Copper-catalyzed cross-coupling of bromozinc-difluoromethylphosphonate with iodo/bromo-aryl triazenes†
Zhang
Feng
,
Yu-Lan
Xiao
and
Xingang
Zhang
*
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: xgzhang@mail.sioc.ac.cn; Fax: (+86)-21-6416-6128; Tel: (+86)-21-5492-5333
First published on 23rd December 2013
Abstract
A versatile method for the preparation of aryldifluoromethylphosphonates has been developed. This represents the first example of copper-catalyzed difluoroalkylation of ArBr and provides a useful and facile access to diverse aryldifluoromethylphosphonates that are difficult to prepare otherwise.
Incorporation of a difluoromethylene group (CF2) into an organic molecule can not only enhance its neighbouring group acidity, but also act as an isopolar–isosteric substitute for oxygen.1 It has been used as one strategy for modification of biologically active compounds, such as enzyme inhibitors and anti-cancer agents.2 Among the gem-difluoroalkylated compounds, aryldifluoromethylphosphonates are an important class of compounds because of their significant bioactivities as protein tyrosine phosphate (PTP) inhibitors.3 However, compared to trifluoromethylation of organic substrates,4 strategies for incorporation of a CF2 group into aromatics have been less explored.5 To date, only a few methods with limited substrate scope have been developed for the preparation of aryldifluoromethylphosphonates.6 Hence, it is highly desirable to develop new methods for their widespread synthetic applications. Herein, we report a copper-catalyzed cross-coupling of bromozincdifluoromethylphosphonate with iodo/bromo-aryltriazenes. This protocol represents a first example of copper-catalyzed difluoroalkylation of ArBr and provides a useful and facile access to diverse aryldifluoromethylphosphonates that are difficult to prepare otherwise.
