Sequential carboxylation/intramolecular cyclization reaction of o-alkynyl acetophenone with CO2†
Wen-Zhen
Zhang
*,
Ling-Long
Shi
,
Chuang
Liu
,
Xu-Tong
Yang
,
Yan-Bo
Wang
,
Yi
Luo
and
Xiao-Bing
Lu
*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian, 116024, P. R. China. E-mail: zhangwz@dlut.edu.cn; lxb-1999@163.com; Fax: +86 411 8498 6256; Tel: +86 411 8498 6257
First published on 3rd March 2014
Abstract
A novel sequential carboxylation/intramolecular cyclization reaction of o-alkynyl acetophenone with CO2 was developed for producing the synthetically important 1(3H)-isobenzofuranylidene acetic acids and esters in good yield and exclusive selectivity toward 5-exo oxygen cyclization under very mild reaction conditions (room temperature and CO2 balloon). This efficient reaction system showed wide functional group compatibility. Also, the computational study successfully explained the exclusive product selectivity toward the 5-exo oxygen cyclization product.
Introduction
Since carbon dioxide (CO2) is an abundant, inexpensive, and attractive C1 feedstock, the development of new reaction systems for catalytic transformation of CO2 into value-added fine chemicals is of great interest and has been a long-standing goal for chemists.1 CO2 can serve as an efficient carboxylative reagent for preparing various carboxylic acids in the transition-metal catalyzed carboxylation of carbon nucleophiles such as organotin,2 organoboron,3 organozinc4 reagents and terminal alkynes,5 which represents a convenient method to access carboxylic acids bearing various functional groups.6 The hydro-, sila- or bora-carboxylation reaction, which concerns the carboxylation of in situ formed organometallic nucleophiles, has also been established as another attractive protocol to synthesize functionalized carboxylic acids and derivatives that were previously inaccessible by reaction of unsaturated compounds and CO2.7 Recently, direct carboxylation of the C–H bond8 or halide electrophiles9 with CO2 was successfully realized. It represents a more economical and straightforward method, since this process completely avoids tedious and costly pre-activation steps.It is well-known that the α C–H bond in some ketones, especially aromatic ketone, can undergo carboxylation reaction with CO2 in the presence of suitable organic or inorganic bases.10 However, the products β-ketocarboxylic acids are inherently unstable due to rapid decarboxylation, and thus relatively difficult to isolate. Therefore, further conversion of the in situ formed β-ketocarboxylic acids into the corresponding stable esters or β-hydroxycarboxylic acids was required after the carboxylation reaction. Usually, those reactions encountered high CO2 pressures and other rigorous reaction conditions. Recently, Yamada and co-workers10i reported the reaction of alkynyl ketone with 1.0 MPa of CO2 in the presence of 20 mol% silver catalyst and 4 equiv. MTBD, in which the generated β-ketocarboxylate was thought to conduct a nucleophilic attack at the activated alkyne moiety through the carboxylate O atom, giving the stable lactone in high selectivity.
