Scalable, efficient total synthesis of (+)-mupirocin H†
Changgui
Zhao
a,
Ziyun
Yuan
a,
Yuanyuan
Zhang
a,
Bin
Ma
a,
Huilin
Li
a,
Shouchu
Tang
a,
Xingang
Xie
*a and
Xuegong
She
*ab
aState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: xiexg@lzu.edu.cn; shexg@lzu.edu.cn; Fax: +86-931-8912582
bState Key Laboratory of Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou, Gansu 730000, P. R. China
First published on 23rd December 2013
Abstract
A scalable, efficient total synthesis of mupirocin H was accomplished in 7 longest linear steps with 39% overall yield. The developed strategy is great progress for the construction of pseudomonic acid analogues and all steps in our strategy could be conducted on a gram scale. The strategy is also suitable for other related molecules.
As early as 1887, Garre found that the fermentation broth of Pseudomonas fluorescens had antimicrobial activity,1 but it was not until the late 1960s that Fuller et al. determined that pseudomonic acids were responsible for the bioactivity.2 In order to avoid ambiguity, the British Pharmacopoeia and World Health Organization use the generic name “mupirocin” instead of “pseudomonic acids”. Mupirocin is a mixture of pseudomonic acids A, B, C and D (Fig. 1) with the principal constituent pseudomonic acid A accounting for 95%. As a polyketide3 antibiotic, it has been widely used clinically for the treatment of skin infections. Due to the unusual structures and important biological activities, the pseudomonic acids have attracted many organic chemists to their total syntheses.4,5 When Simpson et al. studied the biosynthesis of pseudomonic acid from Pseudomonas fluorescens, two new pseudomonic acid analogues named mupirocin W6 and mupirocin H7 were isolated and mupirocin W showed similar bioactivity to the pseudomonic acids.
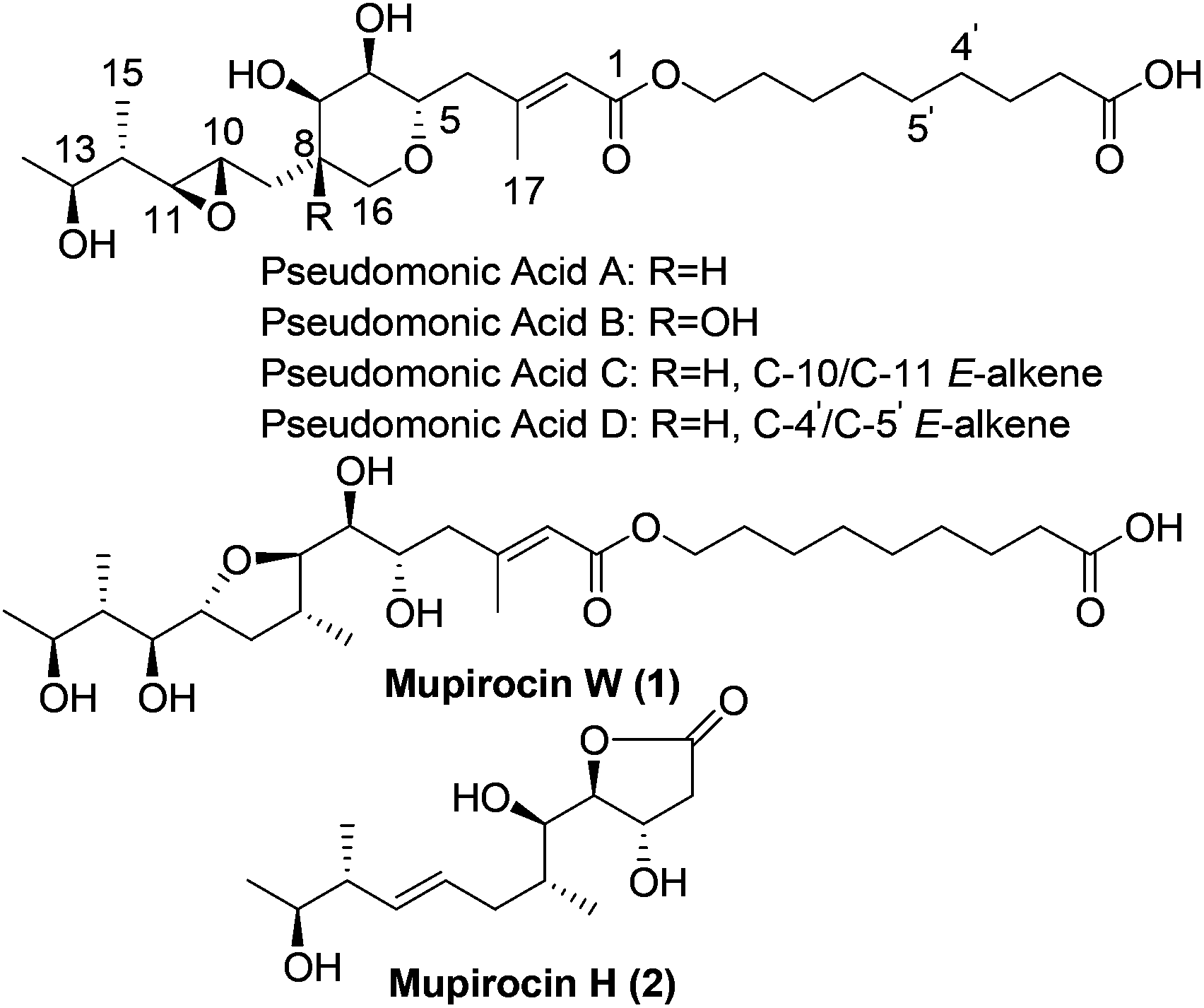 | ||
| Fig. 1 Chemical structures of pseudomonic acids, mupirocin W (1) and mupirocin H (2). | ||