Several years ago, Shibuya reported a method for the preparation of aryldifluoromethylphosphonates through copper-mediated cross-coupling between aryl iodides and bromozinc-difluoromethylphosphonate.6e However, the use of excess copper salt and the limited substrate scope restrict its widespread synthetic applications. From a new practical and academic point of view, a copper-catalyzed process would be attractive. But it remains challenging as the difluoromethylphosphonate copper complex is prone to decompose to a radical or form a dimer.7 Very recently, we reported a first example of the preparation of aryldifluoromethylphosphonates through copper-catalyzed cross-coupling between iodobenzoates and bromozinc-difluoromethylphosphonate, in which the carboxylate ester was used as a directing group.8 Despite the usefulness of this method, the carboxylate ester is not easily removed, thus restricting its wide application in medicinal chemistry. Inspired by this work, we envisioned that if a directing group not only benefits the copper based catalytic cycle, but also serves as a versatile functional group for various transformations, a diversity of aryldifluoromethylphosphonates would thus be easily accessed.
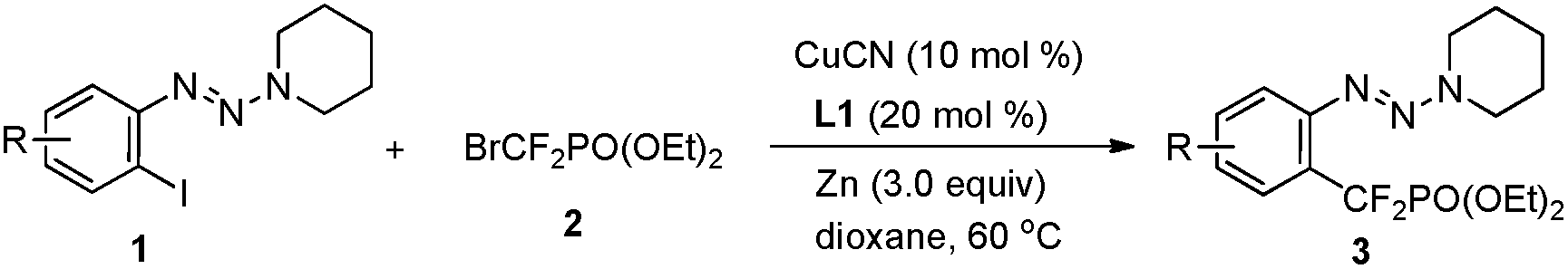
Accordingly, diisopropylamine-derived triazene 1a was chosen as the model substrate, because the triazene moiety can be easily removed or transformed into various functional groups (Table 1).9,10 Furthermore, the nitrogen atom in the triazene moiety may chelate and direct delivery of an organocopper complex, so as a result a copper-catalyzed cross-coupling between aryl halides and BrZnCF2PO(OEt)24 would be possible.11
|
|
||||
|---|---|---|---|---|
| Entry | 1, R | [Cu] | Ligand | 3, Yieldb (%) |
| a Reaction conditions (unless otherwise specified): 1 (0.3 mmol, 1.0 equiv.), 2 (2.0 equiv.), Zn (2.0 equiv.), [Cu] (10 mol%), ligand (20 mol%), dioxane (2.0 mL), 21 h, 60 °C. b NMR yield determined by 19F NMR using fluorobenzene as an internal standard and number in parenthesis is isolated yield. c 3.0 equiv. of 2 and 3.0 equiv. of Zn were used. d 2.5 equiv. of 2, 2.5 equiv. of Zn and 10 mol% of L1 were used. e 2.5 equiv. of 2, 2.5 equiv. of Zn and 20 mol% of L1 were used. f The reaction was conducted in the absence of Zn. | ||||