Generally, carboxylate is considered to be a relatively weak nucleophile. Compared with that in aromatic ketone and the carboxylate moiety of β-ketocarboxylate, the oxygen atom in the keto-moiety and the α carbon atom of β-ketocarboxylate are more liable to be involved in many reactions, partially due to the keto–enol tautomerism.11 In other words, in the reaction of aromatic ketone with CO2 to generate β-ketocarboxylate, the keto-moiety was activated by CO2 insertion (Scheme 1). Based on this insight, we envisioned that with judicious design of the substrate and choice of the catalytic system, the activated keto-moiety in β-ketocarboxylate would conduct an intramolecular cyclization reaction with anchored multiple bonds (Scheme 1), providing a more stable carboxylated product. More importantly, it may offer a novel approach to some intermediates useful for the preparation of biologically active compounds and other fine chemicals.12 Herein, we report the realization of this hypothesis, focusing on the highly efficient synthesis of 1(3H)-isobenzofuranylidene acetic acids and the corresponding esters by sequential carboxylation/intramolecular cyclization reaction of o-alkynyl acetophenone using CO2 as a carboxylative reagent under very mild reaction conditions (low catalyst loading, CO2 balloon, and room temperature).13
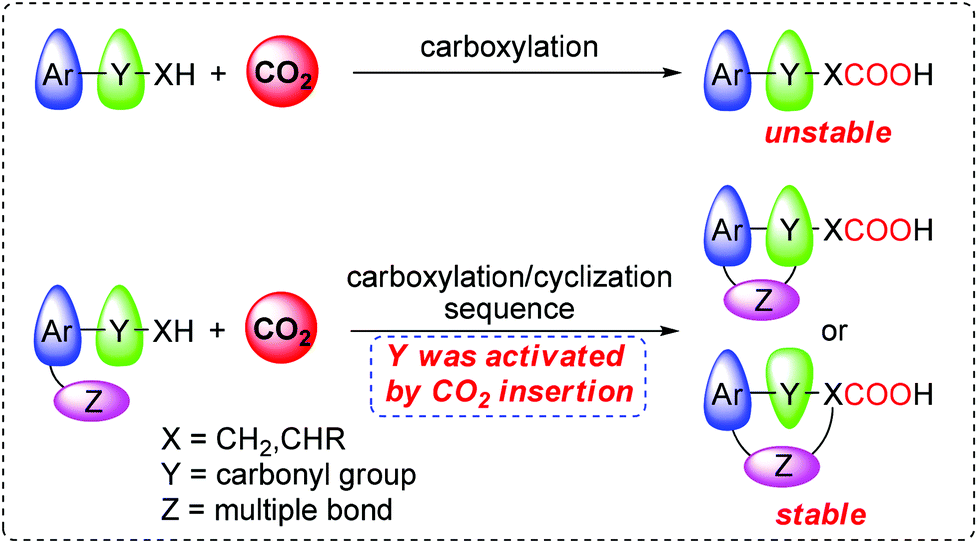 | ||
| Scheme 1 Carboxylation vs. carboxylation/cyclization sequence. | ||
Results and discussion
Indeed, once the β-ketocarboxylate (with the example of o-alkynyl acetophenone as reaction substrate) was formed by CO2 insertion in the presence of a base, there were four possible intramolecular cyclization pathways to give different products (Scheme 2): 5-exo (2 or 3) or 6-endo (4) type cyclization at the oxygen atom, and 5-exo (5) or 6-endo (6) type cyclization at the carbon atom.11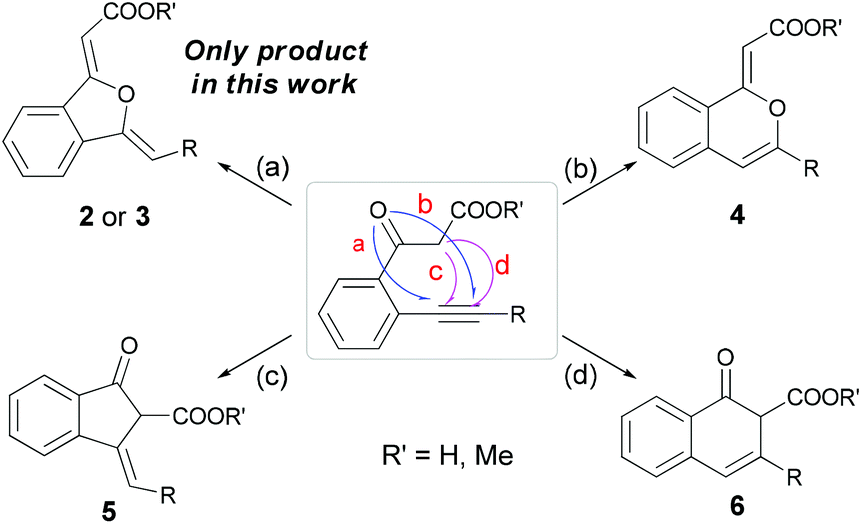 | ||
| Scheme 2 Four possible cyclization pathways. | ||
Optimization of the reaction conditions
Initially, the reaction of 2-(phenylethynyl)phenylethanone (1a) with CO2 was chosen as a model reaction to identify the effective catalytic system and optimize the reaction conditions (Table 1). Since DBU was proven by Jessop and co-workers10f to be an efficient base for the carboxylation reaction of acetophenone, and copper(I) salt was a general π-acid catalyst for alkynes, the CuI/DBU was firstly tested as a catalytic system for this reaction. It is delightedly found that only the 5-exo oxygen cyclization product 1(3H)-isobenzofuranylidene acetic acid 2a was isolated in 83% yield as a stable white solid (entry 1), and no other type of cyclization product was detected. In order to simplify the chromatography separation procedure, further esterification of the resulting carboxylate using MeI (5 equiv.) was performed, affording the corresponding ester derivative 3a in 86% isolation yield (entry 1). DBN, K2CO3 and KOtBu were found to be unsuitable bases (entries 2–4), while MTBD was established as the most efficient base for this reaction system (entry 5). Interestingly, the decrease of catalyst CuI loading from 10 mol% to 2 mol% did not change the yield of the product (entries 5 and 6), while the replacement of CuI with CuBr or CuCl resulted in a decrease in the yield of 3a (entries 7 and 8).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | Cat. (mol%) | Base | Solvent | Yieldb [%] |
