Highly diasteroselective intermolecular 1,3-dipolar cycloaddition reactions of carbonyl ylides with aldimines to afford sterically disfavored cis-oxazolidines†
Xinfang
Xu
,
Xin
Guo
,
Xingchun
Han
,
Liping
Yang
and
Wenhao
Hu
*
Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, Shanghai, 200062, China. E-mail: whu@chem.ecnu.edu.cn; Fax: (+86) 021-62221237
First published on 30th January 2014
Abstract
Rhodium acetate and AgSbF6 co-catalyzed highly diastereoselective 1,3-dipolar cycloaddition reactions of carbonyl ylides with aldimines have been carried out to afford sterically disfavored oxazolidines via a [3 + 2] exo-addition process. Subsequent hydrolysis gave trans-β-amino-α-hydroxyl ester derivatives in high yields.
Carbonyl ylide dipoles are highly reactive and are important intermediates for 1,3-dipolar cycloadditions. They find widespread use in heterocyclic chemistry.1 These ylides are typically generated in situ from carbonyl compounds and metallocarbenoids. The bimolecular version of cycloaddition has been applied with great success in natural product synthesis.2 However, the cycloaddition of an intermolecularly generated carbonyl ylide with various dipolarophiles is still a challenge in terms of efficiency and selectivity.
The 1,3-dipolar cycloaddition reaction between carbonyl ylides and imines is one of the most useful protocols that have been used for the preparation of oxazolidines.3 Subsequent hydrolysis of the N,O-acetal ultimately yields β-amino alcohols, which are found in a vast array of biologically important compounds and are frequently used as building blocks in natural product synthesis.4 Surprisingly, very limited examples of cycloadditions involving imines and intermolecularly generated carbonyl ylide dipoles have been reported,3 although the analogous reactions of carbonyl ylide dipoles with aldehydes,5 alkynes, or alkenes6 have been extensively studied. The reported 1,3-dipolar cycloaddition of carbonyl ylides with benzylimines to give trans-4,5-oxazolidines (Scheme 1, Path A),3 was believed to arise from a sterically preferred metal-free endo-transition state (TS).7 It is worth noting that in this study, imines derived from aromatic amines were inactive dipolarophiles. Presumably, the conjugate resonance of the aromatic ring with the imine double bond stabilizes the imine moiety toward the reaction. It was also reported that Lewis acid additives disrupted the diastereoselectivity of the reaction.3e Here, we wish to report our continuing study on the cooperative-catalysis strategy,8 which facilitates the highly diastereoselective [3 + 2] exo-addition process of carbonyl ylides with imines (Path B).