In 2011, the Chakraborty group reported the first enantioselective total synthesis of mupirocin H in 19 steps with 4.96% overall yield using D-glucose as the chiral source and Julia–Kocienski reaction for construction of the E-olefinic bond.8 In 2012, the Willis group also reported a convergent total synthesis of mupirocin H in 11 steps with 6.9% overall yield using a functionalized lactone transformation strategy.9 As mentioned by Chakraborty, although mupirocin has excellent antibiotic properties, it exhibits poor bioavailability, which restricts its use as a topical antibiotic. Thus, it is highly desirable to develop a general, scalable and divergent strategy that could be applied to synthesis of a large number of the pseudomonic acid analogues offering convenience for further bioactivity studies. Herein we disclose a scalable and efficient total synthesis of mupirocin H using Suzuki–Miyaura and Mukaiyama aldol reactions as key steps. To the best of our knowledge, this is a great advance in the construction of pseudomonic acid analogues and all steps in our developed strategy could be conducted on a gram scale. The strategy is also suitable to other related molecules (Scheme 1).
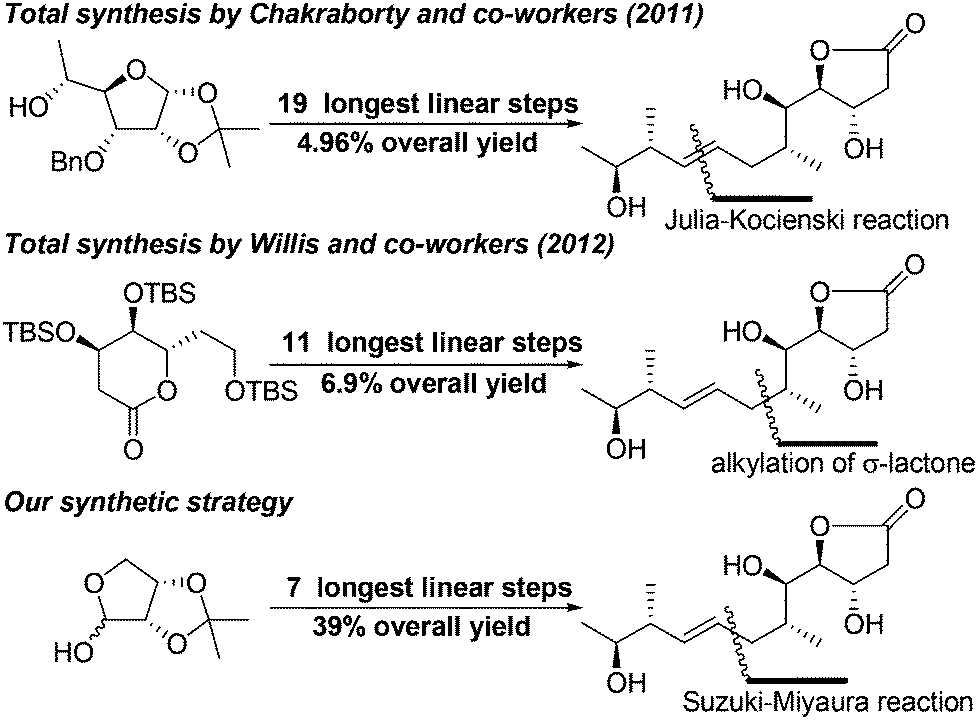 | ||
| Scheme 1 Past synthetic approaches and our synthetic strategy. | ||
Our retrosynthetic approach to mupirocin H (2) is presented in Scheme 2. We rationalized that the γ-lactone moiety could be established by deprotection and lactonization from ester 3. Unlike most of the syntheses performed on pseudomonic acid analogues, our strategy to obtain the E-olefin leaves the formation of the carbon–carbon bond between C7 and C8 through a Suzuki–Miyaura coupling reaction of vinyl iodide 4 and the trialkyl boron reagent 5 to a late stage. The requisite coupling precursor 5 could be generated from aldehyde 7 and 8via a Mukaiyama aldol reaction. For aldehyde 7, it could be obtained from known compound 10. On the other hand, the vinyl iodide 4 could be easily prepared via a Takai reaction from aldehyde 6, which was derived from the known chiral alcohol 9.