| a Unless otherwise noted, the reaction was carried out with 1a (0.20 mmol), catalyst, base (0.40 mmol), solvent (3 mL) at 50 °C for 12 h in 2.0 MPa of CO2, and then reacted with CH3I for 4 h. b Isolated yield. Data given in parentheses are for a reaction in which 2a is the product. c In the absence of CO2. d 2 mol% PPh3 as a ligand. e 1.0 MPa CO2. f 1.0 MPa CO2, 25 °C. g CO2 balloon, 25 °C. DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. DBN = 1,5-diazabicyclo[4.3.0]non-5-ene. MTBD = 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene. IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene. DMAc = N,N-dimethylacetamide. | ||||
| 1 | CuI (10) | DBU | DMF | 86 (83) |
| 2 | CuI (10) | DBN | DMF | 14 |
| 3 | CuI (10) | K2CO3 | DMF | 7 |
| 4 | CuI (10) | KOtBu | DMF | <1 |
| 5 | CuI (10) | MTBD | DMF | 90 |
| 6 | CuI (2) | MTBD | DMF | 90 |
| 7 | CuBr (2) | MTBD | DMF | 65 |
| 8 | CuCl (2) | MTBD | DMF | 75 |
| 9 | AgI (2) | MTBD | DMF | 59 |
| 10 | AgBr (2) | MTBD | DMF | 67 |
| 11 | AgCl (2) | MTBD | DMF | 93 |
| 12 | AgF (2) | MTBD | DMF | 90 |
| 13 | AgOTf (2) | MTBD | DMF | 83 |
| 14 | AgOAc (2) | MTBD | DMF | 92 |
| 15 | AgNO3 (2) | MTBD | DMF | 82 |
| 16 | AgBF4 (2) | MTBD | DMF | 95 |
| 17 | — | MTBD | DMF | 8 |
| 18 | AgBF4 (2) | — | DMF | — |
| 19c | AgBF4 (2) | MTBD | DMF | — |
| 20d | AgBF4 (2) | MTBD | DMF | 51 |
| 21 | AgBF4 (2) | MTBD | DMAc | 88 |
| 22 | AgBF4 (2) | MTBD | DMSO | 10 |
| 23 | AgBF4 (2) | MTBD | THF | 54 |
| 24 | AgBF4 (2) | MTBD | Toluene | 31 |
| 25 | AgBF4 (2) | MTBD | CH2Cl2 | 10 |
| 26e | AgBF4 (2) | MTBD | DMF | 93 |
| 27f | AgBF4 (2) | MTBD | DMF | 96 |
| 28g | AgBF4 (2) | MTBD | DMF | 93 |
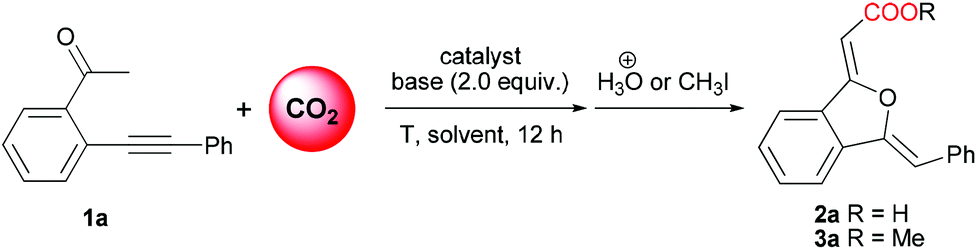
Compared with copper(I) salts, silver(I) salts are much better π-acid catalysts for alkyne activation in some reactions.14 It was found in our experiments that silver(I) salts also showed higher catalytic activity toward this reaction than copper(I) salts, simultaneously maintaining exclusive selectivity toward 5-exo oxygen cyclization products (entries 9 to 16). Among the used silver(I) salts, 2 mol% loading of AgBF4 together with 2 equivalents of MTBD gave the best result (95% yield) (entry 16). It is noteworthy that a very low yield of product 3a was obtained in the absence of any metal catalyst (entry 17), which testified the catalytic role of copper(I) and silver(I) salts. The reaction did not occur in the absence of a base or CO2 (entries 18 and 19), more than 96% of 1a was recovered, clearly demonstrating the crucial role of the base in the carboxylation step and the activation of the keto-moiety by CO2 insertion. On addition of 2 mol% PPh3 to the silver catalytic system as a ligand, a striking decrease in yield was observed (entry 20). Importantly, the silver catalyst system proved to be not sensitive to CO2 pressure (entries 26–28), and a slightly increased yield of 3a was obtained with decreased CO2 pressure (1.0 MPa) and temperature (25 °C) (entry 27). Notably, when the AgBF4-catalyzed reaction was carried out at room temperature using a CO2 balloon, 93% yield of 3a was still obtained (entry 28). Therefore, under very mild reaction conditions, AgBF4-catalyzed carboxylation/intramolecular cyclization reaction of 2-(phenylethynyl)phenylethanone using CO2 as a carboxylative reagent resulted in synthetically important 1(3H)-isobenzofuranylidene acetate in high yield and exclusive product selectivity.
Substrate scope of the reaction
