Access to pyridines via DMAP-catalyzed activation of α-chloro acetic ester to react with unsaturated imines†
Lin
Hao‡
a,
Xingkuan
Chen‡
a,
Shaojin
Chen
a,
Ke
Jiang
b,
Jaume
Torres
b and
Yonggui Robin
Chi
*a
aDivision of Chemistry & Biological Chemistry, School of Physical & Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: robinchi@ntu.edu.sg
bSchool of Biological Sciences, Nanyang Technological University, Singapore 637551, Singapore
First published on 27th January 2014
Abstract
Facile access to trisubstituted pyridines from α-chloro acetic ester and unsaturated imines is achieved. DMAP-catalyzed activation of ester to form an enolate intermediate constitutes a key reaction step. On the application side, the wide availability and low cost of the substrates and catalysts make this method very attractive.
Functionalized pyridines are important building blocks and subunits of natural products and synthetic pharmaceuticals (Fig. 1a).1 Effective catalytic access to pyridines is therefore of considerable research interest. Transition metal-mediated cycloaddition reactions constitute a major effort for the assembly of substituted pyridines.2 Typically, relatively high reaction temperatures are necessary and the residual transition metal left with products may not be desirable in these otherwise very successful metal-based approaches. Organocatalytic methods for pyridine synthesis have recently emerged. The mild reaction conditions and non-toxic nature of the organocatalysts make this approach highly promising, especially for the synthesis of pyridines bearing functional groups or for medical uses. Loh and co-workers reported amine-catalyzed aza-Rauhut–Currier reaction of allenoates and α,β-unsaturated imines (1-azadienes) to generate functionalized pyridines.3 Smith recently reported isothiourea-catalyzed pyridine synthesis by reacting anhydrides with 1-azadienes.4
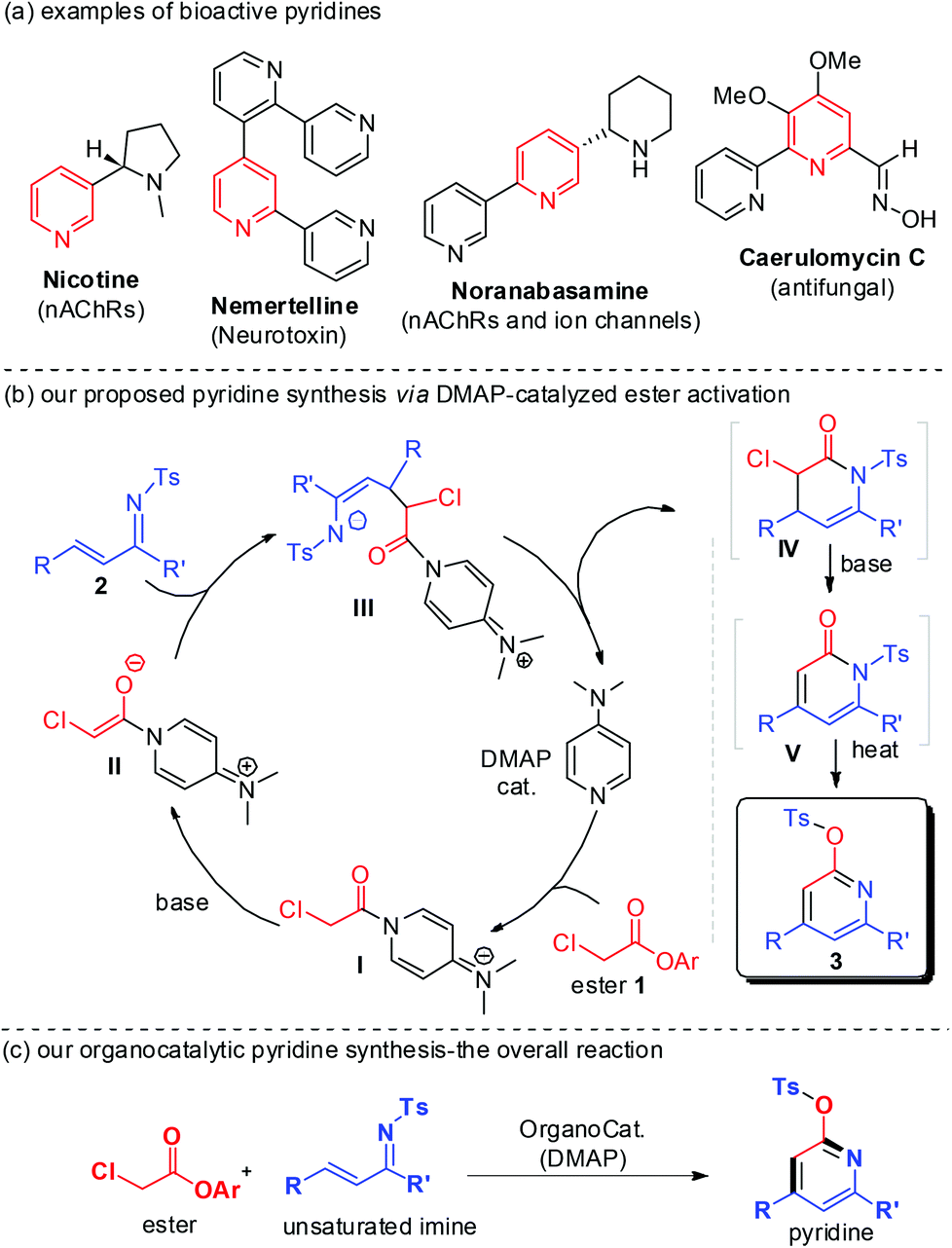 | ||
| Fig. 1 Examples of bioactive pyridines and our organocatalytic approach for pyridine synthesis. | ||