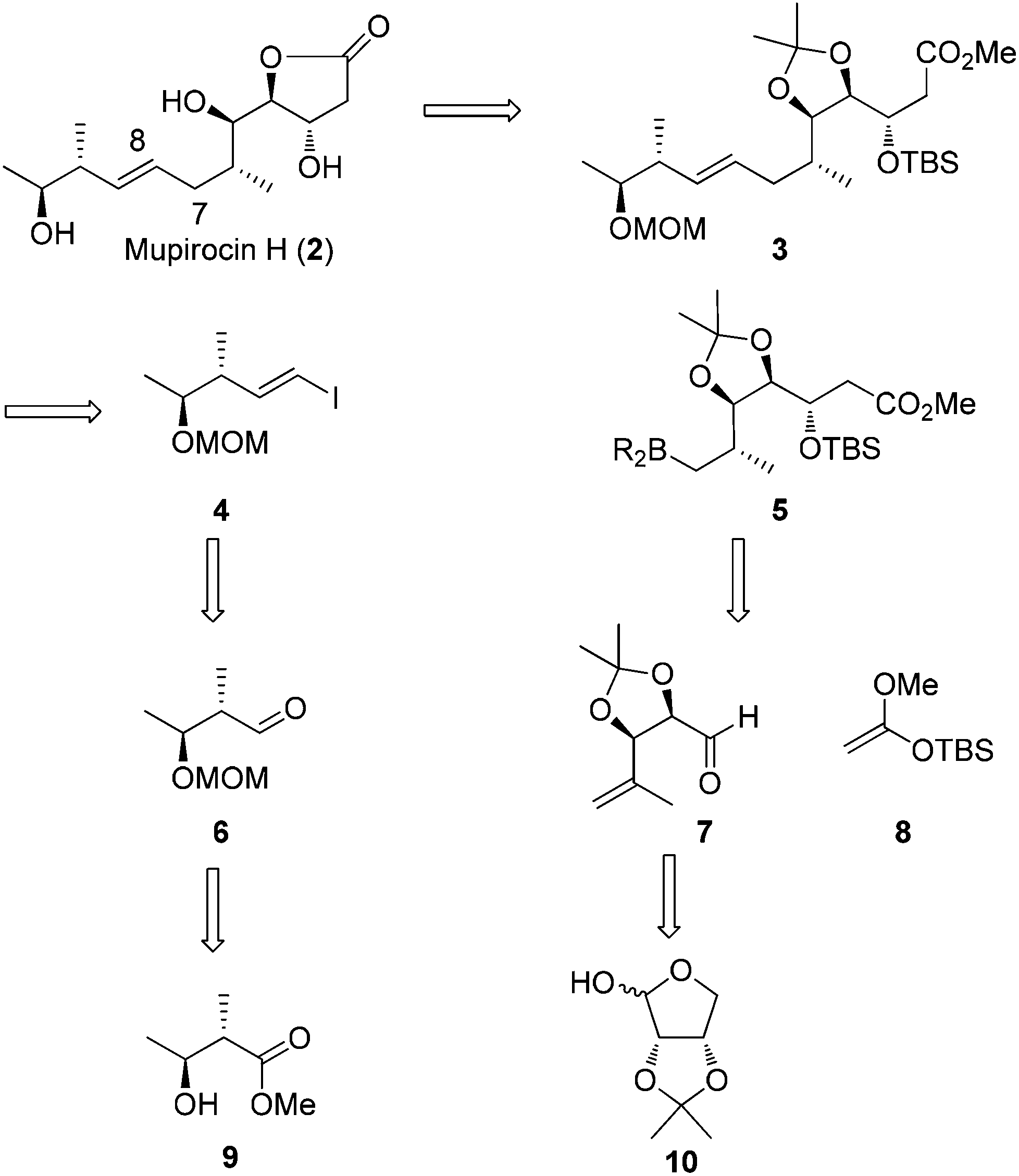 | ||
| Scheme 2 Retrosynthetic analysis of mupirocin H (2). | ||
Our synthesis started with the preparation of segment 4. Protection of the known chiral alcohol 910 followed by LiAlH4 reduction gave the primary alcohol 12 in 83% yield for two steps. Subsequent Swern oxidation of alcohol 12 produced the desired aldehyde 6,11 which was subjected to a Takai reaction to afford the vinyl iodide 4 in 78% overall yield for two steps12 (Scheme 3).