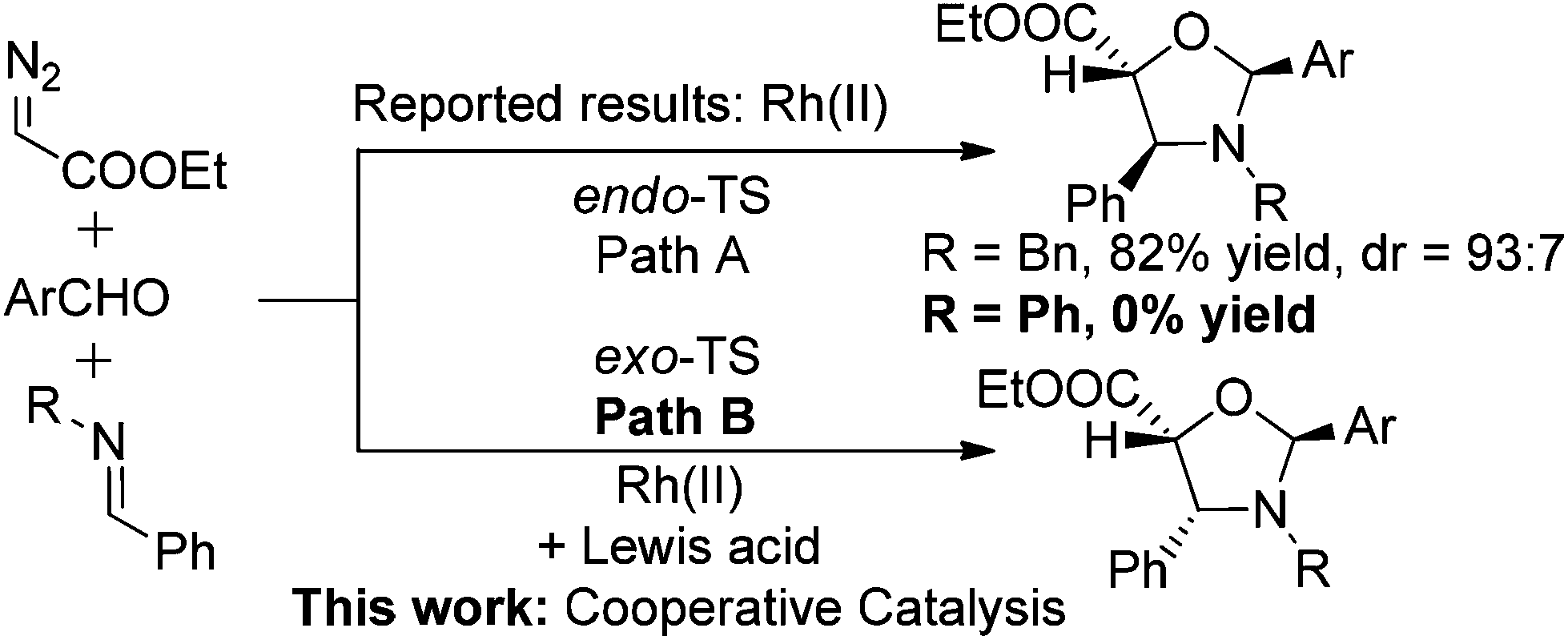 | ||
| Scheme 1 Rhodium-catalyzed 1,3-dipolar cycloaddition of carbonyl ylide dipoles with imines. | ||
In recent years, cooperative catalysis has gained much attention owing to its ability to enhance both selectivity and reactivity in organic reactions.9 In some cases of multi-component reactions (MCRs) in which three or more components are involved in one transition state (TS) to generate two or more chemical bonds simultaneously, cooperative catalysis provides an opportunity to control the reaction selectivity of the multi-bond formation process, as the appropriate combination of compatible co-catalysts can affect the intrinsic reaction kinetics in a designed manner by separately activating the desired component(s).8 In the transition-metal-catalyzed three-component reaction of a diazo compound, which comprises an aldehyde and an aldimine, there are possible side reactions, including aziridination,10 epoxidation,11 Mannich-type addition to the imine,12 aldol-type addition to the aldehyde,13 other carbonyl ylide or azomethine ylide cycloadditions with aldehydes or imines, respectively (Scheme 2),14 and others,15 that may compete with the desired cycloaddition of the carbonyl ylide with the aldimine. We envisioned that the complexity of this reaction system provides us with an opportunity to carry out the co-catalyzed multi-component strategy. In the current situation (Scheme 2), Rh2(OAc)4 is the catalyst of choice to generate the active carbonyl ylide dipole, and it should be possible to find a compatible co-catalyst to activate the aldimine substrate so as to achieve control of chemo- and diastereoselectivity.
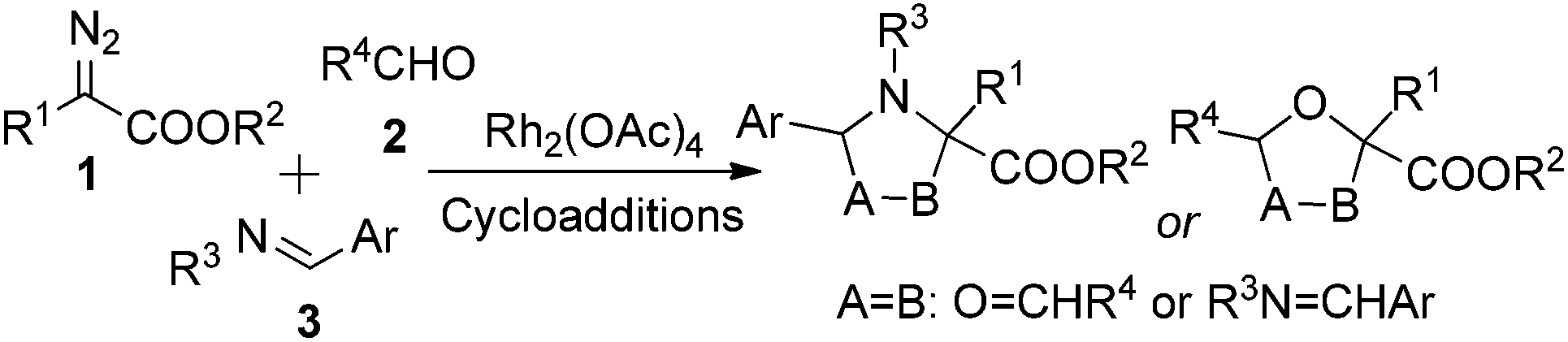 | ||
| Scheme 2 Possible [3 + 2] cycloaddition reactions of diazo compounds with aldehydes and imines. | ||
With the above goal, we began to study the three-component reactions using ethyl diazoacetate (1a, EDA), p-bromophenylaldehyde (2a), and an aniline-derived imine (3a) in the presence of Rh2(OAc)4 (2.0 mol%) to find that only epoxidation occurred (Table 1, entry 1). Next, a variety of Lewis acids were used as co-catalysts to activate the aldimine 3a. We found that in the presence of Yb(OTf)3, the 1,3-dipolar cycloaddition occurred to afford oxazolidine 4a in a 27% isolated yield with very low diastereoselectivity (4,5-cis/4,5-trans = 60/40). This promising outcome, despite the low yield and limited selectivity, clearly demonstrated the activation of imine 3a towards the 1,3-carbonyl dipole by a Lewis acid, and the slight, though noticeable, inversion of diastereoselectivity is comparable to those obtained in the reported results.16
|
|
|||
|---|---|---|---|
