz, 2H), 5.36 (d, J = 4.1 Hz, 1H), 7.47 (s, 1H), 7.49 (s, 1H), 7.50 (s, 1H). 13C NMR (125 MHz, CD3CN) δ 25.24 (q, J = 2.7 Hz), 25.30 (q, J = 2.7 Hz), 71.9 (q, J = 30.0 Hz), 72.0 (q, J = 30.2 Hz), 105.89, 105.92, 126.0 (q, J = 283.0 Hz), 126.3 (q, J = 283.0 Hz), 155.0, 155.1. 19F NMR (282 MHz, CD3CN) −81.1 (d, J = 7.7 Hz), −81.0 (d, J = 8.9 Hz). HRMS-ESI: calc. for C15H15F9NaO6+ (M + Na)+ 485.0617, found 485.0640.
Titrations
One milliliter of dilute solutions (0.1–2.5 mM) of 1 or 2 in different solvents were placed in NMR tubes and capped with septa. More concentrated guest solutions (25–100 mM) containing the same dilute concentration of the initial host in all but three cases were subsequently added sequentially via syringe, and the course of the titration was followed by 1H NMR. The signals of the aliphatic hydroxyl groups and methine hydrogens were monitored and the data were fit with a 1:1 nonlinear binding isotherm using the Excel Solver add-on program to obtain the equilibrium association constants.31
Photoelectron spectroscopy
Ions of interest were generated by electrospray ionization of ∼10−3 M methanol–water solutions containing hexaol 1 or triol 2, and except for the studies on the (M − 1)− anion of the former compound (1a), a small amount of the ammonium salt of OAc−, Cl−, or H2PO4−. A home-built photoelectron spectrometer that has been previously described was used in this work.41 A F2 excimer laser that emits 157 nm (7.867 eV) photons and which was operated at 20 Hz was employed and enabled shot-to-shot background subtraction of the data to be carried out. Photoelectrons were analyzed with a 5.2 m long flight tube and the resulting spectra obtained at 20 K had a resolution of ∼2% or 50 meV at 5 eV.Computations
Electronic structure calculations were carried out with the Gaussian 09 suite of programs.42 To locate local and global minima, systematic conformational searches were carried out using Spartan 08 and the MMFF force field on hexaol 1 and triol 2, both the conjugate bases and their anion-bound complexes.43 Stable structures that were within 7 kcal mol−1 of the lowest energy structure were selected and B3LYP/6-311+G(d,p)27 single point energies of these species were computed. Full optimizations were subsequently carried out on the most stable conformers and the lowest energy species were then minimized using the aug-cc-pVDZ basis set.28 M06-2X/maug-cc-pVT(+d)Z29,30 single point energies were also computed on these final geometries. To obtain the adiabatic and vertical detachment energies (ADEs and VDEs), the corresponding radicals were also optimized starting with the lowest energy anion structures. The ADEs are reported as enthalpies at 0 K, whereas all of the other energies were corrected to 298 K.Conclusions
Two new hydroxyl-based hosts (1 and 2) were synthesized and their 1:1 anion-complexes along with the (M − 1)− ion of 1 were characterized in the gas phase by negative ion photoelectron spectroscopy. The adiabatic detachment energies (ADEs) for all seven species are unusually large due to the stabilization resulting from the intramolecular hydrogen bond networks and the electron withdrawing trifluoromethyl groups. Hexaol 1·X− (X = Cl, OAc, and H2PO4) appears to have the larger anion binding energies in the gas phase, but triol 2 has the bigger ADEs. This is because X˙ interacts more strongly with 1 than 2 since the former species can lead to a more stable phenoxyl radical. Binding constants were also measured in four solvents. The preorganization in 1 arising from the intramolecular hydrogen bond network, which is absent in 2, led to a poorer anion receptor. The binding entropy for hexaol 1 is more favorable than for triol 2, at least in CD3CN where the van't Hoff analysis was carried out, but the enthalpy difference dominates and it is less favorable for the hexaol. Given the flexibility of the cavity in 1 and 2, these hosts are able to bind a variety of anions and the latter species is the strongest aliphatic hydroxyl-based receptor reported to date.