Under the optimized reaction conditions, the scope of sequential carboxylation/intramolecular cyclization reaction of various o-alkynyl acetophenone with CO2 was investigated. As shown in Scheme 3, this silver(I) catalyst system showed wide substrate generality and various functional groups were tolerated, from electron-rich aryl ethers to electron-withdrawn aryltrifluoromethyl and arylfluoro functionalities. Chloro and hydroxyl substituents in the phenylethynyl group were compatible with the reaction conditions. When 2-(3-aminophenylethynyl)phenylethanone 1i was used as a substrate, methoxycarbonylamino substituted 1(3H)-isobenzofuranylidene acetate 3i was obtained in 71% yield, in which the free amino group was converted into methyl carbamate during the esterification procedure after the sequential reaction. Alkyl substituted ethynylphenylethanones 1j and 1k were also found to be suitable substrates.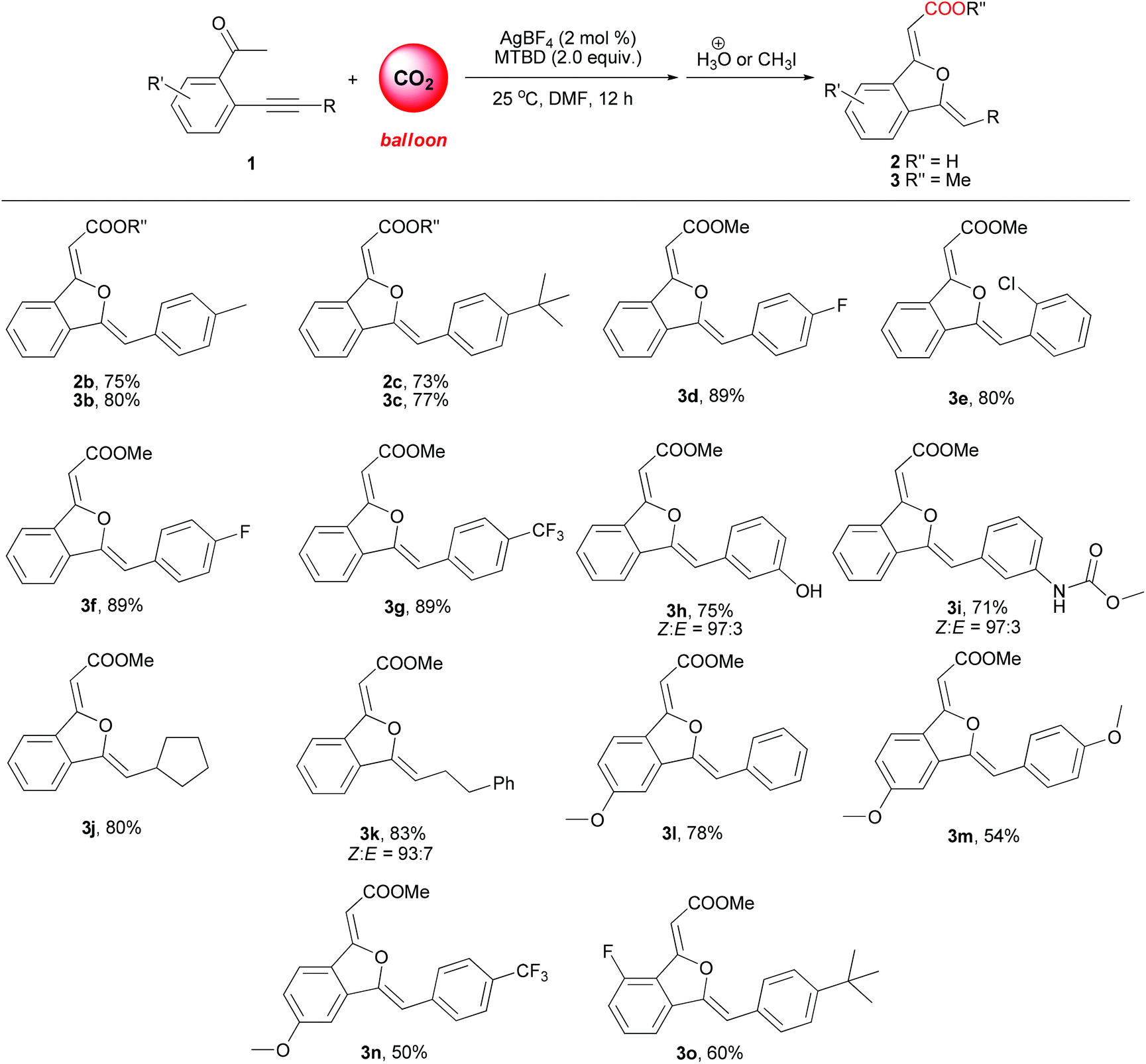 | ||
| Scheme 3 Sequential carboxylation/cyclization reaction of various o-alkynyl ketones using CO2. | ||
When o-alkynyl acetophenones 1b and 1c were used as the substrates and the acid products were targeted, 1(3H)-isobenzofuranylidene acetic acids 2b and 2c were achieved in more than 70% yield. The structures of 2b and 2c were unambiguously determined by X-ray crystallography (Fig. 1),15 which clearly confirmed both Z configurations of the double bond in the product molecules.
 | ||
| Fig. 1 ORTEP plot of 2b (left) and 2c (right) shown with ellipsoids at the 30% probability level; most hydrogen atoms were omitted for clarity. | ||
It was reported by Herndon et al. that interconversion between Z and E (C–C double bond adjacent to the carbonyl group) isomers was observed in alkylidenephthalan derivatives in silica gel column chromatography or in the slightly acidic NMR solvent.16 Except for 3h, 3i and 3k (Scheme 3), all other 1(3H)-isobenzofuranylidene acetic ester products were observed as a single isomer in the 1H NMR spectrum.
Mechanistic studies
Deuterium-labelling experiments (Scheme 4) testified that the α-H of carboxylate and phenyl groups in the 1(3H)-isobenzofuranylidene acetate came from the methyl H in 2-(phenylethynyl)phenylethanone. The obvious deuterium-content difference between the two α-H and substrate indicated that the hydrogen shift was involved in the reaction.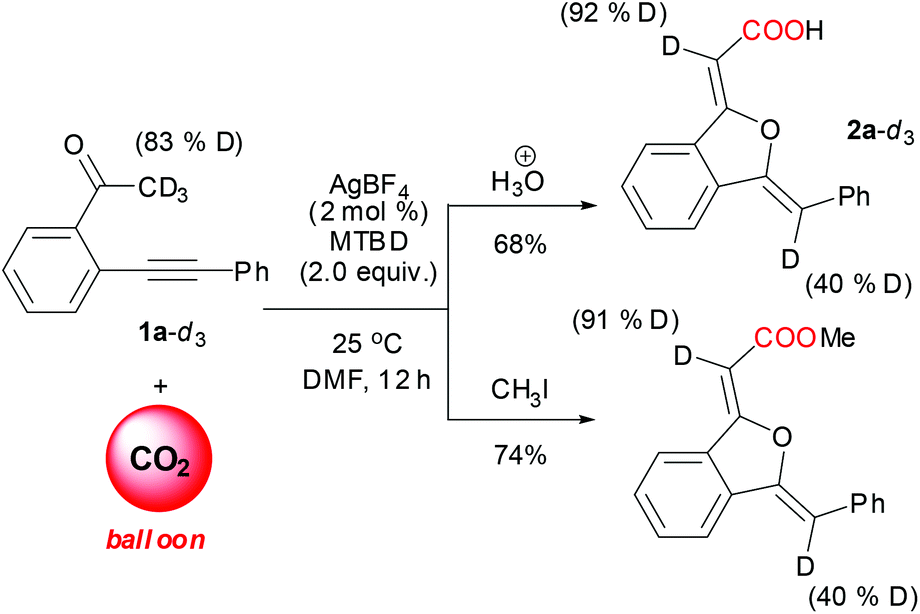 | ||
| Scheme 4 Sequential reaction of deuterated variant of 1a with CO2. | ||
On the basis of the above experiments and insights, a possible reaction mechanism was proposed (Scheme 5). In the presence of a suitable base such as DBU or MTBD, o-alkynyl acetophenone was firstly carboxylated with CO2 to produce β-ketocarboxylate I. There are two possible pathways for the following cyclization reaction (Scheme 5). In Path 1, β-ketocarboxylate I underwent keto–enol tautomerism and the enol oxygen atom of II conducted a nucleophilic attack towards the silver(I)-activated alkyne moiety, cyclization happened and 1(3H)-isobenzofuranylidene acetate IV was formed. Path 2 concerns the direct attack of the keto oxygen atom of intermediate I towards the silver(I)-activated alkyne moiety to generate intermediate V, and the following 1,5-hydrogen shift to give the acetate IV. Finally, acidification or esterification of the resulting carboxylate IV released carboxylic acid or ester. A computational study (Fig. 2) revealed that intermediate III was more stable than intermediate V, and the energy barrier of the cyclization step through intermediate V was much higher than that of intermediate III, indicating that Path 1 might be preferred for this reaction.