| Entry | Co-catalyst | Yieldb (%) | drc (cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans) :trans) |
| a Unless otherwise noted, the reaction was carried out by the addition of 1a (0.22 mmol) in CH2Cl2 (0.5 mL) to a mixture of 2a (0.22 mmol), 3a (0.20 mmol), 4 Å MS (0.1 g) Rh2(OAc)4 (2.0 mol%), and the co-catalyst (10.0 mol%) in 1.5 mL CH2Cl2 under an argon atmosphere for 1 h at 25 °C. b Isolated yield of 4a (based on limiting reagent 3a). c Determined by 1H NMR spectroscopy of the unpurified reaction mixture. d Isolated yield of the corresponding epoxide. e The reaction was carried out at 0 °C. f The reaction was carried out in the absence of Rh2(OAc)4, and no desired product was detected. | |||
| 1 | — | 71d | — |
| 2 | Yb(OTf)3 | 27 | 60:40 |
| 3 | Sc(OTf)3 | 33 | 63:37 |
| 4 | Zn(OTf)2 | 38 | 55:45 |
| 5 | AgOTf | 69 | 67:33 |
| 6 | AgBF4 | 51 | 90:10 |
| 7 | AgPF6 | 73 | 86:14 |
| 8 | AgSbF6 | 76 | >95:5 |
| 9e | AgSbF6 | 81 | >95:5 |
| 10e,f | AgSbF6 | ND | ND |
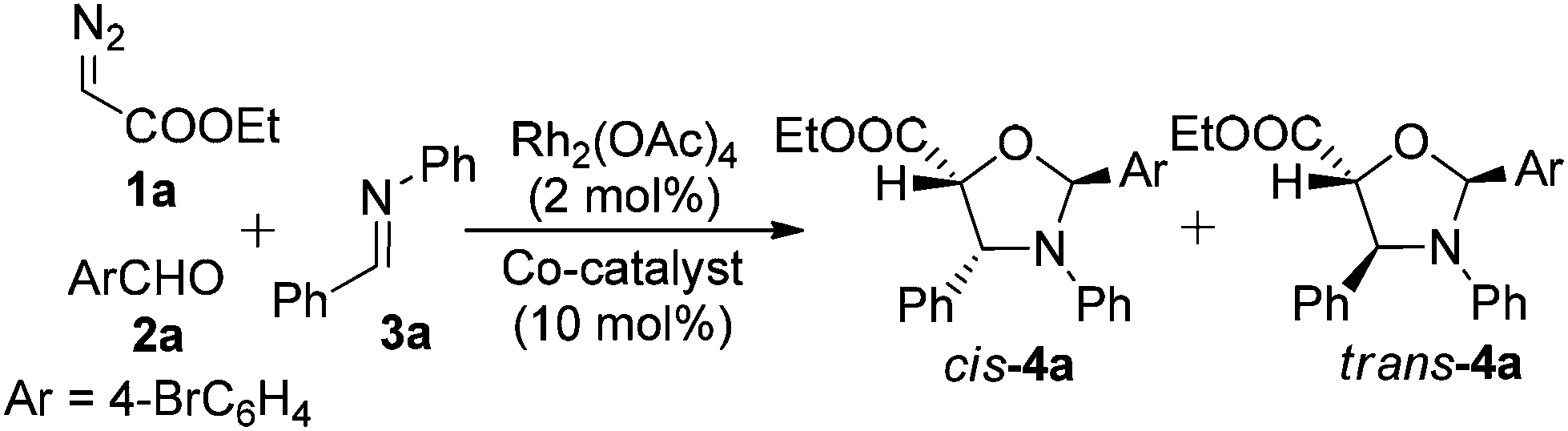
Other Lewis acid co-catalysts were tested: Sc(OTf)3 promoted the reaction in a similar manner to that of Yb(OTf)3, and Zn(OTf)2 effected a 10% improvement of the yield. When AgOTf was employed, a dramatic increase in the yield to 69% and a slight enhancement of the diastereomeric ratio (dr) to 67/33 (4,5-cis/4,5-trans) were observed. A further survey of a variety of silver salts revealed that AgSbF6 was the optimum co-catalyst, affording 4a in a 76% yield and in a cis/trans ratio as high as 95/5 (entry 8). When the reaction was carried out at 0 °C, the yield was further improved to 81% and the high levels of diastereoselectivity were maintained (entry 9). A control reaction was also carried out under the optimal conditions, in the absence of Rh2(OAc)4, which recovered the three starting materials with no trace of 4a being detected (entry 10). These findings rule out the possibility that the silver metal complex alone participates in the metal carbenoid formation and the subsequent carbonyl dipole formations.
The scope of this AgSbF6 and Rh2(OAc)4 co-catalyzed three-component cycloaddition was further explored with EDA and p-bromophenylaldehyde as the 1,3-carbonyl dipole precursor and various aldimines as dipolarophiles, using CH2Cl2 as a solvent and 4 Å MS as a water scavenger. The results are listed in Table 2. Imines containing both electron-poor and electron-rich arenes gave the corresponding products with excellent selectivity (dr > 95:5) and high yields (75–90%). Substrates with either an o or m substituent on Ar4 exhibit slightly decreased reactivity while keeping the same high levels of stereoselective control (entries 6, 7 and 8).