| 1 | 1a, iPr | CuI | Phen | 3aa, 10 |
| 2 | 1a, iPr | CuBr | Phen | 3aa, 14 |
| 3 | 1a, iPr | CuBr2 | Phen | 3aa, 26 |
| 4 | 1a, iPr | CuCN | Phen | 3aa, 28 |
| 5 | 1b, (CH2)5 | CuBr2 | Phen | 3ba, 50 |
| 6 | 1b, (CH2)5 | CuCN | Phen | 3ba, 73 |
| 7 | 1c, (CH2)4 | CuCN | Phen | 3ca, 67 |
| 8 | 1d, Me | CuCN | Phen | 3da, 65 |
| 9 | 1b, (CH2)5 | CuCN | — | 3ba, 61 |
| 10c | 1b, (CH2)5 | CuCN | Phen | 3ba, 79 |
| 11c | 1b, (CH2)5 | CuCN | L1 | 3ba, 97 (91) |
| 12c | 1b, (CH2)5 | CuCN | L2 | 3ba, 88 |
| 13c | 1b, (CH2)5 | CuCN | L3 | 3ba, 90 (82) |
| 14c | 1b, (CH2)5 | CuCN | L4 | 3ba, 75 |
| 15c | 1b, (CH2)5 | CuCN | TMEDA | 3ba, 59 |
| 16d | 1b, (CH2)5 | CuCN | L1 | 3ba, 61 |
| 17e | 1b, (CH2)5 | CuCN | L1 | 3ba, 76 |
| 18c | 1b, (CH2)5 | — | L1 | 3ba, NR |
| 19f | 1b, (CH2)5 | CuCN | L1 | 3ba, NR |
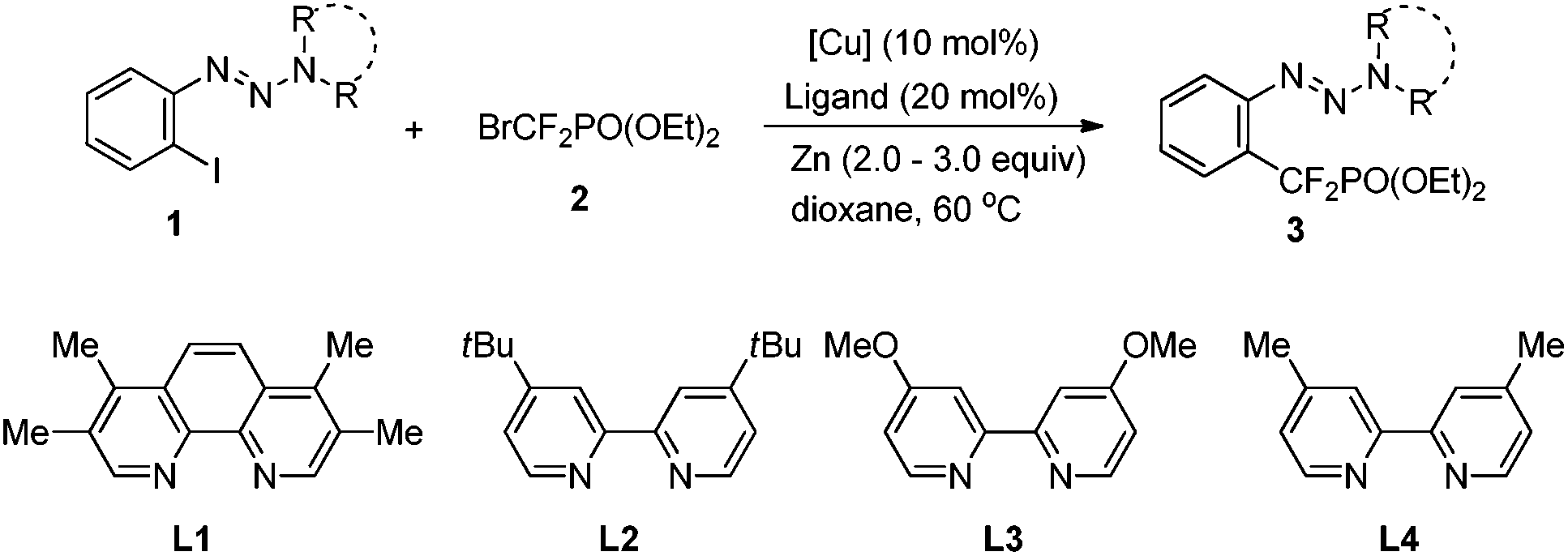
Although a poor yield was obtained when 1a was treated with 2.0 equiv. of bromozinc-difluoromethylphosphonate 4 in the presence of CuI (10 mol%) and 1,10-phenanthroline (Phen) in dioxane at 60 °C (Table 1, entry 1), we were delighted to find that an improved yield (73% determined by 19F NMR) was obtained by employing piperidine-derived triazene 1b as a substrate and CuCN as a catalyst (Table 1, entry 6) (For details see ESI† and Table 1, entries 2–5). Other triazenes 1, such as pyrrolidine-, and dimethylamine-derived triazenes 1c and 1d, showed less effect (Table 1, entries 7–8). The absence of Phen led to a lower yield, thus demonstrating the important role of the diamine ligand in the organocopper complex (Table 1, entry 9). To improve the reaction efficiency further, a series of diamine ligands previously demonstrated to have a beneficial effect on stabilization of organocopper species was investigated (Table 1, entries 11–15). Among the ligands tested, 3,4,7,8-tetramethyl-1,10-phenanthroline