Acknowledgements
We thank Mr Masoud Samet for obtaining the 13C NMR spectrum of 1. Generous support from the National Science Foundation and the Minnesota Supercomputer Institute for Advanced Computational Research are gratefully acknowledged. The U. S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences and Biosciences supported the photoelectron spectra work (XBW), which was performed at the EMSL, a national scientific user facility sponsored by DOE's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory, which is operated by Battelle for DOE.Notes and references
- J. L. Sessler, P. A. Gale and W. S. Cho, in Anion Receptor Chemistry (Monographs in Supramolecular Chemistry), ed. J. F. Stoddart, RSC, Cambridge, 2006 Search PubMed.
- P. A. Gale, Acc. Chem. Res., 2006, 39, 465–475 CrossRef CAS PubMed.
- K. Bowman-James, Acc. Chem. Res., 2005, 38, 671–678 CrossRef CAS PubMed.
- T. Ooi and K. Maruoka, Angew. Chem., Int. Ed., 2007, 46, 4222–4266 CrossRef CAS PubMed.
- (a) A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348–357 RSC; (b) V. Gorteau, G. Bollot, J. Mareda, A. Perez-Velasco and S. Matile, J. Am. Chem. Soc., 2006, 128, 14788–14789 CrossRef CAS PubMed.
- M. J. Welsh, B. W. Ramsey, F. Accurso and G. R. Cutting, in The Metabolic and Molecular Basis of Inherited Disease, ed. C. R. Scriver, A. L. Beaudet, W. S. Sly and D. Valle, McGraw-Hill, New York, 2001 Search PubMed.
- F. M. Ashcroft, Ion Channels and Disease Channelopathies, Academic Press, San Diego, 2000 Search PubMed.
- (a) T. J. Jentsch, C. A. Hubner and J. C. Fuhrmann, Nat. Cell Biol., 2004, 6, 1039–1047 CrossRef CAS PubMed; (b) T. J. Jentsch, T. Maritzen and A. A. Zdebik, J. Clin. Invest., 2005, 115, 2039–2046 CrossRef CAS PubMed.
- (a) J. T. Davis, O. Okunola and R. Quesada, Chem. Soc. Rev., 2010, 39, 3843–3862 RSC; (b) P. R. Brotherhood and A. P. Davis, Chem. Soc. Rev., 2010, 39, 3633–3647 RSC; (c) G. W. Gokel and N. Barkey, New J. Chem., 2009, 33, 947–963 RSC; (d) P. A. Gale, Acc. Chem. Res., 2011, 44, 216–226 CrossRef CAS PubMed.
- (a) K. Kavallieratos, S. R. de Gala, D. J. Austin and R. H. Crabtree, J. Am. Chem. Soc., 1997, 119, 2325–2326 CrossRef CAS; (b) K. Kavallieratos, C. M. Bertao and R. H. Crabtree, J. Org. Chem., 1999, 64, 1675–1683 CrossRef CAS PubMed; (c) J. L. Sessler, S. Camiolo and P. A. Gale, Coord. Chem. Rev., 2003, 240, 17–55 CrossRef CAS; (d) M. J. Chmielewski and J. Jurczak, Chem.–Eur. J., 2005, 11, 6080–6094 CrossRef CAS PubMed; (e) D. Esteban-Gomez, L. Fabbrizzi and M. Licchelli, J. Org. Chem., 2005, 70, 5717–5720 CrossRef CAS PubMed; (f) S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 7882–7894 CrossRef CAS PubMed; (g) S. E. Garcia-Garrido, C. Caltagirone, M. E. Light and P. A. Gale, Chem. Commun., 2007, 1450–1452 RSC.
- (a) J. M. Coteron, F. Hacket and H. J. Schneider, J. Org. Chem., 1996, 61, 1429–1435 CrossRef CAS; (b) A. P. Davis, J. J. Perry and R. S. Wareham, Tetrahedron Lett., 1998, 39, 4569–4572 CrossRef CAS; (c) K. Kano, N. Tanaka and S. Negi, Eur. J. Org. Chem., 2001, 3689–3694 CrossRef CAS; (d) D. K. Smith, Org. Biomol. Chem., 2003, 1, 3874–3877 RSC; (e) K. J. Winstanley and D. K. Smith, J. Org. Chem., 2007, 72, 2803–2815 CrossRef CAS PubMed; (f) S. I. Kondo, N. Okada, R. Tanaka, M. Yamamura and M. Unno, Tetrahedron Lett., 2009, 50, 2754–2757 CrossRef CAS PubMed.