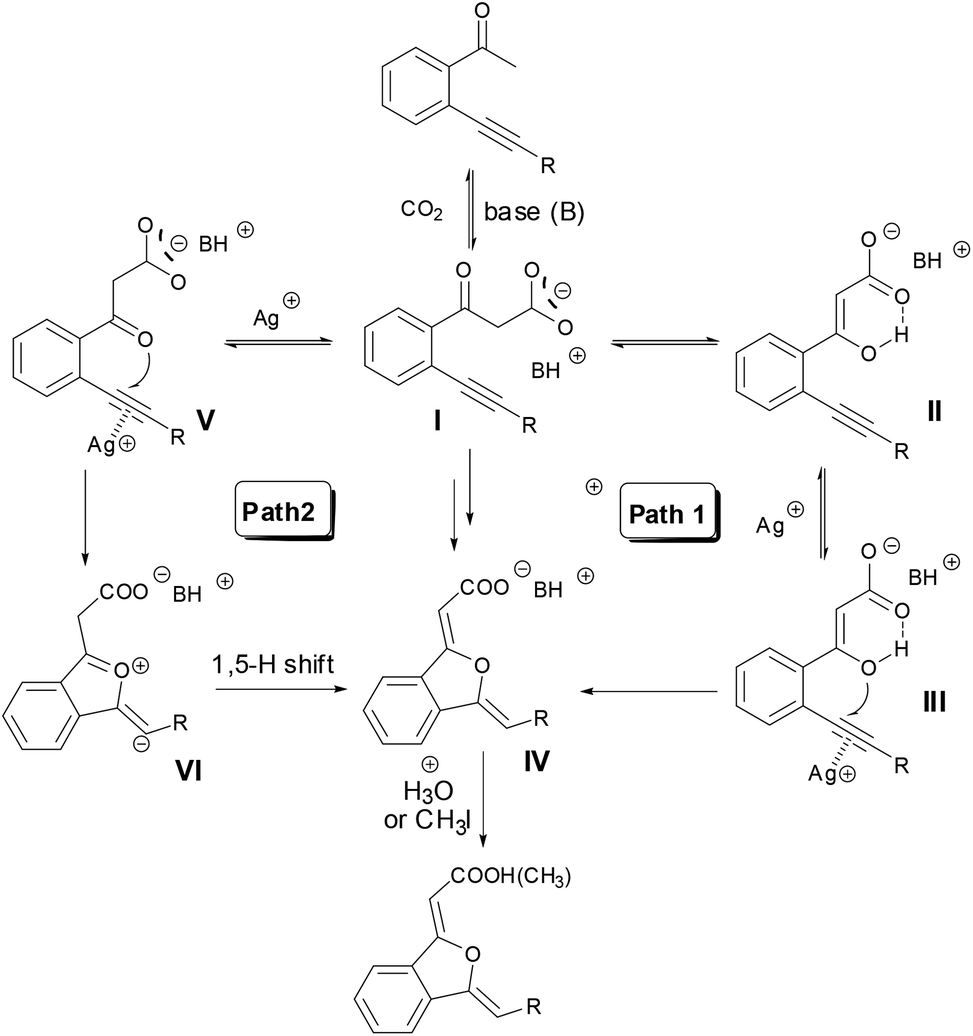 | ||
| Scheme 5 Possible mechanism of the sequential carboxylation/cyclization reaction of various o-alkynyl ketones using CO2. | ||
 | ||
| Fig. 2 The transition state structures and the solvation free energy computed by the ωB97XD method. | ||
Also, a computational study was carried out to explain the exclusive product selectivity toward the 5-exo oxygen cyclization product. Firstly, many DFT methods such as B3LYP, B3PW91, and TPSSh were used to find the TS which leads to 5-exo oxygen cyclization,17 but all these methods failed to find the TS. So a flexible scan of the potential energy surface (PES) of the formed C–O bond was carried out (ESI, Fig. S1†). It turned out that there is no energy barrier when the O atom approaches the C atom during 5-exo oxygen cyclization, which also indicates why the TS could not be found. However, the TS for the other three types of cyclization were all located, and the formation of those TS has moderate energy barriers. The above DFT study using these methods explained the exclusive product selectivity toward the 5-exo oxygen cyclization product.
Then, the ωB97XD function, which was suggested to be an efficient method for predicting barrier heights in the weak interaction, was used.18 As shown in Fig. 2, all the TS were located, and the 5-exo oxygen cyclization showed the lowest energy barrier, which therefore clearly explained why the reaction showed exclusive product selectivity toward 5-exo oxygen cyclization.
Conclusions
In conclusion, we have developed a new reaction system using CO2 as an carboxylative reagent under very mild reaction conditions (room temperature and CO2 balloon), in which a sequential carboxylation/intramolecular cyclization reaction of o-alkynyl acetophenone was successfully carried out to produce stable, synthetically important 1(3H)-isobenzofuranylidene acetic acids and esters in good yield and exclusive selectivity toward 5-exo oxygen cyclization. This efficient reaction system showed wide functional group compatibility. The computational study using different DFT methods found that the transition state for 5-exo oxygen cyclization showed the lowest energy barrier, which clearly explained the exclusive product selectivity toward 5-exo oxygen cyclization.Experimental section
General information
Unless otherwise stated, all manipulations were performed using standard Schlenk techniques under a dry nitrogen or CO2 atmosphere. DMF, DMAc and DMSO were distilled from CaH2 at 60 °C under reduced pressure and stored over 4A molecular sieves. o-Alkynyl ketones were synthesized through a Sonogashira reaction of the corresponding 2-bromoketones with terminal alkynes according to a reported procedure.19 Carbon dioxide (99.999%), commercially available silver(I) and copper(I) salts, and all other reagents were used without further purification.General procedure for sequential carboxylation/cyclization reaction
Taking the sequential carboxylation/intramolecular cyclization reaction of o-alkynyl ketone 1a as an example: A 50 mL oven dried Schlenk flask containing a stir bar was charged with 1a (44 mg, 0.20 mmol), MTBD (61 mg, 0.4 mmol), AgBF4 (0.8 mg, 0.004 mmol) and 3.0 mL dry DMF under argon. After purging the flask with CO2 two times, the reaction mixture was stirred at 25 °C with a CO2 balloon attached. After 12 h, CH3I (142 mg, 1.0 mmol) was injected into the flask and the reaction mixture was stirred at 25 °C for 4 h. The reaction mixture was then diluted with water (30 mL) and extracted with ethyl acetate (3 × 30 mL). The combined organic layers were washed with water and brine, dried over Na2SO4 and filtered. The solvent was removed under vacuum. The product 3a (53 mg, 0.19 mmol, 95% yield) was isolated as a white solid by column chromatography on silica gel (ethyl acetate–petroleum ether: 1:10).