L1 showed the best catalytic effect with a 91% isolated yield of 3b obtained (Table 1, entry 11).12 We supposed that the increasing electron-density at the copper center resulting from the chelation of diamine ligand L1 with copper may stabilize the difluoromethylphosphonate copper complex and facilitate the catalytic cycle.13 No desired product was obtained without Cu or Zn species (Table 1, entries 18–19), thus demonstrating that the reaction proceeds via a copper involved catalytic cycle, and the generation of the nucleophile BrZnCF2PO(OEt)24 is essential for the reaction.
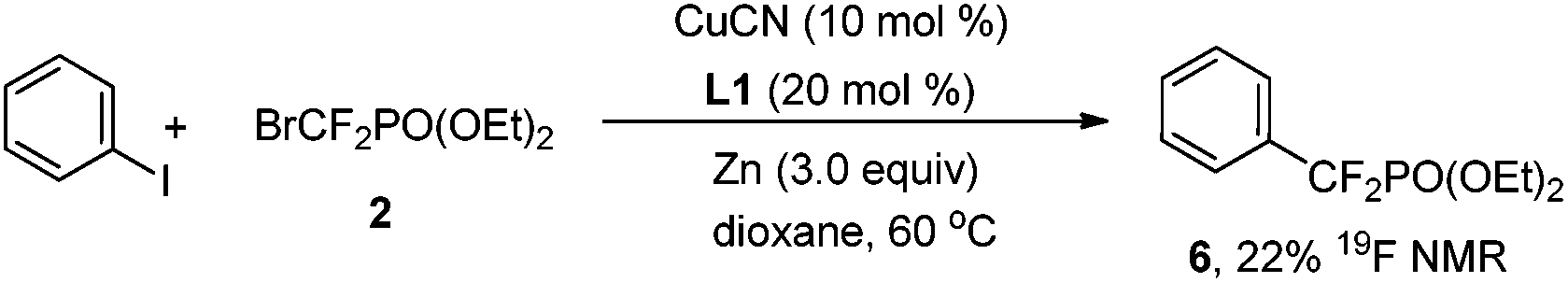
To demonstrate the pivotal role of the triazene moiety in this copper-catalyzed reaction, iodobenzene was further examined (eqn (1)). However, only 22% yield (determined by 19F NMR) of 6 was obtained when the reaction was carried out under the optimal reaction conditions. Thus, this finding implied that the triazene moiety indeed functions as a directing group and facilitates the present catalytic cycle.
 | (1) |
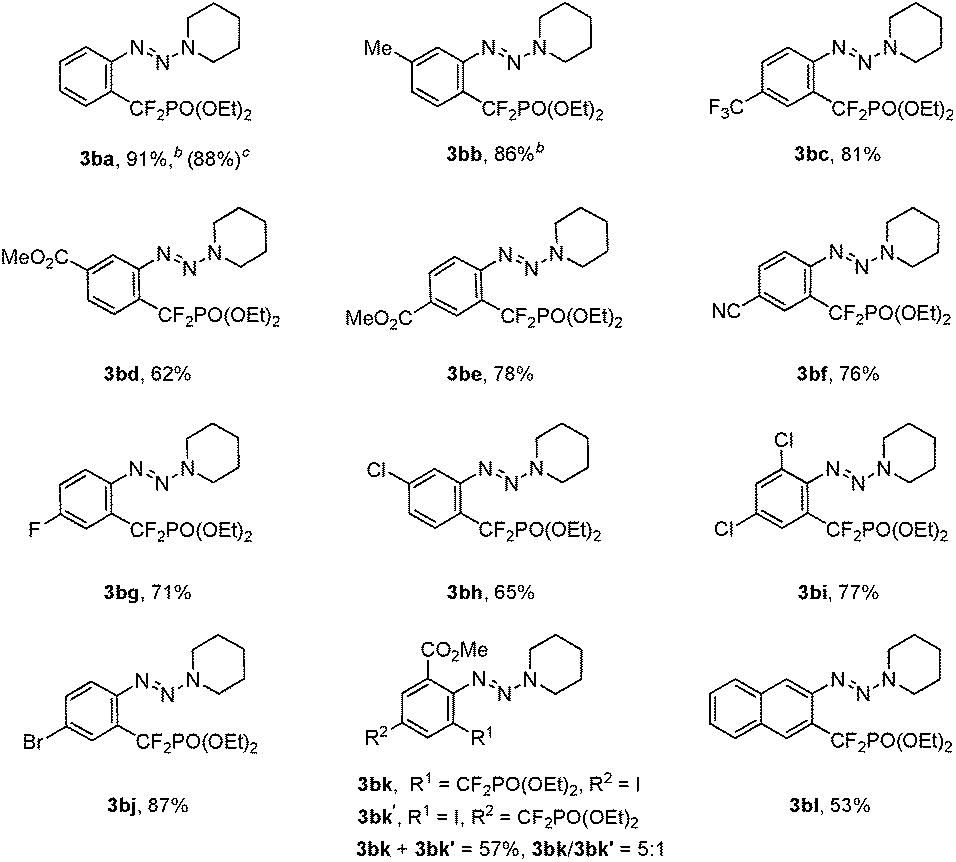
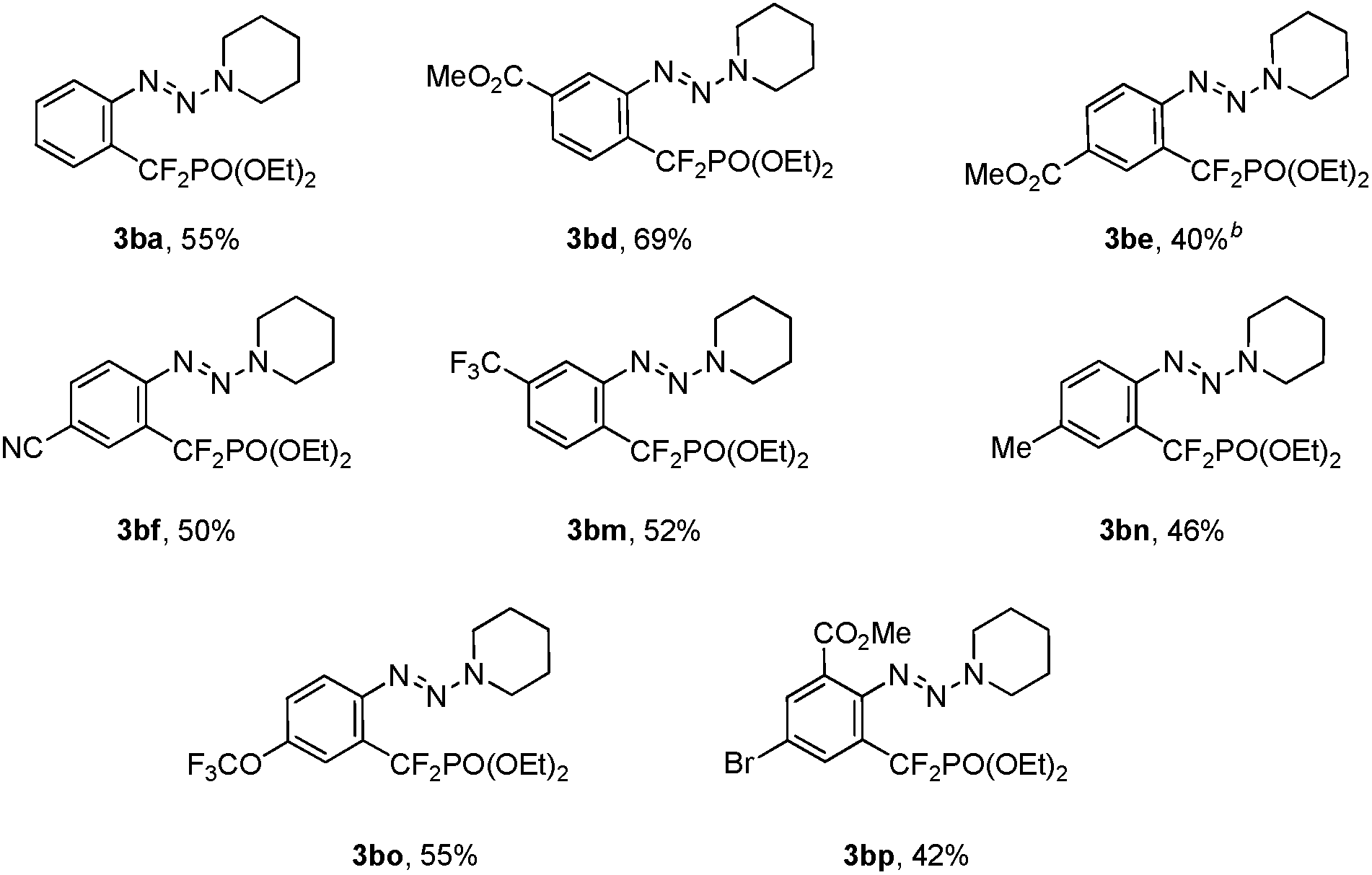
To ascertain the scope of this method, a variety of iodoaryl triazenes were investigated, and the representative results are illustrated in Table 2. Generally, the electronic effect of the substituents on the aromatic ring does not influence the reaction efficiency, and moderate to good yields of 3 were obtained. It is noteworthy that important functional groups such as ester, nitrile, chloride, bromide are compatible with the reaction conditions (3bd–3bj), thus providing opportunities for downstream transformations. Furthermore, good chemo- and regio-selectivities can be observed for diiodide substituted aryltriazene 1bk, in which the iodide ortho to the triazene moiety is the primary reaction site (3bk). The usefulness of this method is also demonstrated by the facile access to 3ba on a gram scale, thus indicating the good reliability of the process.