- (a) V. Hedge, P. Madhukar, J. D. Madura and R. P. Thummel, J. Am. Chem. Soc., 1990, 112, 4549–4550 CrossRef; (b) V. Hedge, C. Y. Hung, P. Madhukar, R. Cummingham, T. Hopfner and R. P. Thummel, J. Am. Chem. Soc., 1993, 115, 872–878 CrossRef; (c) T. W. Bell, Z. Hou, S. C. Zimmerman and P. A. Thiessen, Angew. Chem., Int. Ed., 1995, 34, 2163–2165 CrossRef CAS; (d) T. W. Bell, N. M. Hext and A. B. Khasanov, Pure Appl. Chem., 1998, 70, 2371–2377 CrossRef CAS.
- P. V. Santacroce, J. T. Davis, M. E. Light, P. A. Gale, J. C. Iglesias-Sanchez, P. Prados and R. Quesada, J. Am. Chem. Soc., 2007, 129, 1886–1887 CrossRef CAS PubMed.
- S. Lee, Y. Hua, H. Park and A. H. Flood, Org. Lett., 2010, 12, 2100–2102 CrossRef CAS PubMed.
- (a) J.-L. Jestin and F. Pecorari, Foldamers: Structure, Properties and Applications, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (b) I. Saraogi and A. D. Hamilton, Chem. Soc. Rev., 2009, 38, 1726–1743 RSC.
- (a) Y. Hamuro, S. J. Geib and A. D. Hamilton, Angew. Chem., Int. Ed., 1994, 33, 446–448 CrossRef; (b) V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS PubMed; (c) B. Gong, H. Zeng, J. Zhu, L. Yua, Y. Han, S. Cheng, M. Furukawa, R. D. Parra, A. Y. Kovalevsky, J. L. Mills, E. Skrzypczack-Jankun, S. Martinovic, R. D. Smith, C. Zheng, T. Szyperski and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11583–11588 CrossRef CAS PubMed.
- (a) R. W. Sinkeldam, M. H. C. J. vanHoutem, G. Koeckelberghs, J. A. J. M. Vekemans and E. W. Meijer, Org. Lett., 2006, 8, 383–385 CrossRef CAS PubMed; (b) A. Zhang, Y. Han, K. Yamato, X. C. Zeng and B. Gong, Org. Lett., 2006, 8, 803–806 CrossRef CAS PubMed.
- (a) C. Li, G.-T. Wang, H.-P. Yi, X.-K. Jiang, Z.-T. Li and Z. R.-X. Wang, Org. Lett., 2007, 9, 1797–1800 CrossRef CAS PubMed; (b) W. Cai, G.-T. Wang, Y.-X. Xu, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2008, 130, 6936–6937 CrossRef CAS PubMed.
- (a) V. Amendola, D. Esteban-Gomez, L. Fabbrizzi and M. Licchelli, Acc. Chem. Res., 2006, 39, 343–353 CrossRef CAS PubMed; (b) C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernandez, R. Perez-Tomas and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed.
- (a) M. Boiocchi, L. Del Boca, D. E. Gomez, L. Fabbrizzi, M. Licchelli and E. Monzani, J. Am. Chem. Soc., 2004, 126, 16507–16514 CrossRef CAS PubMed; (b) G. Bergamaschi, M. Boiocchi, E. Monzani and V. Amendola, Org. Biomol. Chem., 2011, 9, 8276–8283 RSC.
- A. Shokri, X. B. Wang and S. R. Kass, J. Am. Chem. Soc., 2013, 135, 9525–9530 CrossRef CAS PubMed.
- A. Shokri, A. Abedin, A. Fattahi and S. R. Kass, J. Am. Chem. Soc., 2012, 134, 10646–10650 CrossRef CAS PubMed.