We are interested in the organocatalytic activation of readily available carboxylic esters for facile access to useful molecules.5 We have recently developed N-heterocyclic carbene (NHC)-catalyzed activation of esters5a–e and unsaturated esters,5f,g including unusual β-sp3 carbon activation5e of saturated esters. Herein, we report an organocatalytic activation of esters to form enolate intermediates by using DMAP as an organocatalyst (Fig. 1b). The reaction of α-chloro acetic ester with unsaturated imines effectively forms substituted pyridines (Fig. 1c). The ester and imine substrates can be easily prepared, and the DMAP organocatalyst is commercially available at low cost ($ 0.05 per gram from Alibaba).
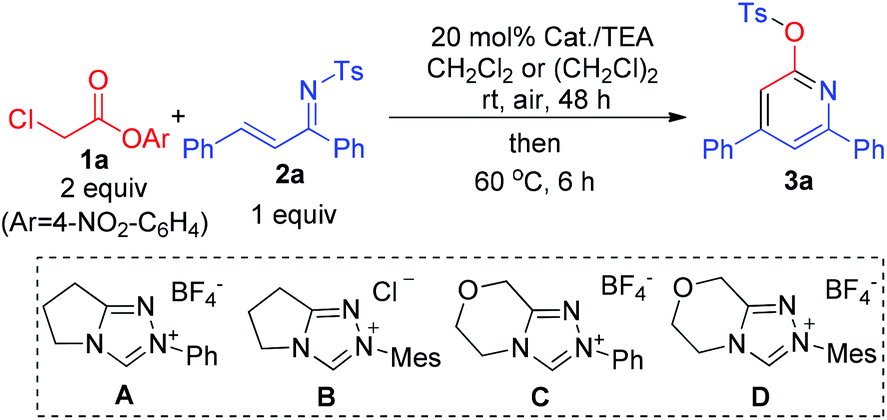
Experimentally, ester 1a prepared from inexpensive and commercially available α-chloro acetic acid ($ 0.73 per kg from Alibaba) was chosen as our substrate to react with 2a as a model unsaturated imine. No formation of pyridine product 3a was observed when bases (such as Cs2CO3, DIPEA, etc.; in the absence of DMAP) were used for a direct α-CH deprotonation of ester 1a to react with 2a (Table 1, entry 1). N-heterocyclic carbene (NHC) organocatalysts, found effective in our earlier studies on ester activation,5 were then evaluated. The use of triazolium-based NHC catalysts A–D led to 3a with low to moderate yields (entries 2–5). Although further development of NHC catalysts could likely lead to effective reactions, we decided to move towards a different direction in search of a cheaper organocatalyst. At last, we found that by using DMAP6 as an ester-activating catalyst and triethylamine (TEA) as a base, pyridine 3a could be obtained in 54% isolated yield for a reaction carried out in CH2Cl2 at room temperature and 24 hours (entry 6). An improved yield (66%) could be obtained when the reaction time was prolonged to 48 hours (entry 7). A switch of solvent from CH2Cl2 to (CH2Cl)2 led to 3a with 75% yield (entry 8). Increasing the reaction temperature led to a decreased yield (entry 9).