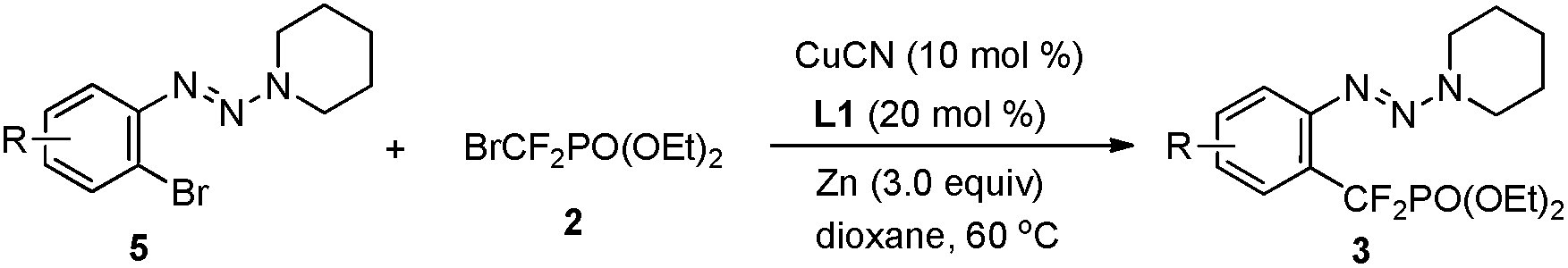
In addition to the demonstrated scope of this strategy, a variety of bromoaryl triazenes 5 were tested (Table 3). To our delight, under the optimized reaction conditions, the corresponding desired products 3 still could be obtained in moderate yields with excellent functional group compatibility. In particular, the intact bromide in 3bp provides a good platform for further functionalization (3bp), thus illustrating the advantages of this strategy. To the best of our knowledge, this is the first example of copper-catalyzed difluoroalkylation of an aryl bromide. Even for transition-metal-catalyzed trifluoromethylation of aryl bromides, only rare examples have been reported so far.14 Therefore we believe that these intriguing results would prompt much wider examination of copper-catalyzed fluoroalkylation of aryl bromides.
The importance and usefulness of this strategy can also be highlighted by the rapid access to diverse aryldifluoromethylphosphonates from the simple compound 3ba through transformations of the triazene moiety. It should be mentioned that these newly resulting aryldifluoromethylphosphonates are difficult to obtain otherwise. As shown in Scheme 1, the triazene moiety of 3ba can be easily removed with BF3·Et2O in dioxane.9 The formation of new C–C bonds through transformation of the triazene moiety also proceeded smoothly.15 For example, olefination of 3ba with styrene in the presence of Pd(OAc)2 (10 mol%) and BF3·Et2O in EtOH afforded alkenylated aryldifluoromethylphosphonate 7 in a high yield. The conversion of the triazene moiety into iodide,16 followed by a Sonogashira reaction, afforded compound 8 with high efficiency. Furthermore, arylation of 3ba with different arylboronic acids also proceeded smoothly.