Synthesis and reaction of 2-(phenylethynyl)phenylethanone-methyl-d3
2-(Phenylethynyl)phenylethanone-methyl-d3 was synthesized using the following protocol:20 Under a nitrogen atmosphere, a mixture of 2-(phenylethynyl)phenylethanone (0.55 g, 2.5 mmol), NaOH (8 mg, 0.2 mmol) and D2O (99.9% D) (4.0 mL, 0.2 mol) was stirred at room temperature for 48 h. The reaction mixture was then diluted with dry diethyl ether (20 mL). The aqueous layer was extracted with diethyl ether (3 × 10 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated. Then, the crude product was deuterated again using the same procedure. The final residue was chromatographed using ether–EtOAc (40/1) to afford 2-(phenylethynyl)phenylethanone-methyl-d3 (74% atom D, yield 72%). This product (74% atom D) was deuterated again using the same procedure to give 2-(phenylethynyl)phenylethanone-methyl-d3 (83% atom D, yield 74%). The procedures for sequential carboxylation/cyclization reaction of deuterated substrates are the same as the above general procedure. The 1H NMR spectra of two 2-(phenylethynyl)phenylethanone-methyl-d3 and the corresponding acid, ester products are included in ESI.†Computational study
DFT calculations were performed using the Gaussian 09 package. The 6-31+G(d) all-electron basis set was used for the C, H, O and Cl atoms, and the LanL2DZ basis set and associated effective core potential (ECP) was used for the Ag atom. Frequency calculations were performed to identify the geometrically optimized stationary points as minima (zero imaginary frequency) or transition states (one imaginary frequency) and to obtain the thermodynamic data. Intrinsic reaction coordinates (IRC) were carried out to identify transition states indeed connecting two relevant minima. The single-point calculations were performed with the 6-311++G(2d,p) basis set for the C, H, O and Cl atoms, and the SDD basis set and associated ECP for the Ag atoms. The solvation effect was considered by performing SCRF single-point calculations with the conductor polarizable continuum model (CPCM). DMF was employed as the solvent in the CPCM calculations.Product characterization
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21172026 and U1162101), the Fundamental Research Funds for the Central Universities (DUT12LK47), and Scientific Research Fund of Liaoning Provincial Education Department (no. L2012024). X.-B. L. gratefully acknowledges the Chang Jiang Scholars Program (no. T2011056) from the Ministry of Education of the People's Republic of China.Notes and references
- For a review, see: (a) J. Louie, Curr. Org. Chem., 2005, 9, 605 CrossRef CAS; (b) M. Mori, Eur. J. Org. Chem., 2007, 4981 CrossRef CAS; (c) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (d) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC; (e) A. Correa and R. Martin, Angew. Chem., Int. Ed., 2009, 48, 6201 CrossRef CAS PubMed; (f) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347 RSC; (g) A. Behr and G. Henze, Green Chem., 2011, 13, 25 RSC; (h) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435 RSC; (i) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed; (j) X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462 RSC; (k) W.-Z. Zhang and X.-B. Lu, Chin. J. Catal., 2012, 33, 745 CrossRef CAS; (l) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC; (m) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC.
- For the carboxylation of organotin reagents with CO2, see: (a) M. Shi and K. M. Nicholas, J. Am. Chem. Soc., 1997, 119, 5057 CrossRef CAS; (b) R. J. Franks and K. M. Nicholas, Organometallics, 2000, 19, 1458 CrossRef CAS; (c) R. Johansson and O. F. Wendt, Dalton Trans., 2007, 488 RSC; (d) J.-G. Wu and N. Hazari, Chem. Commun., 2011, 47, 1069 RSC; (e) T. Mita, J. Chen, M. Sugawara and Y. Sato, Angew. Chem., Int. Ed., 2011, 50, 1393 CrossRef CAS PubMed; (f) X. Feng, A. Sun, S. Zhang, X. Yu and M. Bao, Org. Lett., 2013, 15, 108 CrossRef CAS PubMed.
- For the carboxylation of organoboron reagents with CO2, see: (a) K. Ukai, M. Aoki, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2006, 128, 8706 CrossRef CAS PubMed; (b) J. Takaya, S. Tadami, K. Ukai and N. Iwasawa, Org. Lett., 2008, 10, 2697 CrossRef CAS PubMed; (c) T. Ohishi, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 5792 CrossRef CAS PubMed; (d) H. Ohmiya, M. Tanabe and M. Sawamura, Org. Lett., 2011, 13, 1086 CrossRef CAS PubMed; (e) T. Ohishi, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 8114 CrossRef CAS PubMed; (f) X. Zhang, W.-Z. Zhang, L.-L. Shi, C.-X. Guo, L.-L. Zhang and X.-B. Lu, Chem. Commun., 2012, 48, 6292 RSC.
- For the carboxylation of organozinc reagents with CO2, see: (a) H. Ochiai, M. Jang, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 2681 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 7826 CrossRef CAS PubMed; (c) K. Kobayashi and Y. Kondo, Org. Lett., 2009, 11, 2035 CrossRef CAS PubMed.