|
|
|||
|---|---|---|---|
| Entry | Ar3/Ar4 (3) | Yieldb (%) | drc |
| a Unless otherwise noted, the reaction was carried out by the addition of 1a (0.22 mmol) in CH2Cl2 (0.5 mL) to a mixture of 2a (0.22 mmol), 3 (0.20 mmol), 4 Å MS (0.1 g) Rh2(OAc)4 (2.0 mol%), and AgSbF6 (10.0 mol%) in 1.5 mL CH2Cl2 under an argon atmosphere for 1 h at 0 °C. b Isolated yield of 4. c Determined by 1H NMR spectroscopy of the unpurified reaction mixture. | |||
| 1 | Ph/Ph (3a) | 81 (4a) | >95:5 |
| 2 | Ph/4-BrC6H4 (3b) | 87 (4b) | >95:5 |
| 3 | Ph/4-ClC6H4 (3c) | 83 (4c) | >95:5 |
| 4 | Ph/3,4-2ClC6H3 (3d) | 82 (4d) | >95:5 |
| 5 | 4-ClC6H4/4-BrC6H4 (3e) | 76 (4e) | >95:5 |
| 6 | PMP/4-BrC6H4 (3f) | 90 (4f) | >95:5 |
| 7 | PMP/3-BrC6H4 (3g) | 78 (4g) | >95:5 |
| 8 | PMP/2-BrC6H4 (3h) | 75 (4h) | >95:5 |
| 9 | PMP/PMP (3i) | 89 (4i) | >95:5 |
| 10 | PMP/4-MeC6H4 (3j) | 78 (4j) | >95:5 |
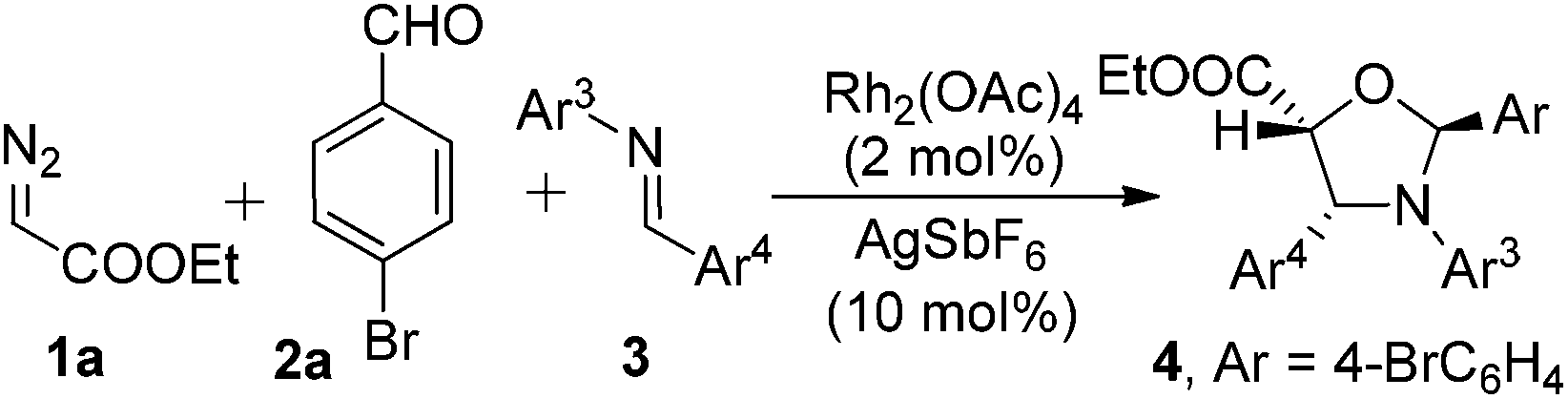
The relative stereochemistry of the major product was determined to be 2,5-trans and 4,5-cis via single-crystal X-ray analysis of 4f16 and the 1H NMR coupling data of J(4,5) of the corresponding major product is consistent with the 4,5-cis relative stereochemistry.17,18
Deprotection of the oxazolidines under acidic conditions offered the valuable trans-amino alcohol 5a in a high yield, substantiating a facile entry to this class of compounds.18,19 In the meantime, the stereochemistry of the minor isomer was deduced as 4,5-trans, since a diastereomeric mixture of 4a gave a diastereomeric mixture of 5a in the same dr, indicating that the relative stereochemistry of 4a is derived only from the 4,5-position.18
Compared to the previously reported Lewis acid (LA)-free endo-TS-favored addition,3 the dominant products were exo-TS adducts in this LA co-catalyzed three-component reaction. It is proposed that the cationic silver pre-organizes the thermally stable trans rhodium-free ylide7 and its cycloaddition partner in an exo-TS assembly, as depicted in Scheme 3, through coordination with both the ester carbonyl oxygen and the imine nitrogen.20 Thus, the ester group is brought to the same side of Ar4, and Ar2 is spontaneously forced to the same side as Ar3, leading to the formation of the exo-cycloaddition product. The intriguing effects of the LA in this system were underscored by the inversion of the trans/cis selection as well as the activation of the otherwise inert dipolarophiles.