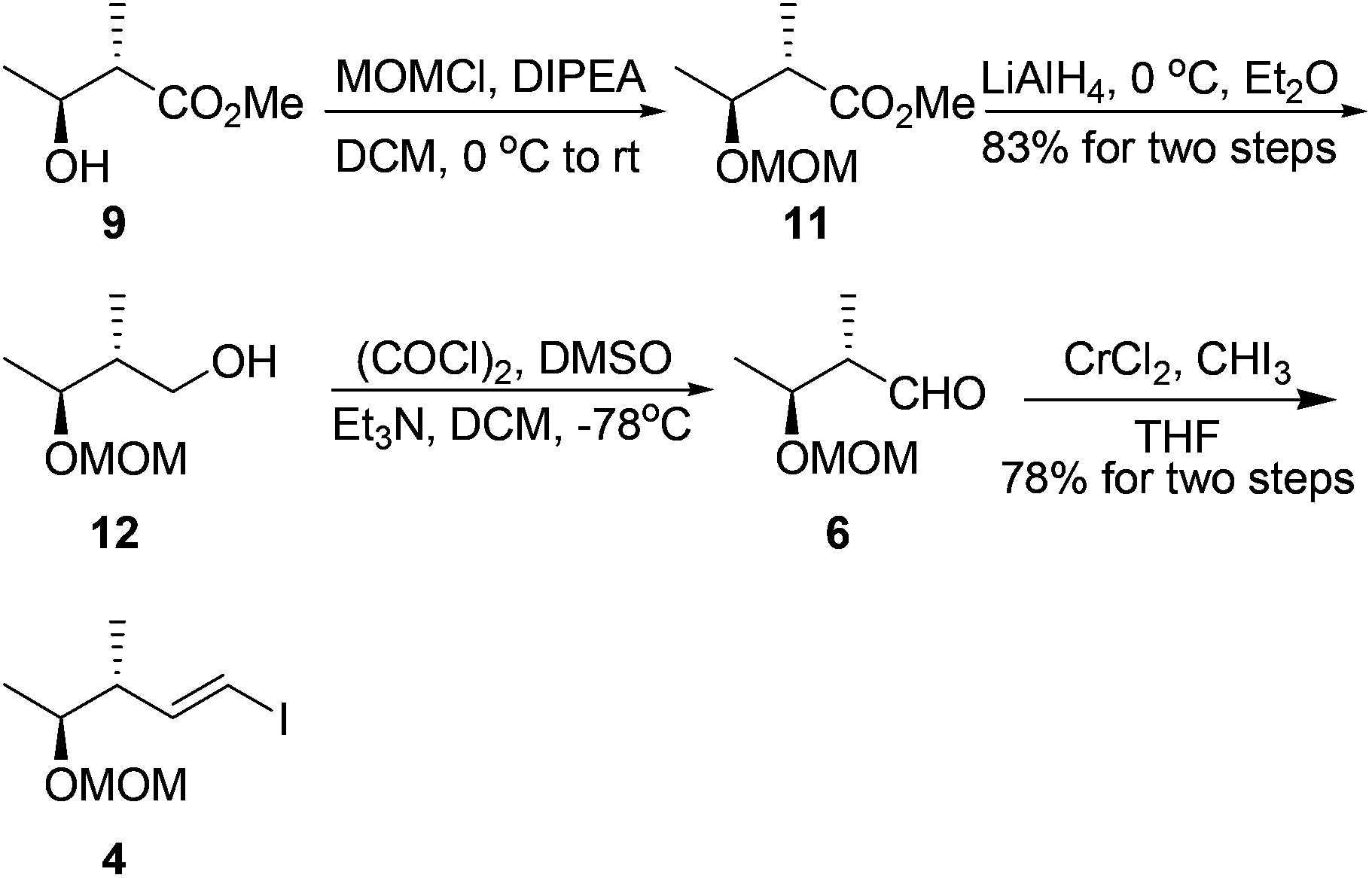 | ||
| Scheme 3 Synthesis of vinyl iodide 4. | ||
Having succeeded in construction of building block 4, we then turned our attention to synthesizing segment 5. Our synthesis began with the known 2,3-O-isopropylidene-D-erythrose 10,13 oxidation of the aldehyde 10 using Anelli conditions gave erythronolactone 13 in 71% yield.14 Reaction of MeLi with 13 afforded the protected 1-deoxyribulose 14 in 96% yield.15 Subsequent Wittig reaction of 14 with methylene triphenylphosphorane gave the olefinic product 15 in 99% yield.16 Swern oxidation of alcohol 15 produced the desired aldehyde 7 which was used directly in the next step without further purification. Exposure of the crude aldehyde 7 to ZnI2 and O-silylated ketene acetal 8 gave a (anti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :syn over 10:1) separable mixture of diastereomers in 80% combined yield for two steps17 (Scheme 4).
:syn over 10:1) separable mixture of diastereomers in 80% combined yield for two steps17 (Scheme 4).