|
|
|||
|---|---|---|---|
| Entry | Conditions | Solvent | Yieldb [%] |
| a General conditions (unless otherwise specified): 1a (0.20 mmol), 2a (0.10 mmol), solvent (0.40 mL), reacted at room temperature for 48 h and then at 60 °C for 6 h. In all the above cases, the conversion of ester 1a was >90%. b Isolated yield (except entries 1–3, which were estimated via1H NMR analysis). c Bases such as DIPEA (5 eq.), TEA (5 eq.), DBU (2 eq.), TBD (2 eq.), Cs2CO3 (2 eq.) and K2CO3 (2 eq.) were tested. DIPEA = N,N-diisopropylethylamine, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene, TBD = 1,5,7-triazabicyclo-[4.4.0]dec-5-ene, TEA = triethyl amine. | |||
| 1 | 2–5 equiv. basesc (24 h) | CH2Cl2 | <5 |
| 2 | NHC A (24 h) | CH2Cl2 | 10 |
| 3 | NHC B (24 h) | CH2Cl2 | 4 |
| 4 | NHC C (24 h) | CH2Cl2 | 36 |
| 5 | NHC D (24 h) | CH2Cl2 | 53 |
| 6 | DMAP (24 h) | CH2Cl2 | 54 |
| 7 | DMAP (48 h) | CH2Cl2 | 66 |
| 8 | DMAP (48 h) | (CH2Cl)2 | 75 |
| 9 | DMAP (60 °C, 24 h) | (CH2Cl)2 | 55 |
Mechanistically (as illustrated in Fig. 1b), the reaction of substrate ester 1 and imine 2 initially formed a lactam IV (unstable, not isolable) that underwent E2-elimination to afford adduct V (isolable). N- to O-tosyl transfer7 of V effectively led to pyridine 3a at an elevated temperature. All the transformations from 1a and 2a to pyridine 3a were performed in a “single-step” operation. Notably, when only a base catalyst (a weak base such as TEA or a strong base such as DBU or NaOCH3) was used, no pyridine product could be formed. The addition of a DMAP organocatalyst to the ester substrate (to form intermediate I) and subsequent formation of DMAP-bound ester enolate intermediate II are necessary for this reaction. In other words, the DMAP catalyst not only facilitates ester α-CH deprotonation but also helps to modulate the reactivity of the DMAP-bound enolate intermediate in this reaction.