- A. Shokri, J. Schmidt, X. B. Wang and S. R. Kass, J. Am. Chem. Soc., 2012, 134, 2094–2099 CrossRef CAS PubMed.
- Z. Tian, A. Fattahi, L. Lis and S. R. Kass, J. Am. Chem. Soc., 2009, 131, 16984–16988 CrossRef CAS PubMed.
- A. J. Lampkins, O. Abdul-Rahim, H. Li and R. K. Castellano, Org. Lett., 2005, 7, 4471–4474 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef PubMed.
- (a) Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2008, 112, 1095–1099 CrossRef CAS PubMed; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS; (c) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- E. Papajak and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 597–601 CrossRef CAS.
- K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, Wiley-Interscience, New York, 1987, p. 432 Search PubMed.
- (a) Z. L. Zhong, A. Ikeda and S. Shinkai, In Calixarenes, ed. Z. Asfari, V. Bohmer, J. Harrowfield and J. Vicens, Kluwer Academic Publishers, Dodrecht, 2001 Search PubMed; (b) P. Molenveld, J. F. J. Engbersen and D. N. Reinhoudt, Chem. Soc. Rev., 2000, 75–86 RSC.
- (a) R. Boyce, G. Li, P. Nestler, T. Suenaga and W. C. Still, J. Am. Chem. Soc., 1994, 116, 7955–7956 CrossRef CAS; (b) T. A. P. Davies, J. J. Perry and R. P. Williams, J. Am. Chem. Soc., 1997, 119, 1793–1794 CrossRef.
- (a) D. S. Kemp and K. S. Petrakis, J. Org. Chem., 1981, 46, 5140–5143 CrossRef CAS; (b) P. Kocis, O. Issakova, N. F. Sepetov and M. Leibl, Tetrahedron Lett., 1995, 36, 6623–6626 CAS; (c) R. D. Rodgers and M. W. Brechbiel, Inorg. Chem., 2001, 40, 493–498 CrossRef.
- B. Bogdanov, M. Peschke, D. S. Tonner, J. E. Szulejko and T. B. McMahon, Int. J. Mass Spectrom., 1999, 187, 707–725 Search PubMed.
- R. F. Gunion, M. K. Gilles, M. L. Polak and W. C. Lineberger, Int. J. Mass Spectrom. Ion Processes, 1992, 117, 601–620 CrossRef CAS.
- (a) H. Iwamoto, K. Niimi, T. Haino and Y. Fukazawa, Tetrahedron, 2009, 65, 7259–7267 CrossRef CAS PubMed; (b) S. L. Tobey and E. V. Anslyn, J. Am. Chem. Soc., 2003, 125, 14807–14815 CrossRef CAS PubMed; (c) T. Hayashi, T. Miyahara, N. Koide, Y. Kato, H. Masuda and H. Ogoshi, J. Am. Chem. Soc., 1997, 119, 7281–7290 CrossRef CAS.
- For hydrophobic effect review see: (a) N. T. Southall, K. A. Dill and A. D. J. Haymet, J. Phys. Chem. B, 2002, 106, 521–533 CrossRef CAS; (b) W. Blokzijl and J. B. F. N. Engberts, Angew. Chem., Int. Ed., 1993, 32, 1545–1579 CrossRef; (c) D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- R. M. Izatt, J. S. Bradshaw, K. Pawlak, R. L. Bruening and B. J. Tarbet, Chem. Rev., 1992, 92, 1261–1354 CrossRef CAS.
- D. T. Sawyer and J. L. Roberts, in Experimental electrochemistry for chemists, John Wiley & Sons, Inc., 1974 Search PubMed.
- X. B. Wang and L. S. Wang, Rev. Sci. Instrum., 2008, 79, 073108 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Spartan'08 for Macintosh, Wavefunction, Inc., Irvine, CA Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, computed xyz coordinates and energies, and representative binding data for 1 and 2. See DOI: 10.1039/c3qo00009e |
| ‡ Upon removal of HCl from 1·Cl˙ the resulting alkoxy radical underwent a hydrogen atom shift to form a phenoxyl radical upon carrying out a B3LYP/aug-cc-pVDZ optimization. |
| This journal is © the Partner Organisations 2014 |