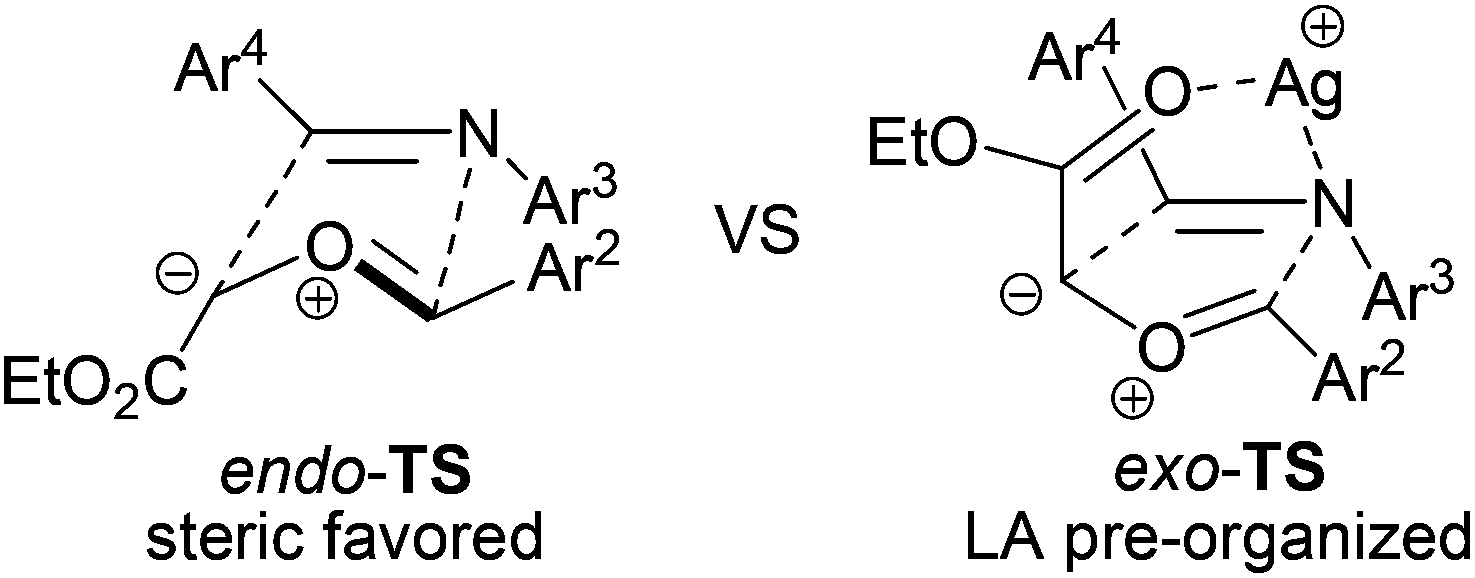 | ||
| Scheme 3 Proposed effect of the co-catalyst in the addition process. | ||
Chiral diazo compound 1b, derived from L-menthol, was employed to introduce the chirality in this system (Scheme 4). Its reactions with 2a and 3 furnished the chiral products 6a–c in good yields (62–73%). Only two diastereomers out of the eight possible isomers were observed in the cycloaddition reaction. In order to clarify the stereochemistry of the adducts, subsequent reduction and hydrolysis of a crude product mixture of 6a were carried out to yield the known amino alcohol 8 in 80% ee, as determined by chiral HPLC, indicating a 90:10 dr in the cycloaddition step.17 To ensure that no racemization occurred during the transformation, purified 6a was used to carry out the same transformation to give 8 in 100% ee. The physical and spectrometric data of 8 ([α]20D = +4.0°) agree with the data reported for (2S,3S)-8 {lit [α]20D = +4.0° for (2S,3S)-8 (c = 1, EtOH)}.21 Thus, the absolute configuration of the major isomer 6a was deduced as 2R,4S,5S, and the minor diastereomer of 6a from the cycloaddition as 2S,4R,5R. At this stage, by analogy with 6a, the absolute configurations of all the major cycloaddition products 6a–c were finally assigned, as shown in Scheme 4.
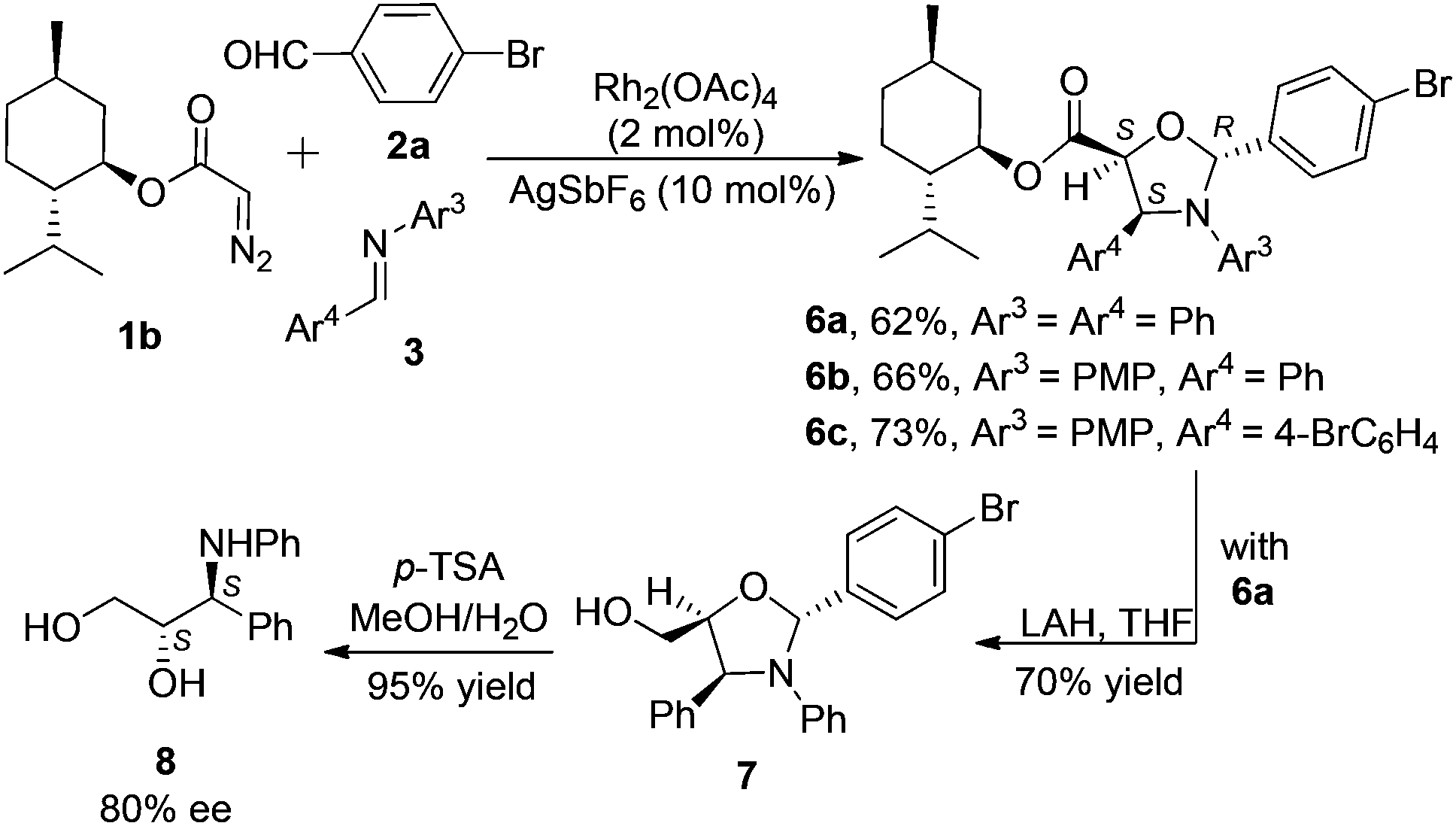 | ||
| Scheme 4 Asymmetric cycloadditions and subsequent transformations. | ||
In summary, we have presented intermolecular LA co-catalyzed highly diastereoselective 1,3-dipolar cycloadditions of carbonyl ylides with inert aldimines. This multi-component reaction allowed facile access to cis-oxazolidines and gave chiral α-hydroxyl-β-amino ester derivatives in good yields with a high level of control of the stereoselectivity. The otherwise sterically disfavored exo-TS operates in this system because of the effect of a cationic Lewis acid co-catalyst that presumably occurs through a dual functional mechanism of coordination and activation of [3 + 2] cycloaddition partners. Further work will focus on developing an enantioselective version of this methodology.