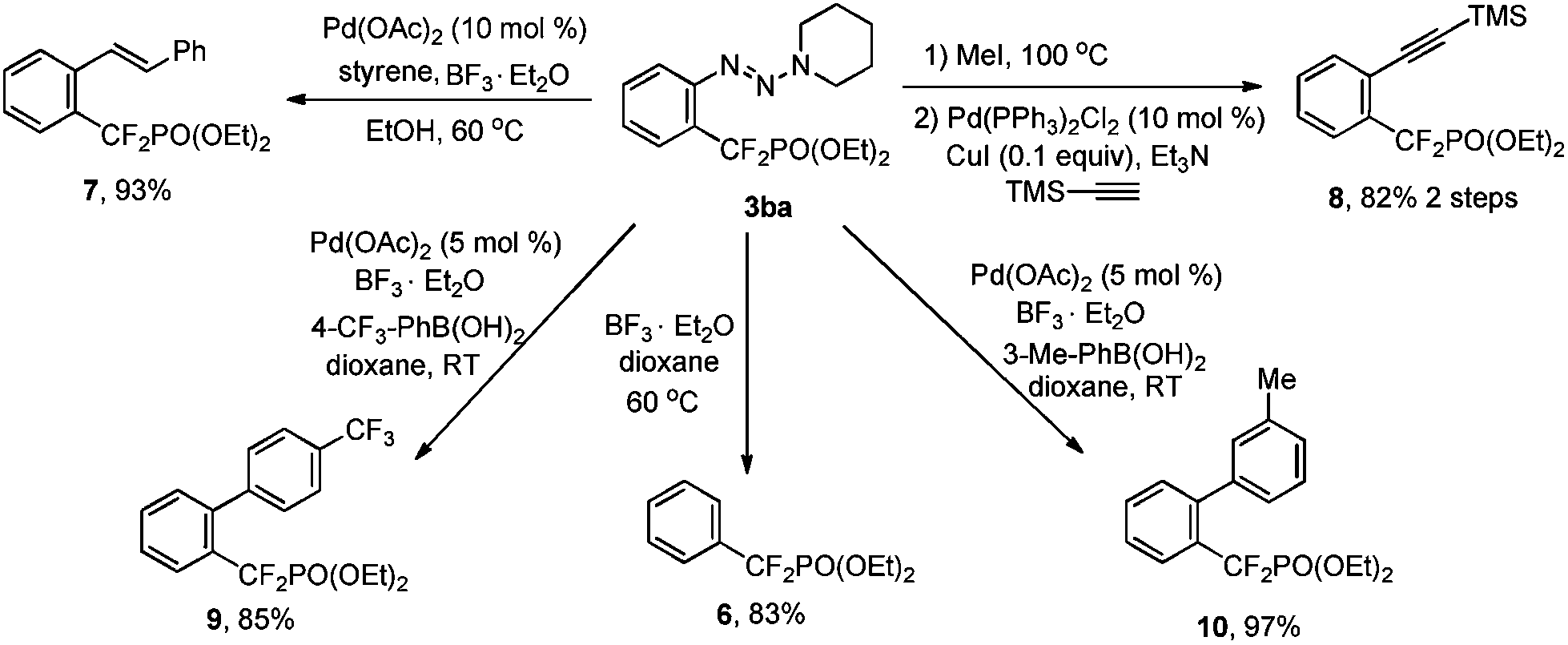 | ||
| Scheme 1 Synthesis of diversified aryldifluoromethylphosphonates through transformation of the triazene moiety. | ||
To demonstrate the importance and versatility of this method further, selective functionalization strategies were performed to access highly functionalized molecules that are of great interest in the life sciences. As shown in Scheme 2, selective arylation of compound 3bj with arylboronate 1117 in the presence of Pd(OAc)2 (5 mol%) and PPh3 (10 mol%) afforded compound 12 with high chemoselectivity. Subsequently, conversion of the triazene moiety into an alkene catalyzed by Pd(OAc)2 (10 mol%) and BF3·Et2O afforded bioactive compound derivative 13 in high efficiency. Alternatively, through tuning the reaction conditions, the difunctionalization of compound 3bj can also be started with a triazene moiety. After replacing the triazene moiety with an aryl group through a Suzuki reaction, another different aryl group can also be introduced at the bromide site. Thus, these strategies provide a vigorous tool for medicinal chemistry.
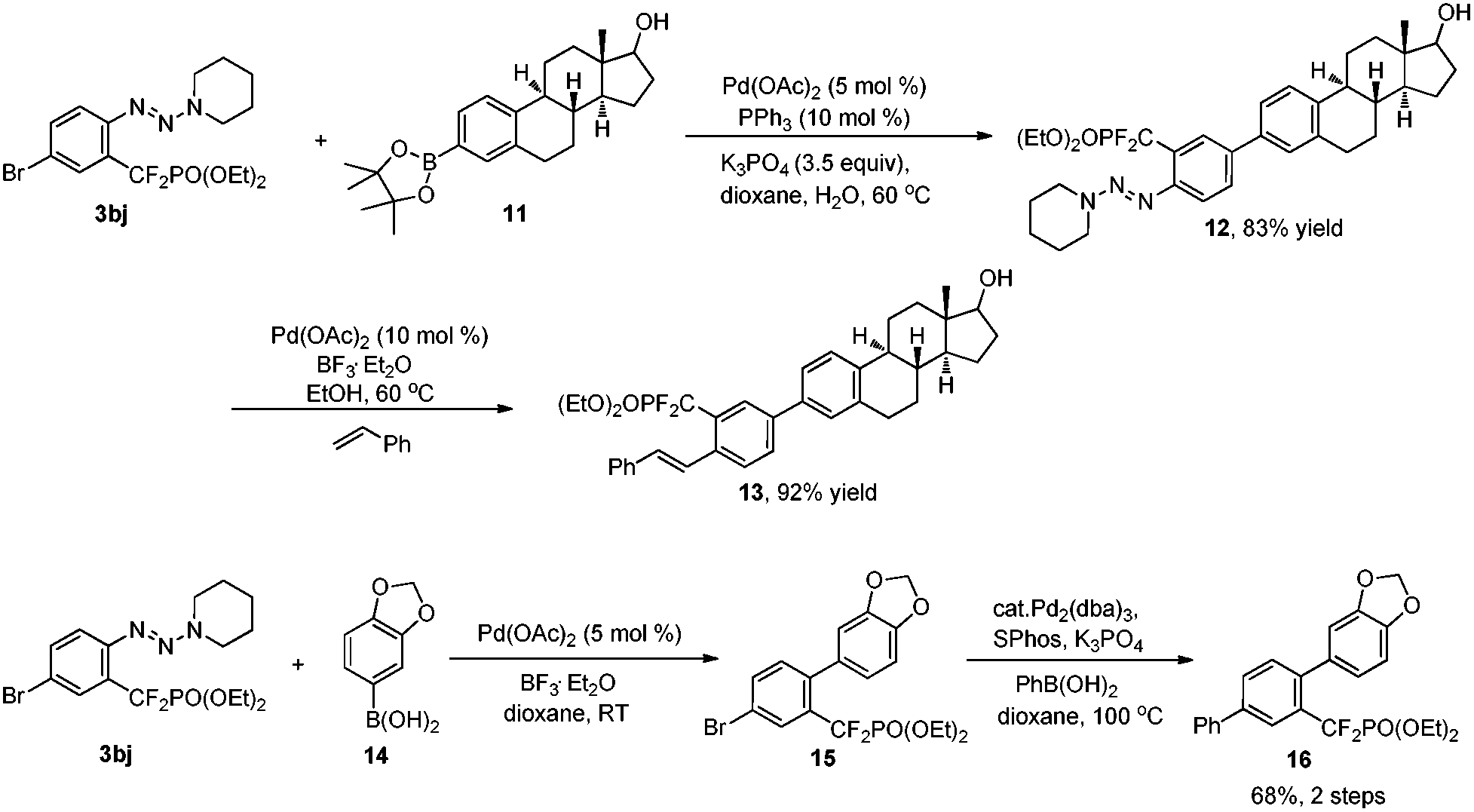 | ||
| Scheme 2 Selective difunctionalization of aryldifluoromethylphosphonate 3bj. | ||
Conclusions
In conclusion, we have developed an efficient and versatile method for the preparation of aryldifluoromethylphosphonates through copper-catalyzed difluoroalkylation of iodo/bromo-aryl triazenes. This represents the first example of copper-catalyzed difluoroalkylation of ArBr. The triazene moiety not only dramatically improves the reaction efficiency, but also provides a highly valuable platform to access diverse aryldifluoromethylphosphonates that are difficult to obtain otherwise. The notable features of this protocol are its high reaction efficiency, simple