Examples of the unsaturated imine substrates that worked well under the optimal conditions (Table 1, entry 8) are shown in Scheme 1. When the R and/or R′ groups on the imines bear electron-donating substituents, the corresponding pyridine products (3b–e) were obtained with moderate to good yields. Replacing the phenyl group by a bulkier napthyl substituent was tolerated (3f, 3g). The use of electron-deficient imines bearing electron-withdrawing substituents afforded products with higher yields (3h–j). The imines containing heterocyclic rings (3k–n) such as furanyl or pyridinyl units were also suitable substrates. The pyridine products (3k–n) that resulted from these imines are amenable to further transformations such as Suzuki couplings to prepare terpyridine derivatives.8 The sulfonyl tosyl group on the imine nitrogen can be replaced by a methyl (3p) or p-methoxybenzenesulfonyl (3q) unit. As a note, the use of chloro acetic esters with an α-alkyl substituent (e.g. ethyl or benzyl unit) led to no pyridine products under current conditions.
 | (1) |
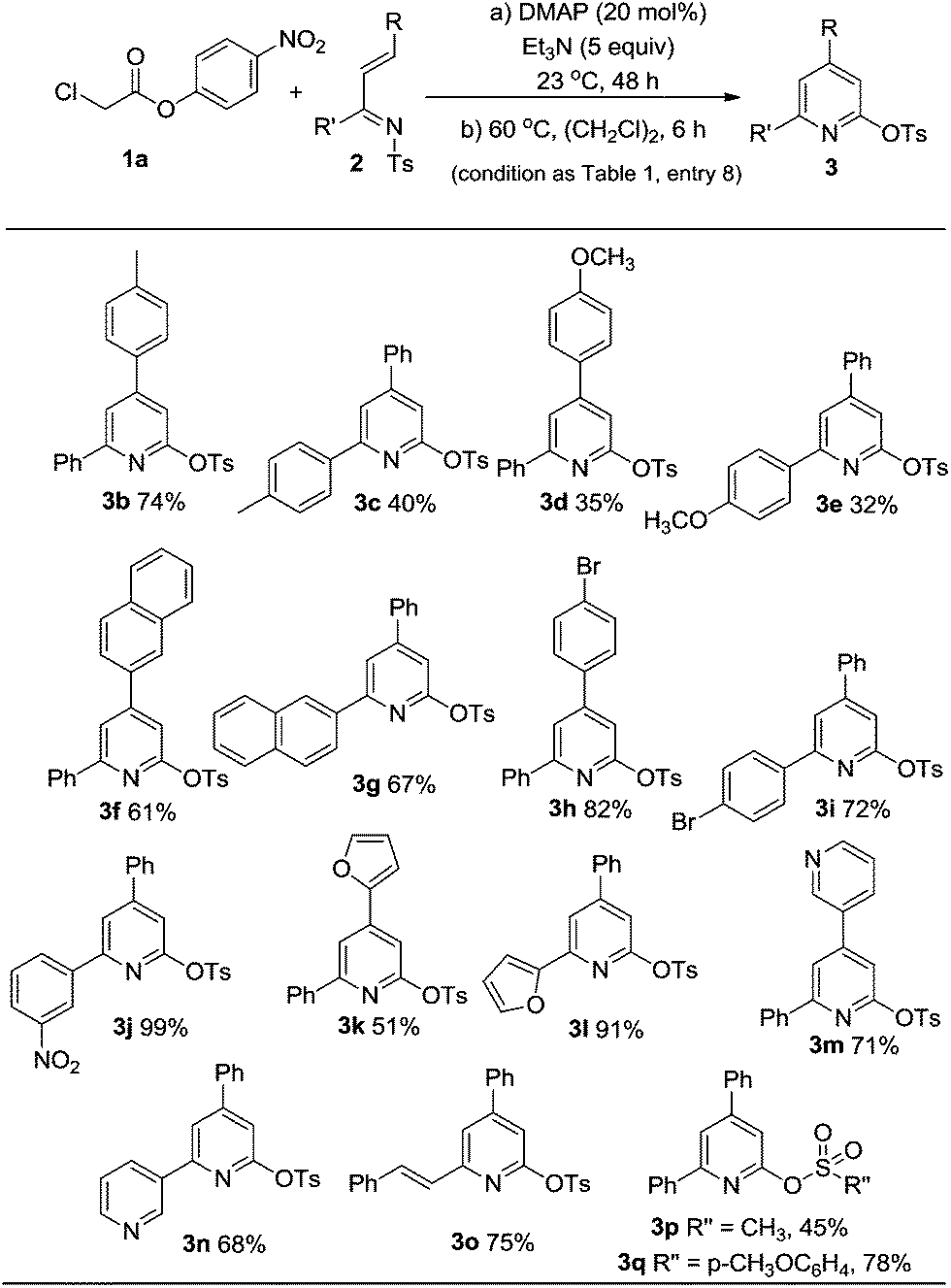 | ||
| Scheme 1 Substrate scope. | ||
The tosylate unit in the pyridine products (3a–o) is a versatile reactive group in cross-coupling reactions such as Suzuki couplings,9 Heck couplings,10 Kumada couplings11 and metal-catalyzed amination reactions.12 For example, amino pyridine 4a, a selective 5-HT2A/5-HT1A receptor ligand,13 could be readily prepared by coupling 3a with N-methylpiperazine (eqn (1)).