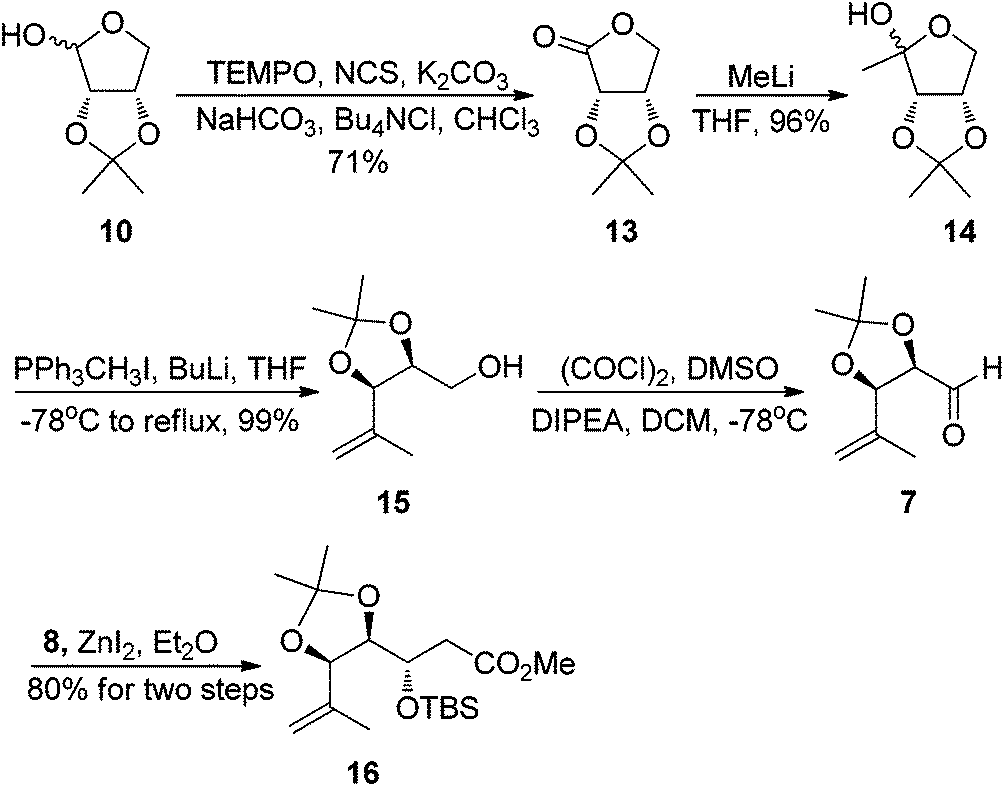 | ||
| Scheme 4 Synthesis of segment 16. | ||
The observed diastereoselection may arise from the si face addition via a Felkin–Anh type transition state. The enhancement of the anti-selectivity in the presence of ZnI2 can be explained by chelation with β-oxygen as in the transition state A, the re face of the aldehyde becomes much more crowded than the si face18 (Fig. 2).
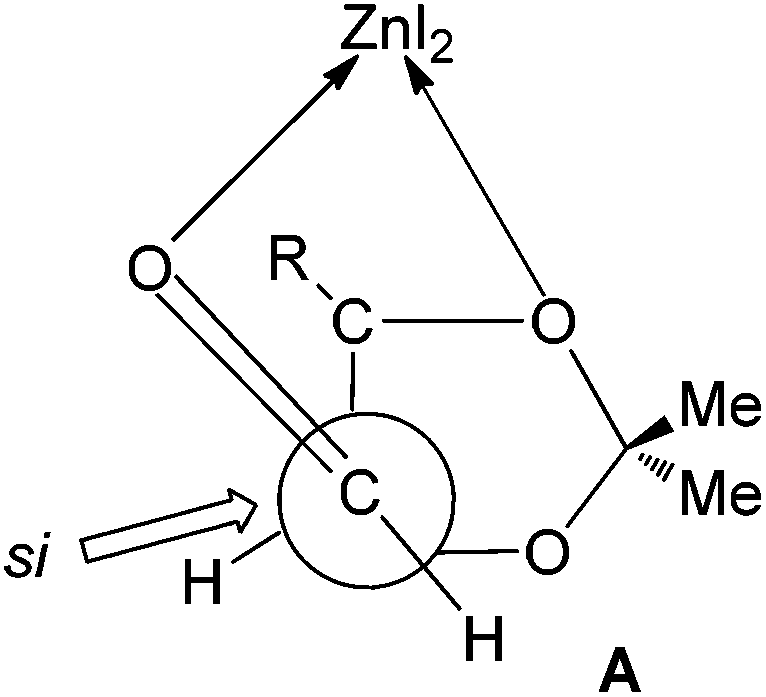 | ||
| Fig. 2 Transition states for Mukaiyama aldol reactions. | ||
With the two requisite fragments in hand, our attention was directed to their linkage using the Suzuki–Miyaura coupling reaction.19 Pleasingly, the linkage between vinyl iodide 4 and hydroboration product generated in situ from 16 proceeded well to produce 3 in 82% yield. The desired stereogenic center of the C6 methyl group20 and configuration of the E-olefinic bond (deduced from coupling constant J = 15.2 Hz in 1H NMR spectrum of the natural product) were established stereoselectively during this process.