catalytic system, excellent functional group compatibility, and high versatility with tunable reaction conditions to give diverse structures. We believe that this protocol should not only be useful for drug discovery and development, but also prompt research in copper-catalyzed fluoroalkylation of aryl bromides.Acknowledgements
The National Basic Research Program of China (973 Program (no. 2012CB821600)), the NSFC (nos 21172242 and 21332010), and SIOC are gratefully acknowledged for funding this work.Notes and references
- (a) C. M. Blackburn, D. A. England and F. Kolkmann, J. Chem. Soc., Chem. Commun., 1981, 930 RSC; (b) G. M. Blackburn, D. E. Kent and F. Kolkmann, J. Chem. Soc., Perkin Trans. 1, 1984, 1119 RSC.
- (a) L. W. Hertel, J. S. Kroin, J. W. Missner and J. M. Tustin, J. Org. Chem., 1988, 52, 2406 CrossRef; (b) F. Akahoshi, A. Ashimori, H. Sakashita, T. Yoshimura, M. Eda, T. Imada, M. Nakajima, N. Mitsutomi, S. Kuwahara, T. Ohtsuka, C. Fukaya, M. Miyazaki and N. Nakamura, J. Med. Chem., 2001, 44, 1297 CrossRef CAS PubMed; (c) A. A. Makarov, P. O. Tsvetkov, C. Villard, D. Esquieu, B. Pourroy, J. Fahy, D. Braguer, V. Peyrot and D. Lafitte, Biochemistry, 2007, 46, 14899 CrossRef CAS PubMed.
- For reviews, see: (a) Z.-Y. Zhang, Acc. Chem. Res., 2003, 36, 385 CrossRef CAS PubMed; (b) T. R. Burke and K. Lee, Acc. Chem. Res., 2003, 36, 426 CrossRef CAS PubMed; (c) V. D. Romanenko and V. P. Kukhar, Chem. Rev., 2006, 106, 3868 CrossRef CAS PubMed . For recent papers, see: ; (d) S. Hikishima, M. Hashimoto, L. Magnowska, A. Bzowska and T. Yokomatsu, Bioorg. Med. Chem. Lett., 2010, 18, 2275 CAS; (e) P. K. Mandal, W. S.-L. Liao and J. S. McMurray, Org. Lett., 2009, 11, 3394 CrossRef CAS PubMed; (f) S. Mitra and A. M. Barrios, ChemBioChem, 2008, 9, 1216 CrossRef CAS PubMed.