Conclusions
In summary, we have developed a DMAP-catalyzed synthesis of trisubstituted pyridines. Activation of the ester substrate by DMAP to form a DMAP-bound enolate intermediate is a key step in this process. A readily available and inexpensive organocatalyst and an ester substrate were used. The pyridine adducts obtained with our methods are amenable to effective transformations to bioactive molecules. Further development in activating readily available raw materials such as esters and acids as chemical feedstock is being pursued in our laboratory. In addition, this study is expected to encourage exploration of DMAP and its analogs as organocatalysts for the activation of carboxylic esters and their derivatives.Acknowledgements
We thank the Singapore National Research Foundation (NRF), Singapore Economic Development Board (EDB), GlaxoSmithKline (GSK) and Nanyang Technological University (NTU) for generous financial support. Torres acknowledges funding of the National Research Foundation grant NRF-CRP4-2008-02.Notes and references
- For reviews, see: (a) F. W. Bergstrom, Chem. Rev., 1944, 35, 77 CrossRef CAS; (b) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2003, 103, 3119 CrossRef CAS PubMed; (c) V. C. Gibson, C. Redshaw and G. A. Solan, Chem. Rev., 2007, 107, 1745 CrossRef CAS PubMed ; For selected examples, see: ; (d) R. B. Martin and J. A. Lissfelt, J. Am. Chem. Soc., 1956, 78, 938 CrossRef CAS; (e) T. Tokuyama and J. W. Daly, Tetrahedron, 1983, 39, 41 CrossRef CAS; (f) M. P. Cruskie Jr., J. A. Zoltewicz and K. A. Abboud, J. Org. Chem., 1995, 60, 7491 CrossRef; (g) F. Trécourt, B. Gervais, M. Mallet and G. Quéguiner, J. Org. Chem., 1996, 61, 1673 CrossRef PubMed; (h) T. T. Denton, X. Zhang and J. R. Cashman, Biochem. Pharmacol., 2004, 67, 751 CrossRef CAS PubMed.
- (a) J. A. Varela and C. Saá, Chem. Rev., 2003, 103, 3787 CrossRef CAS PubMed; (b) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed; (c) B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1085 RSC; (d) M. D. Hill, Chem.–Eur. J., 2010, 16, 12052 CrossRef CAS PubMed.
- Z. Shi and T.-P. Loh, Angew. Chem., Int. Ed., 2013, 52, 8584 CrossRef CAS PubMed.
- D. G. Stark, L. C. Morrill, P.-P. Yeh, A. M. Z. Slawin, T. J. C. O'Riordan and A. D. Smith, Angew. Chem., Int. Ed., 2013, 52, 11642 CrossRef CAS PubMed.