The excellent diastereoselective of the C6 methyl group could be explained by the two possible transition states depicted in Fig. 3. Transition state S2 is favored over the diastereomeric transition state S1 since the nonbonding interactions between allylic diacetonide protected the hydroxyl group and boron substituents.20a,b
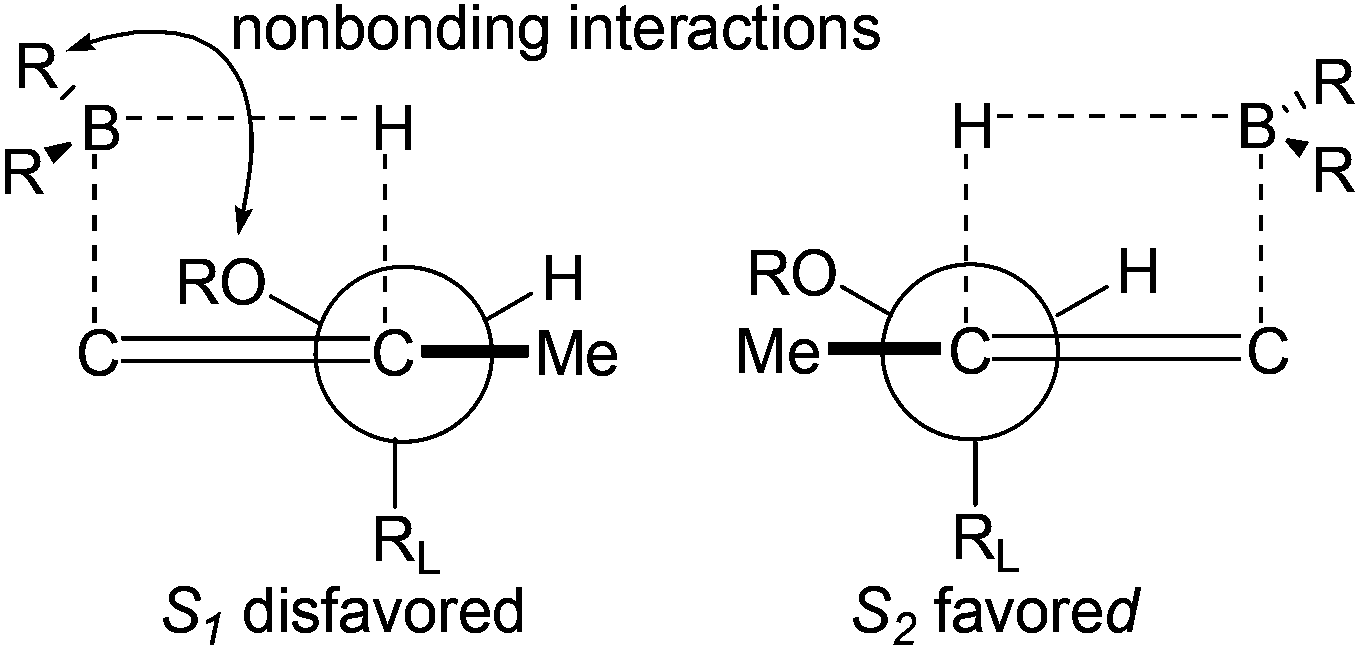 | ||
| Fig. 3 Transition states for hydroboration of alkene 16. | ||
At this point, the key core and all of the six stereogenic centers has been established, and the only task that remained was to remove the diacetonide, MOM and TBS protecting groups and γ-lactonization. To our delight, the process could be achieved by treatment of 3 with 2 N HCl in THF all in one pot to yield the natural product mupirocin H (2) in 86% yield.211H and 13C NMR spectra and other physical data of our synthetic compound were all in good agreement with those of the natural mupirocin H (2) (Scheme 5).