- (a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (c) F.-L. Qing, Chin. J. Org. Chem., 2012, 32, 815 CrossRef CAS.
- For selected examples: (a) X. Zhang, H. Xia, X. Dong, J. Jin, W.-D. Meng and F.-L. Qing, J. Org. Chem., 2003, 68, 9026 CrossRef CAS PubMed; (b) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465 RSC; (c) K. Fujikawa, Y. Fujioka, A. Kobayashi and H. Amii, Org. Lett., 2011, 13, 5560 CrossRef CAS PubMed; (d) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524 CrossRef CAS PubMed; (e) G. K. S. Prakash, S. K. Ganesh, J.-P. Jones, A. Kulkarni, K. Masood, J. K. Swabeck and G. A. Olah, Angew. Chem., Int. Ed., 2012, 51, 12090 CrossRef CAS PubMed; (f) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494 CrossRef CAS PubMed.
- (a) M. S. Smyth and T. R. Burke, Tetrahedron Lett., 1994, 35, 551 CrossRef CAS; (b) D. Solas, R. L. Hale and D. V. Patel, J. Org. Chem., 1996, 61, 1537 CrossRef CAS; (c) S. D. Taylor, C. C. Kotoris, A. N. Dinaut and M.-J. Chen, Tetrahedron, 1998, 54, 1691 CrossRef CAS; (d) W. Qiu and D. J. Burton, Tetrahedron Lett., 1996, 37, 2745 CrossRef CAS; (e) T. Yokomatsu, T. Murano, K. Suemune and S. Shibuya, Tetrahedron, 1997, 53, 815 CrossRef CAS; (f) X. Jiang, L. Chu and F.-L. Qing, New J. Chem., 2013, 37, 1736 RSC.
- T. Yokomatsu, K. Suemune, T. Murano and S. Shibuya, J. Org. Chem., 1996, 61, 7207 CrossRef CAS.
- Z. Feng, F. Chen and X. Zhang, Org. Lett., 2012, 14, 1938 CrossRef CAS PubMed.
- For transformation of triazenes, see: reviews, (a) D. B. Kimball and M. M. Haley, Angew. Chem., Int. Ed., 2002, 41, 3338 CrossRef CAS; (b) S. Brase, Acc. Chem. Res., 2004, 37, 805 CrossRef CAS PubMed ; For selected papers, ; (c) T. B. Patrick, R. P. Willaredt and D. J. DeGonia, J. Org. Chem., 1985, 50, 2232 CrossRef CAS; (d) M. L. Gross, D. H. Blank and W. M. Welch, J. Org. Chem., 1993, 58, 2104 CrossRef CAS.
- Iodo/bromo-aryl triazenes 1 and 5 can be easily prepared from ortho-iodoaniline, see ESI.†.
- For the use of triazene as a directing group, see: (a) A. Hafner and S. Brase, Angew. Chem., Int. Ed., 2012, 51, 3713 CrossRef CAS PubMed; (b) C. Wang, H. Chen, Z. Wang, J. Chen and Y. Huang, Angew. Chem., Int. Ed., 2012, 51, 7242 CrossRef CAS PubMed; (c) A. Hafner, A. Bihlmeier, M. Nieger, W. Klopper and S. Brase, J. Org. Chem., 2013, 78, 7938 CrossRef CAS PubMed.
- Due to the formation of some protonated HCF2PO(OEt)2, 3.0 equiv. of 2 and 3.0 equiv. of Zn were needed.
- For reviews of copper-catalyzed cross-coupling reactions, see: (a) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13 RSC; (b) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed; (c) K. Kunz, U. Scholz and D. Ganzer, Synlett, 2003, 2428 CrossRef CAS; (d) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed; (e) D. W. Ma and Q. A. Cai, Acc. Chem. Res., 2008, 41, 1450 CrossRef CAS PubMed; (f) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed.
- B. S. Samant and G. W. Kabalka, Chem. Commun., 2011, 47, 7236 RSC.
- (a) S. Brase and M. Schroen, Angew. Chem., Int. Ed., 1999, 38, 1071 CrossRef CAS; (b) T. Saeki, E.-C. Son and K. Tamao, Org. Lett., 2004, 6, 617 CrossRef CAS PubMed.
- P. Wautelet, J. Le Moigne, V. Videva and P. Turek, J. Org. Chem., 2003, 68, 8025 CrossRef CAS PubMed.
- For the synthesis of arylboronate 11, see: E. Ascic, S. T. Le Quement, M. Ishoey, M. Daugaard and T. E. Nielsen, ACS Comb. Sci., 2012, 14, 253 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, and analytical data for all new compounds. See DOI: 10.1039/c3qo00044c |
| This journal is © the Partner Organisations 2014 |