- (a) L. Hao, Y. Du, H. Lv, X. Chen, H. Jiang, Y. Shao and Y. R. Chi, Org. Lett., 2012, 14, 2154 CrossRef CAS PubMed; (b) L. Hao, C. Chuen, R. Ganguly and Y. R. Chi, Synlett, 2013, 1197 CAS; (c) L. Hao, S. Chen, J. Xu, B. Tiwari, Z. Fu, T. Li, J. Lim and Y. R. Chi, Org. Lett., 2013, 15, 4956 CrossRef CAS PubMed; (d) S. Chen, L. Hao, Y. Zhang, B. Tiwari and Y. R. Chi, Org. Lett., 2013, 15, 5822 CrossRef CAS PubMed; (e) Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, Nature Chem., 2013, 5, 835 CrossRef CAS PubMed; (f) J. Cheng, Z. Huang and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 8592 CrossRef CAS PubMed; (g) J. Xu, Z. Jin and Y. R. Chi, Org. Lett., 2013, 15, 5028 CrossRef CAS PubMed.
- (a) U. Ragnarsson and L. Grehn, Acc. Chem. Res., 1998, 31, 494 CrossRef CAS; (b) R. Murugan and E. F. V. Scriven, Aldrichimica Acta, 2003, 36, 21 CAS; (c) G. C. Fu, Acc. Chem. Res., 2004, 37, 542 CrossRef CAS PubMed.
- (a) M. Hamer and E. P. Lira, J. Heterocycl. Chem., 1972, 9, 215 CrossRef CAS; (b) K. Afarinkia and F. Mahmood, Tetrahedron Lett., 1998, 39, 493 CrossRef CAS; (c) A. Perry, A. Nelson, R. Hodgson and A. Kennedy, Synlett, 2007, 1043 Search PubMed.
- For selected reviews, see: (a) E. Baranoff, J.-P. Collin, L. Flamigni and J.-P. Sauvage, Chem. Soc. Rev., 2004, 33, 147 RSC; (b) E. C. Constable, Chem. Soc. Rev., 2007, 36, 246 RSC; (c) I. Eryazici, C. N. Moorefield and G. R. Newkome, Chem. Rev., 2008, 108, 1834 CrossRef CAS PubMed; (d) L. Flamigni, J.-P. Collin and J.-P. Sauvage, Acc. Chem. Res., 2008, 41, 857 CrossRef CAS PubMed; (e) S. Hayami, Y. Komatsu, T. Shimizu, H. Kamihata and Y. H. Lee, Coord. Chem. Rev., 2011, 255, 1981 CrossRef CAS PubMed; (f) J. Husson and M. Knorr, Beilstein J. Org. Chem., 2012, 8, 379 CrossRef CAS PubMed; (g) J. Husson and M. Knorr, J. Heterocycl. Chem., 2012, 49, 453 CrossRef CAS.
- J. Yang, S. Liu, J.-F. Zheng and J. S. Zhou, Eur. J. Org. Chem., 2012, 6248 CrossRef CAS.
- (a) S. b. Noël, C. Pinel and L. Djakovitch, Org. Biomol. Chem., 2006, 4, 3760 RSC; (b) T. M. Gøgsig, A. T. Lindhardt, M. Dekhane, J. Grouleff and T. Skrydstrup, Chem.–Eur. J., 2009, 15, 5950 CrossRef PubMed; (c) T. M. Gogsig, J. Kleimark, S. O. N. Lill, S. Korsager, A. T. Lindhardt, P. O. Norrby and T. Skrydstrup, J. Am. Chem. Soc., 2012, 134, 443 CrossRef PubMed; (d) L. Qin, X. Ren, Y. Lu, Y. Li and J. S. Zhou, Angew. Chem., Int. Ed., 2012, 51, 5915 CrossRef CAS PubMed.
- A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 8704 CrossRef CAS PubMed.
- C.-Y. Gao and L.-M. Yang, J. Org. Chem., 2008, 73, 1624 CrossRef CAS PubMed.
- M. H. Paluchowska, A. J. Bojarski, R. Bugno, S. Charakchieva-Minol and A. Wesolowska, Arch. Pharm., 2003, 336, 104 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c3qo00045a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2014 |