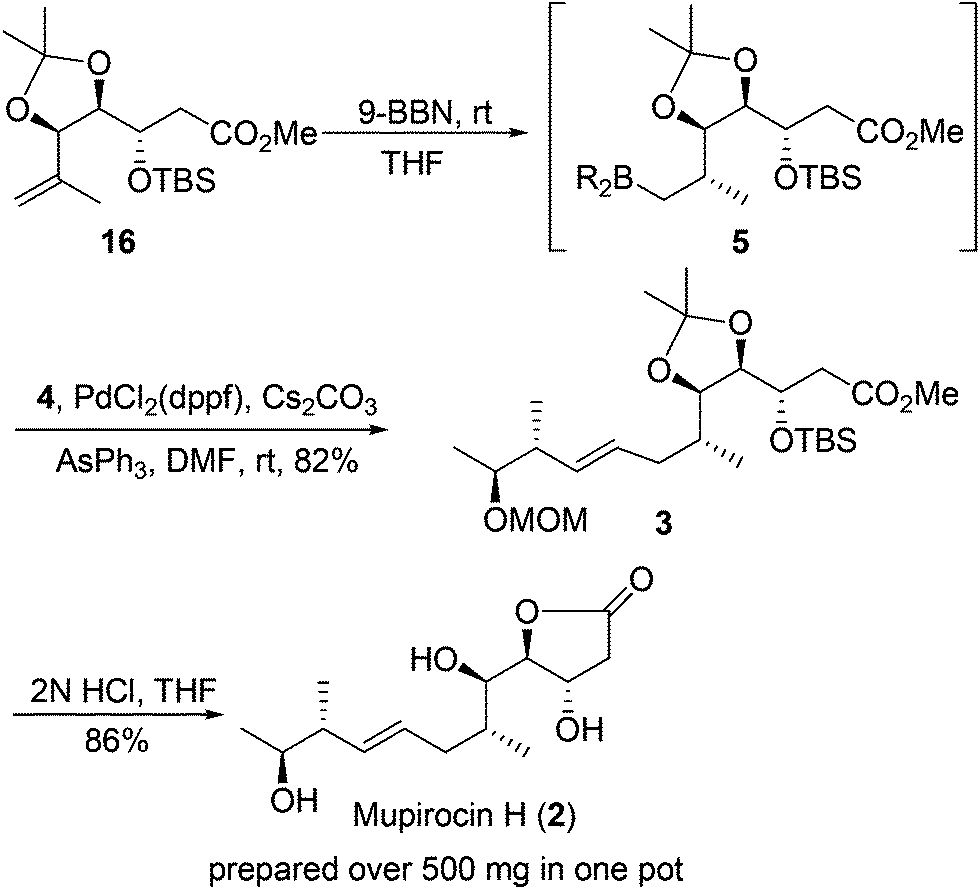 | ||
| Scheme 5 Suzuki–Miyaura coupling reaction and accomplish the synthesis of (+)-mupirocin H (2). | ||
Conclusions
In summary, a scalable and efficient approach for total synthesis of mupirocin H (2) has been achieved in 7 steps (longest linear sequence) with 39% overall yield. The synthetic sequence features a Suzuki–Miyaura coupling reaction for construction of the required E-double bond and a Mukaiyama aldol reaction for establishing the chiral center of C-3. Due to its high efficiency and overall yield, the developed pathway would enable large scale preparation of mupirocin H (2), offering convenience for further studies. The developed strategy is also suitable to synthesize other pseudomonic acid analogues.Acknowledgements
We are grateful for the financial support by the NSFC (21125207, 21102062, 21072086, 21102064), the MOST (2010CB833203), PCSIRT (IRT1138), the FRFCU (lzujbky-2013-49, lzujbky-2013-ct02), and Program 111.Notes and references
- A. Badder and C. Garre, Corresp.-Bl, Schweiz. Aerzte, 1887, 17, 385 Search PubMed.
- A. T. Fuller, G. Mellows, M. Woolford, G. T. Banks, K. D. Barrow and E. B. Chain, Nature, 1971, 234, 416 CrossRef CAS.
- (a) C. J. Arthur, A. Szafranska, S. E. Evans, S. C. Findlow, S. G. Burston, P. Owen, I. Clark-Lewis, T. J. Simpson, J. Crosby and M. P. Crump, Biochemistry, 2005, 44, 15414 CrossRef CAS PubMed; (b) P. Wattana-amorn, C. Williams, E. Płoskoń, R. J. Cox, T. J. Simpson, J. Crosby and M. P. Crump, Biochemistry, 2010, 49, 2186 CrossRef CAS PubMed; (c) P. T. Seden, J. Charmant and C. L. Willis, Org. Lett., 2008, 10, 1637 CrossRef CAS PubMed; (d) C. S. Barry, J. D. Elsworth, P. T. Seden, N. Bushby, J. R. Harding, R. W. Alder and C. L. Willis, Org. Lett., 2006, 8, 3319 CrossRef CAS PubMed; (e) C. M. Thomas, J. Hothersall, C. L. Willis and T. J. Simpson, Nat. Rev. Microbiol., 2010, 8, 281 CrossRef CAS PubMed.
- (a) Y. J. Class and P. DeShong, Chem. Rev., 1995, 95, 1843 CrossRef CAS and references cited therein; (b) C. McKay, T. J. Simpson, C. L. Willis, A. K. Forrest and P. J. O'Hanlon, Chem. Commun., 2000, 1109 RSC; (c) L. Innis, J. M. Plancher and I. E. Markó, Org. Lett., 2006, 8, 6111 CrossRef PubMed.
- (a) J. Wu, J. Hothersall, C. Mazzetti, Y. O'Connell, J. Shields, R. J. Cox, J. Crosby, T. J. Simpson, C. M. Thomas and C. L. Willis, Chem. Biol., 2008, 9, 1500 CAS; (b) C. T. Calderone, Nat. Prod. Rep., 2008, 25, 845 RSC.