We are grateful for the financial support from the NSFC (21125209 and 21332003), the MOST of China (2011CB808600) and STCSM (12JC1403800), and support from Ministry of Education of China (20100076110005).
Notes and references
- For review: (a) M. C. McMills and D. Wright, in The Chemistry of Heterocyclic Compounds, ed. A. Padwa and W. H. Pearson, John Wiley & Sons, Hoboken, NJ, 2002, vol. 59, pp. 253–314 Search PubMed; (b) S. Muthusamy and J. Krishnamurthi, in Topics in Heterocyclic Chemistry, ed. B. U. W. Maes, Springer-Verlag, Berlin, Heidelberg, 2008, vol. 12, pp. 147–192. For application in total synthesis Search PubMed; (c) Y. Sugano, F. Kikuchi, A. Toita, S. Nakamura and S. Hashimoto, Chem.–Eur. J., 2012, 18, 9682 CrossRef CAS PubMed; (d) S. Nakamura, Y. Sugano, F. Kikuchi and S. Hashimoto, Angew. Chem., Int. Ed., 2006, 45, 6532 CrossRef CAS PubMed.
- For recent progress in [3 + 2] cycloadditions of carbonyl ylide dipoles: (a) A. Padwa, Prog. Heterocycl. Chem., 2009, 20, 20 CAS; (b) J. Zhou, Y. Liang, C. Deng, H. Zhou, Z. Wang, X. Sun, J. Zheng, Z. Yu and Y. Tang, Angew. Chem., Int. Ed., 2011, 50, 7874 CrossRef CAS PubMed; (c) C. Qin and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 14516 CrossRef CAS PubMed; (d) J. M. Mejía-Oneto and A. Padwa, Helv. Chim. Acta, 2008, 91, 285 CrossRef; (e) V. Navickas, D. B. Ushakov, M. E. Maier, M. Ströbele and H. J. Meyer, Org. Lett., 2010, 12, 3418 CrossRef CAS PubMed; (f) C. H. Kim, K. P. Jang, S. Y. Choi, Y. K. Chung and E. Lee, Angew. Chem., Int. Ed., 2008, 47, 4009 CrossRef CAS PubMed.
- (a) A. Padwa, P. Laura and A. S. Mark, J. Org. Chem., 1999, 64, 4079 CrossRef CAS; (b) H. Suga, Y. Ebiura, K. Fukushima, A. Kakehi and T. Baba, J. Org. Chem., 2005, 70, 10782 CrossRef CAS PubMed; (c) M. Sengodagounder, K. Janagiraman and S. Eringathodi, Synlett, 2005, 3002 Search PubMed . For intermolecular versions, see: ; (d) S. Torssell, M. Kienle and P. Somfai, Angew. Chem., Int. Ed., 2005, 44, 3096 CrossRef CAS PubMed; (e) S. Torssell and P. Somfai, Adv. Synth. Catal., 2006, 348, 2421 CrossRef CAS.