- For the carboxylation of terminal alkynes with CO2, see: (a) Y. Fukue, S. Oi and Y. Inoue, J. Chem. Soc., Chem. Commun., 1994, 2091 RSC; (b) N. Eghbali, J. Eddy and P. T. Anastas, J. Org. Chem., 2008, 73, 6932 CrossRef CAS PubMed; (c) L. J. Goossen, N. Rodriguez, F. Manjolinho and P. P. Lange, Adv. Synth. Catal., 2010, 352, 2913 CrossRef CAS; (d) D. Yu and Y. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20184 CrossRef CAS PubMed; (e) D. Yu and Y. Zhang, Green Chem., 2011, 13, 1275 RSC; (f) W.-Z. Zhang, W.-J. Li, X. Zhang, H. Zhou and X.-B. Lu, Org. Lett., 2010, 21, 4748 CrossRef PubMed; (g) X. Zhang, W.-Z. Zhang, X. Ren, L.-L. Zhang and X.-B. Lu, Org. Lett., 2011, 13, 2402 CrossRef CAS PubMed; (h) M. Arndt, E. Risto, T. Krause and L. J. Goossen, ChemCatChem, 2012, 4, 484 CrossRef CAS; (i) D. Yu, M. X. Tan and Y. Zhang, Adv. Synth. Catal., 2012, 354, 969 CrossRef CAS; (j) K. Inamoto, N. Asano, K. Kobayashi, M. Yonemoto and Y. Kondo, Org. Biomol. Chem., 2012, 10, 1514 RSC; (k) X. Zhang, W.-Z. Zhang, L.-L. Shi, C. Zhu, J.-L. Jiang and X.-B. Lu, Tetrahedron, 2012, 68, 9085 CrossRef CAS PubMed; (l) F. Manjolinho, M. Arndt, K. Goossen and L. J. Goossen, ACS Catal., 2012, 2, 2014 CrossRef CAS.
- For the transition-metal-free incorporation of CO2, see: (a) H. Yoshida, H. Fukushima, J. Ohshita and A. Kunai, J. Am. Chem. Soc., 2006, 128, 11040 CrossRef CAS PubMed; (b) H. Zhou, W.-Z. Zhang, C.-H. Liu, J.-P. Qu and X.-B. Lu, J. Org. Chem., 2008, 73, 8039 CrossRef CAS PubMed; (c) S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322 CrossRef CAS PubMed; (d) Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194 CrossRef CAS PubMed; (e) G. Chen, C. Fu and S. Ma, Org. Lett., 2009, 11, 2900 CrossRef CAS PubMed; (f) O. Vechorkin, N. Hirt and X. Hu, Org. Lett., 2010, 12, 3567 CrossRef CAS PubMed; (g) S. Minakata, I. Sasaki and T. Ide, Angew. Chem., Int. Ed., 2010, 49, 1309 CrossRef CAS PubMed; (h) G. Chen, C. Fu and S. Ma, Org. Biomol. Chem., 2011, 9, 105 RSC; (i) C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187 CrossRef CAS PubMed; (j) K. Inamoto, N. Asano, Y. Nakamura, M. Yonemoto and Y. Kondo, Org. Lett., 2012, 14, 2622 CrossRef CAS PubMed; (k) W.-J. Yoo, M. G. Capdevila, X. Du and S. Kobayashi, Org. Lett., 2012, 14, 5326 CrossRef CAS PubMed; (l) L.-L. Zhao, S.-Y. Wang, X.-P. Xu and S.-J. Ji, Chem. Commun., 2013, 49, 2569 RSC; (m) Y.-B. Wang, Y.-M. Wang, W.-Z. Zhang and X.-B. Lu, J. Am. Chem. Soc., 2013, 135, 11996 CrossRef CAS PubMed.
- For the hydro-, sila- or bora-carboxylation of unsaturated compounds with CO2, see: (a) J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2008, 130, 15254 CrossRef CAS PubMed; (b) C. M. Williams, J. B. Johnson and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14936 CrossRef CAS PubMed; (c) S. Li, W. Yuan and S. Ma, Angew. Chem., Int. Ed., 2011, 50, 2578 CrossRef CAS PubMed; (d) T. Fujihara, T. Xu, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2011, 50, 523 CrossRef CAS PubMed; (e) M. North, Angew. Chem., Int. Ed., 2009, 48, 4104 CrossRef CAS PubMed; (f) Y. Zhang and S. N. Riduan, Angew. Chem., Int. Ed., 2011, 50, 6210 CrossRef CAS PubMed; (g) M. D. Greenhalgh and S. P. Thomas, J. Am. Chem. Soc., 2012, 134, 11900 CrossRef CAS PubMed; (h) S. Li and S. Ma, Org. Lett., 2011, 13, 6046 CrossRef CAS PubMed; (i) S. Li and S. Ma, Adv. Synth. Catal., 2012, 354, 2387 CrossRef CAS; (j) L. Zhang, J. Cheng, B. Carry and Z. Hou, J. Am. Chem. Soc., 2012, 134, 14314 CrossRef CAS PubMed; (k) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487 CrossRef CAS PubMed; (l) S. Li, B. Miao, W. Yuan and S. Ma, Org. Lett., 2013, 15, 977 CrossRef CAS PubMed; (m) T. G. Ostapowicz, M. Schmitz, M. Krystof, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2013, 52, 12119 CrossRef CAS PubMed.
- For the carboxylation of the C–H bond with CO2, see: (a) I. I. F. Boogaerts and S. P. Nolan, J. Am. Chem. Soc., 2010, 132, 8858 CrossRef CAS PubMed; (b) L. Zhang, J. Cheng, T. Ohishi and Z. Hou, Angew. Chem., Int. Ed., 2010, 49, 8670 CrossRef CAS PubMed; (c) I. I. F. Boogaerts, G. C. Fortman, M. R. L. Catherine and S. P. Nolan, Angew. Chem., Int. Ed., 2010, 49, 8674 CrossRef CAS PubMed; (d) H. Mizuno, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 1251 CrossRef CAS PubMed; (e) D. M. Dalton and T. Rovis, Nat. Chem., 2010, 2, 710 CrossRef CAS PubMed; (f) I. I. F. Boogaerts and S. P. Nolan, Chem. Commun., 2011, 47, 3021 RSC; (g) K. Sasano, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 10954 CrossRef CAS PubMed.