- S. M. Cooper, R. J. Cox, J. Crosby, M. P. Crump, J. Hothersall, W. Laosripaiboon, T. J. Simpson and C. M. Thomas, Chem. Commun., 2005, 1179 RSC.
- J. Wu, S. M. Cooper, R. J. Cox, J. Crosby, M. P. Crump, J. Hothersall, T. J. Simpson, C. M. Thomas and C. L. Willis, Chem. Commun., 2007, 2040 RSC.
- S. Udawant and T. Chakraborty, J. Org. Chem., 2011, 76, 6331 CrossRef CAS PubMed.
- R. W. Scott, C. Mazzetti, T. J. Simpson and C. L. Willis, Chem. Commun., 2012, 48, 2639 RSC.
- M. Jacolot, M. Jean, N. Levoin and P. Weghe, Org. Lett., 2012, 14, 58 CrossRef CAS PubMed.
- A. J. Mancuso and D. Swern, Synthesis, 1981, 165 CrossRef CAS.
- K. Takai, K. Nitta and K. Utimoto, J. Am. Chem. Soc., 1986, 108, 7408 CrossRef CAS.
- L. Nagarapu, S. Karnakanti and R. Bantu, Tetrahedron, 2012, 68, 5829 CrossRef CAS PubMed.
- (a) P. L. Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem., 1987, 52, 2559 CrossRef; (b) J. Einhorn, C. Einhorn, F. Ratajczak and J.-L. Pierre, J. Org. Chem., 1996, 61, 7452 CrossRef CAS PubMed.
- S. Jenkinson, N. Jones, A. Moussa, A. J. Stewartb, T. Heinzc and G. Fleeta, Tetrahedron Lett., 2007, 48, 4441 CrossRef CAS PubMed.
- For a review, see: B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863 CrossRef CAS.
- (a) T. Mukaiyama, K. Narasaka and K. Banno, Chem. Lett., 1973, 1011 CrossRef CAS; (b) T. Mukaiyama, K. Banno and K. Narasaka, J. Am. Chem. Soc., 1974, 96, 7503 CrossRef CAS; (c) Y. Kita, O. Tamura, F. Itoh, H. Yasuda, H. Kishino, Y. Ke and Y. Tamura, J. Org. Chem., 1988, 53, 3 CrossRef; (d) T. Chakraborty and A. Chattopadhyay, J. Org. Chem., 2008, 73, 3578 CrossRef CAS PubMed; (e) K. Pulukuri and T. Chakraborty, Org. Lett., 2012, 14, 2858 CrossRef CAS PubMed.
- (a) M. Chgrest, H. Felkin and N. Prudent, Tetrahedron Lett., 1968, 2199 CrossRef; (b) N. T. Anh and O. Eisentein, Nouv. J. Chim., 1977, I, 61 Search PubMed.
- For selected reviews, see: (a) A. Suzuki, Angew. Chem., 2011, 123, 6854 ( Angew. Chem., Int. Ed. , 2011 , 50 , 6722 ) CrossRef; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (c) S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., 2001, 113, 4676 ( Angew. Chem., Int. Ed. , 2001 , 40 , 4544 ) CrossRef.
- (a) K. N. Houk, N. G. Rondan, U. Wu, J. T. Metz and M. N. Paddon-Row, Tetrahedron, 1984, 40, 2257 CrossRef CAS; (b) D. A. Evans, A. M. Ratz, B. E. Huff and G. S. Sheppard, J. Am. Chem. Soc., 1995, 117, 3448 CrossRef CAS; (c) I. Paterson and J. A. Chennon, Tetrahedron Lett., 1992, 33, 797 CrossRef CAS; (d) D. A. Evans and D. M. Fitch, J. Org. Chem., 1997, 62, 454 CrossRef CAS; (e) H. Sato, Y. S. Kim and M. Shibasaki, Tetrahedron Lett., 1999, 40, 2973 CrossRef CAS; (f) P. Phukan, S. Sasmal and M. E. Maier, Eur. J. Org. Chem., 2003, 1733 CrossRef CAS; (g) O. Loiseleur, G. Koch and T. Wagner, Org. Process Res. Dev., 2004, 8, 597 CrossRef CAS; (h) F.-M. Zhang, L. Peng, H. Li, A.-J. Ma, J.-B. Peng, J.-J. Guo, D. Yang, S.-H. Hou and Y.-Q. Tu, Angew. Chem., 2012, 121, 11004 ( Angew. Chem., Int. Ed. , 2012 , 51 , 10846 ) CrossRef.
- (a) M. Ahmed, B. P. Berry, T. J. Hunter, D. J. Tomcik and G. A. O'Doherty, Org. Lett., 2005, 7, 745 CrossRef CAS PubMed; (b) H. Li, J. Zheng, S. Xu, D. Ma, C. Zhao, B. Fang, X. Xie and X. She, Chem.–Asian J., 2012, 7, 2519 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3qo00038a |
| This journal is © the Partner Organisations 2014 |