- (a) S. Kobayashi, H. Ishitani and M. Ueno, J. Am. Chem. Soc., 1998, 120, 431 CrossRef CAS; (b) J. Kobayashi, M. Nakamura, Y. Mori, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2004, 126, 9192 CrossRef CAS PubMed; (c) X. Liu, L. Lin and X. Feng, Acc. Chem. Res., 2011, 44, 574 CrossRef CAS PubMed; (d) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef CAS PubMed; (e) S. Sato, M. Tetsuhashi, K. Sekine, H. Miyachi, M. Naito, Y. Hashimotoa and H. Aoyama, Bioorg. Med. Chem., 2008, 16, 4685 CrossRef CAS PubMed; (f) D. L. Vaux and A. Strasser, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2239 CrossRef CAS; (g) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef PubMed.
- (a) Z. Chen, Z. Tian, J. Zhang, J. Ma and J. Zhang, Chem.–Eur. J., 2012, 18, 8591 CrossRef CAS PubMed; (b) H. Tsutsui, N. Shimada, T. Abe, M. Anada, M. Nakajima, S. Nakamur, H. Nambu and S. Hashimoto, Adv. Synth. Catal., 2007, 349, 521 CrossRef CAS; (c) A. DeAngelis, P. Panne, G. P. A. Yap and J. M. Fox, J. Org. Chem., 2008, 73, 1435 CrossRef CAS PubMed; (d) Z. Chen, L. Wei and J. Zhang, Org. Lett., 2011, 13, 1170 CrossRef CAS PubMed.
- (a) J. Zhang, Z. Chen, H. Wu and J. Zhang, Chem. Commun., 2012, 48, 1817 RSC; (b) A. Padwa, J. P. Snyder, E. Curtis, S. M. Sheehan, K. J. Worsencroft and C. O. Kappe, J. Am. Chem. Soc., 2000, 122, 8155 CrossRef CAS; (c) K. Takeda, T. Oohara, N. Shimada, H. Nambu and S. Hashimoto, Chem.–Eur. J., 2011, 17, 13992 CrossRef CAS PubMed; (d) D. M. Hodgson, D. Angrish and A. H. Labande, Chem. Commun., 2006, 627 RSC; (e) S. A. Bonderoff and A. Padwa, Org. Lett., 2013, 15, 4114 CrossRef CAS PubMed; (f) A. D. Angelis, M. T. Taylor and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 1101 CrossRef PubMed; (g) R. Liu, M. Zhang and J. Zhang, Chem. Commun., 2011, 47, 12870 RSC.
- M. P. Doyle, D. C. Forbes, M. N. Protopopova, S. A. Stanley, M. M. Vasbinder and K. R. Xavier, J. Org. Chem., 1997, 62, 7210 CrossRef CAS PubMed.
- (a) W. Hu, X. Xu, J. Zhou, W. Liu, H. Huang, J. Hu, L. Yang and L. Gong, J. Am. Chem. Soc., 2008, 130, 7782 CrossRef CAS PubMed; (b) X. Xu, J. Zhou, L. Yang and W. Hu, Chem. Commun., 2008, 6564 RSC; (c) C. Jing, D. Xing, Y. Qian, T. Shi, Y. Zhao and W. Hu, Angew. Chem., Int. Ed., 2013, 52, 9282 CrossRef PubMed; (d) J. Jiang, H. Xu, J. Xi, B. Ren, F. Lv, X. Guo, L. Jiang, Z. Zhang and W. Hu, J. Am. Chem. Soc., 2011, 133, 8428 CrossRef CAS PubMed; (e) M. Terada and Y. Toda, Angew. Chem., Int. Ed., 2012, 51, 2093 CrossRef CAS PubMed; (f) L. Ren, X. Lian and L. Gong, Chem.–Eur. J., 2013, 19, 3315 CrossRef CAS PubMed.
- For reviews, see: (a) J. Zhou, Chem.–Asian J., 2010, 5, 422 CrossRef CAS PubMed; (b) Y. J. Park, J. W. Park and C. H. Jun, Acc. Chem. Res., 2008, 41, 222 CrossRef CAS PubMed; (c) S. Ko, B. Kang and S. Chang, Angew. Chem., Int. Ed., 2005, 44, 455 CrossRef CAS PubMed; (d) E. V. Beletskiy, C. Sudheer and C. J. Douglas, J. Org. Chem., 2012, 77, 5884 CrossRef CAS PubMed; (e) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633 RSC; (f) M. Jeganmohan, S. Bhuvaneswari and C. H. Cheng, Angew. Chem., Int. Ed., 2009, 48, 391 CrossRef CAS PubMed; (g) X. Wu, M. Li and L. Gong, Acta Chim. Sin., 2013, 71, 1091 CAS.
- (a) T. Hashimoto, N. Uchiyama and K. Maruoka, J. Am. Chem. Soc., 2008, 130, 14380 CrossRef CAS PubMed; (b) X. Zeng, X. Zeng, Z. Xu, M. Lu and G. Zhong, Org. Lett., 2009, 11, 3036 CrossRef CAS PubMed; (c) S. P. Bew, S. A. Fairhurst, D. L. Hughes, L. Legentil, J. Liddle, P. Pesce, S. Nigudkar and M. A. Wilson, Org. Lett., 2009, 11, 4552 CrossRef CAS PubMed; (d) Z. Lu, Y. Zhang and W. D. Wulff, J. Am. Chem. Soc., 2007, 129, 7185 CrossRef CAS PubMed.