- For the direct carboxylation of electrophiles with CO2, see: (a) A. Correa and R. Martin, J. Am. Chem. Soc., 2009, 131, 15974 CrossRef CAS PubMed; (b) T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106 CrossRef CAS PubMed; (c) T. Leon, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221 CrossRef CAS PubMed.
- For the α-carboxylation of carbonyl compounds with CO2, see: (a) E. J. Corey and R. H. K. Chen, J. Org. Chem., 1973, 38, 4086 CrossRef CAS; (b) E. Haruki, M. Arakawa, N. Matsumura, Y. Otsuji and E. Imoto, Chem. Lett., 1974, 427 CrossRef CAS; (c) H. Sakurai, A. Shirahata and A. Hosomi, Tetrahedron Lett., 1980, 21, 1967 CrossRef CAS; (d) Y. Hirai, T. Aida and S. Inoue, J. Am. Chem. Soc., 1989, 111, 3062 CrossRef CAS; (e) K. Chiba, H. Tagaya, S. Miura and M. Karasu, Chem. Lett., 1992, 923 CrossRef CAS; (f) B. J. Flowers, R. Gautreau-Service and P. G. Jessop, Adv. Synth. Catal., 2008, 350, 2947 CrossRef CAS; (g) E. J. Beckmana and P. Munshi, Green Chem., 2011, 13, 376 RSC; (h) B. R. Van Ausdall, N. F. Poth, V. A. Kincaid, A. M. Arif and J. Louie, J. Org. Chem., 2011, 76, 8413 CrossRef CAS PubMed; (i) K. Sekine, T. Ishida and T. Yamada, Angew. Chem., Int. Ed., 2012, 51, 6989 CrossRef PubMed.
- For reviews, see: (a) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS PubMed; (b) J.-M. Weibel, A. L. Blanc and P. Pale, Chem. Rev., 2008, 108, 3149 CrossRef CAS PubMed; (c) M. Alvarez-Corral, M. Munoz-Dorado and I. Rodriguez-Garcia, Chem. Rev., 2008, 108, 3174 CrossRef CAS PubMed; (d) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed; (e) P. Belmont and E. Parker, Eur. J. Org. Chem., 2009, 6075 CrossRef CAS; (f) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS PubMed; (g) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed.
- (a) A. Zamilpa, M. Herrera-Ruiz, E. del Olmo, J. L. Lopez-Perez, J. Tortoriello and A. San Feliciano, Bioorg. Med. Chem. Lett., 2005, 15, 3483 CrossRef CAS PubMed; (b) L. M. Bedoya, E. del Olmo, R. Sancho, B. Barboza, M. Beltran, A. E. Garcia-Cadenas, S. Sanchez-Palomino, J. L. Lopez-Perez, E. Munoz, A. San Feliciano and J. Alcami, Bioorg. Med. Chem. Lett., 2006, 16, 4075 CrossRef CAS PubMed; (c) K. Kobayashi, K. Hashimoto, S. Fukamachi and H. Konishi, Synthesis, 2008, 1094 CrossRef CAS PubMed.
- During the preparation of this manuscript, a similar study using silver catalysis was reported: K. Sekine, A. Takayanagi, S. Kikuchi and T. Yamada, Chem. Commun., 2013, 49, 11320 RSC.
- (a) W. Yamada, Y. Sugawara, H.-M. Cheng, T. Ikeno and T. Yamada, Eur. J. Org. Chem., 2007, 2604 CrossRef CAS; (b) Y. Sugawara, W. Yamada, S. Yoshida, T. Ikeno and T. Yamada, J. Am. Chem. Soc., 2007, 129, 12902 CrossRef CAS PubMed; (c) S. Yoshida, K. Fukui, S. Kikuchi and T. Yamada, Chem. Lett., 2009, 786 CrossRef CAS; (d) S. Yoshida, K. Fukui, S. Kikuchi and T. Yamada, J. Am. Chem. Soc., 2010, 132, 4072 CrossRef CAS PubMed; (e) S. Kikuchi, S. Yoshida, Y. Sugawara, W. Yamada, H.-M. Cheng, K. Fukui, K. Sekine, I. Iwakura, T. Ikeno and T. Yamada, Bull. Chem. Soc. Jpn., 2011, 84, 698 CrossRef CAS; (f) T. Ishida, S. Kikuchi, T. Tsubo and T. Yamada, Org. Lett., 2013, 15, 848 CrossRef CAS PubMed; (g) T. Ishida, S. Kikuchi and T. Yamada, Org. Lett., 2013, 15, 3710 CrossRef CAS PubMed.
- CCDC 940369 (2b) and CCDC 940368 (2c) contain the supplementary crystallographic data for this paper.
- S. Duan, K. Cress, K. Waynant, E. Ramos-Miranda and J. W. Herndon, Tetrahedron, 2007, 63, 2959 CrossRef CAS PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS; (c) P. M. W. Gill, B. G. Johnson, J. A. Pople and M. J. Frisch, Int. J. Quantum Chem., Quantum Chem. Symp., 1992, 26, 319 CrossRef CAS; (d) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 6615 RSC.
- (a) M. Dell'Acqua, G. Abbiati, A. Arcadi and E. Rossi, Org. Biomol. Chem., 2011, 9, 7836 RSC; (b) N. Chernyak, S. I. Gorelsky and V. Gevorgyan, Angew. Chem., Int. Ed., 2011, 50, 2342 CrossRef CAS PubMed.
- X. Zhao, J. Jing, K. Lu, Y. Zhang and J.-B. Wang, Chem. Commun., 2010, 46, 1724 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra. CCDC 940368 and 940369. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3qo00047h |
| This journal is © the Partner Organisations 2014 |