- (a) W. Liu, B. Lv and L. Gong, Angew. Chem., Int. Ed., 2009, 48, 6503 CrossRef CAS PubMed; (b) Z. Li, J. Zhang, W. Hu, Z. Chen and X. Yu, Synlett, 2005, 1711 CAS; (c) X. Xu, W. Hu, P. Y. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2011, 50, 11152 CrossRef CAS; (d) A. E. Russell, J. Brekan, L. Gronenberg and M. P. Doyle, J. Am. Chem. Soc., 2004, 69, 5269 CAS.
- (a) T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2007, 129, 10054 CrossRef CAS PubMed; (b) Y. Zhang and J. Wang, Chem. Commun., 2009, 5350 RSC.
- (a) B. M. Trost, S. Malhotra and B. A. Fried, J. Am. Chem. Soc., 2009, 131, 1674 CrossRef CAS PubMed; (b) F. Benfatti, S. Yilmaz and P. G. Cozzi, Adv. Synth. Catal., 2009, 351, 1763 CrossRef CAS; (c) M. L. Kantam, V. Balasubrahmanyam, K. B. S. Kumar, G. T. Venkanna and F. Figueras, Adv. Synth. Catal., 2007, 349, 1887 CrossRef CAS; (d) W. Yao and J. Wang, Org. Lett., 2003, 5, 1527 CrossRef CAS PubMed.
- [3 + 2]-Cycloaddition of azomethine ylide, see: (a) X. Wu, L. Li and J. Zhang, Chem. Commun., 2011, 47, 7824 RSC; (b) G. Song, D. Chen, Y. Su, K. Han, C. Pan, A. Jia and X. Li, Angew. Chem., Int. Ed., 2011, 50, 7791 CrossRef CAS PubMed; (c) M. P. Doyle, M. Yan, W. Hu and L. S. Gronenberg, J. Am. Chem. Soc., 2003, 125, 4692 CrossRef CAS PubMed; (d) X. Jing, C. He, D. Dong, L. Yang and C. Duan, Angew. Chem., Int. Ed., 2012, 51, 10127 CrossRef CAS PubMed; (e) F. Shi, S. Luo, Z. Tao, L. He, J. Yu, S. Tu and L. Gong, Org. Lett., 2011, 13, 4680 CrossRef CAS PubMed.
- (a) D. Chen, P. Wu and L. Gong, Org. Lett., 2013, 15, 3958 CrossRef CAS PubMed; (b) W. Li, X. Liu, X. Hao, X. Hu, Y. Chu, W. Cao, S. Qin, C. Hu, L. Lin and X. Feng, J. Am. Chem. Soc., 2011, 133, 15268 CrossRef CAS PubMed; (c) T. Hashimoto, H. Miyamoto, Y. Naganawa and K. Maruoka, J. Am. Chem. Soc., 2009, 131, 11280 CrossRef CAS PubMed; (d) Z. Cao, X. Wang, C. Tan, X. Zhao, J. Zhou and K. Ding, J. Am. Chem. Soc., 2013, 135, 8197 CrossRef CAS PubMed.
- The relative structure of 4,5-cis-4a was confirmed by single-crystal X-ray diffraction analysis of its bromo-derivative 4f, and CCDC 970866 contains the supplementary crystallographic data for 4f.
- The J(4,5) = 6.0–7.5 Hz coupling constant is consistent with 4,5-cis relative stereochemistry, see: M. Murakami, H. Ito and Y. Ito, J. Org. Chem., 1993, 58, 6766 CrossRef CAS.
- For details, see ESI.†.
- For compound 5, see: (a) S. B. Pujala and A. K. Chakraborti, J. Org. Chem., 2007, 72, 3713 CrossRef PubMed; (b) Z. Guo, T. Shi, J. Jiang, L. Yang and W. Hu, Org. Biomol. Chem., 2009, 7, 5028 RSC; (c) J. Wang, F. D. Rochon, Y. Yang, L. Hua and M. M. Kayser, Tetrahedron: Asymmetry, 2007, 18, 1115 CrossRef CAS PubMed.
- A similar chelation model with a Ag salt has been reported in the reaction of azomethine ylide with aldehyde: B. Seashore-Ludlow, S. Torsell and P. Somfai, Eur. J. Org. Chem., 2010, 3927 CrossRef CAS.
- (a) J. Yoshimura, Y. Ohgo and T. Sato, J. Am. Chem. Soc., 1964, 86, 3858 CrossRef CAS; (b) R. C. Harden, T. J. Hodgkinson, A. McKillop, W. G. Prowse and M. W. J. Urquhart, Tetrahedron, 1997, 53, 21 CrossRef CAS; (c) P. D. Buttero, G. Molteni and M. Roncoroni, Tetrahedron Lett., 2006, 47, 2209 CrossRef PubMed; (d) K. Surendra, N. S. Krishnaveni and K. R. Rao, Synlett, 2005, 506 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 970866 (4f) and other spectra. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qo00040k |
| This journal is © the Partner Organisations 2014 |