 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzoate dioxygenase from Ralstonia eutropha B9 – unusual regiochemistry of dihydroxylation permits rapid access to novel chirons†
Julia A.
Griffen
a,
Sarah J.
Kenwright
a,
Sarah
Abou-Shehada
a,
Scott
Wharry
b,
Thomas S.
Moody
*b and
Simon E.
Lewis
*a
aCentre for Sustainable Chemical Technologies, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: S.E.Lewis@bath.ac.uk
bDepartment of Biocatalysis and Isotope Chemistry, Almac Group Ltd, 20 Seagoe Industrial Estate, Craigavon, BT63 5QD, UK. E-mail: tom.moody@almacgroup.com
First published on 8th January 2014
Abstract
Oxidation of benzoic acid by a microorganism expressing benzoate dioxygenase leads to the formation of an unusual ipso, ortho arene cis-diol in sufficient quantities to be useful for synthesis. This homochiral diol possesses an array of differentiated functionality which can be exploited to access diverse highly oxygenated structures by concise synthetic sequences.
Introduction
Of the various oxygenases that have found use in biocatalysis, it is arguably the arene dioxygenases which add the most value in terms of the synthetic versatility of the products they produce.1 The direct transformation of an aromatic ring into a dearomatised cyclohexadiene diol (Scheme 1) is a reaction that has very little precedent in organic chemistry2 and is therefore appealing to access rapidly uncharted chemical space. In wild-type organisms, these arene cis-diols are usually fleeting metabolic intermediates.3 However, mutants in which the subsequent enzyme in the metabolic pathway is blocked are able to accumulate these diols and they can be isolated in synthetically useful quantities.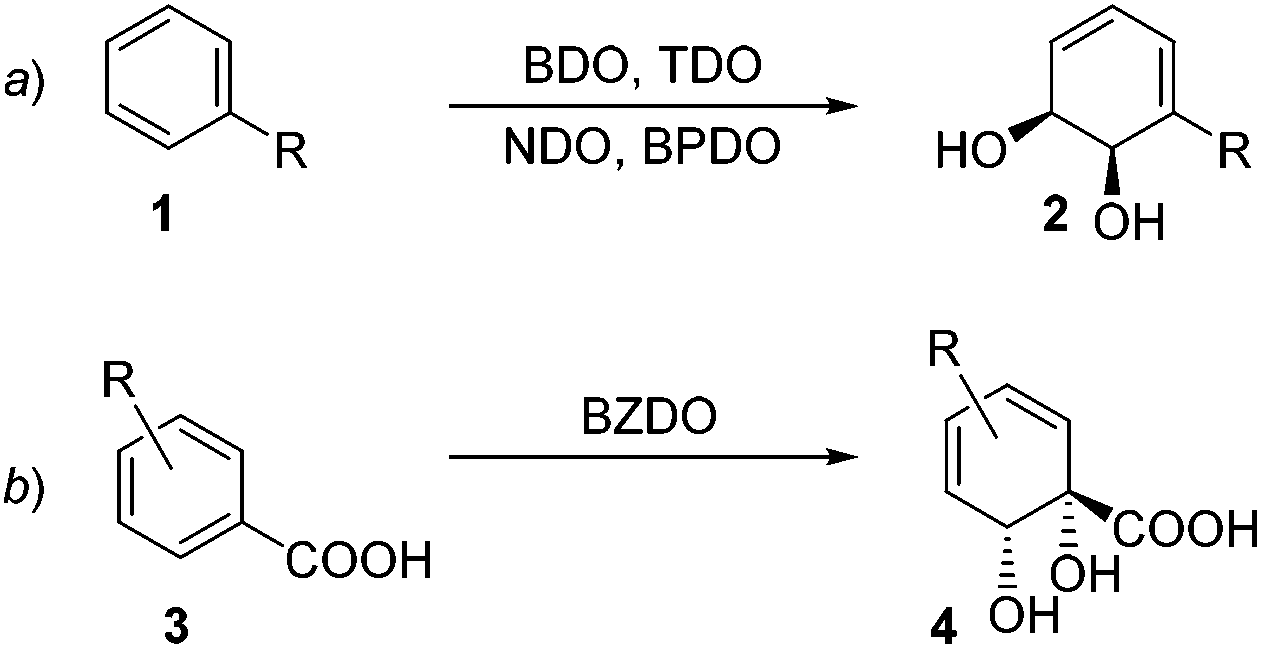 | ||
| Scheme 1 Regio- and stereoselectivity of dioxygenases. | ||
The densely-packed, diverse functionality in these chirons finds ready application in different areas such as synthesis of natural products,4 pharmaceuticals,5 carbohydrates,6 polymers7 and dyes.8 To date, in excess of 400 arene cis-diol products have been reported. The majority of these are produced by organisms expressing toluene dioxygenase (TDO), naphthalene dioxygenase (NDO) and biphenyl dioxygenase (BPDO) enzymes, which are Rieske type non-heme iron oxygenases.1e,9 These metabolise substituted aromatic substrates in a regio- and stereoselective fashion. A robust predictive model has been developed for such transformations,10 with the sense of enantioinduction being consistent across organisms and substrates (Scheme 1a, ortho,meta oxygenation). However, organisms expressing benzoate dioxygenase (BZDO) enzymes dihydroxylate benzoic acids in a process that proceeds with both different regioselectivity and also the opposite absolute sense of enantioinduction. For example, Ralstonia eutropha B911 (formerly known as Alcaligenes eutrophus B9), Pseudomonas putida U10312 and Pseudomonas putida KTSY01 (pSYM01)13 oxidise benzoic acid to benzoate 1,2-cis dihydrodiol 4 (Scheme 1b, ipso,ortho oxygenation).
Diol acid 4 is a highly versatile chiral pool starting material and many transformations of this building block can be envisaged (Fig. 1). Despite this, 4 has been comparatively underutilised to date in synthesis, in comparison with arene cis-diols of type 2.4i,q,t,5h,i,6a,e,o,14–17 In the current work, we describe the synthesis of a library of cyclohexyl chirons from 4, both minimally and more extensively functionalised. This serves to showcase further the versatility of 4 and we anticipate these new building blocks will find diverse applications in synthesis and catalysis. With regards to handling and storage, it should be noted that although 4 is prone to exothermic decomposition by rearomatisation, as are all arene cis-diols, it may be stored in pure form in excess of a year at −78 °C without appreciable decomposition occurring. Additionally, storage of 4 as its mixed sodium/potassium salt has been described and reportedly leads to enhanced stability.16b Production of 4 on a multihundred gram scale is possible without recourse to specialised equipment.15 The absolute configuration and enantiopurity of 4 have been demonstrated through chemical correlation and by X-ray crystallographic analysis of a derivative.14,15
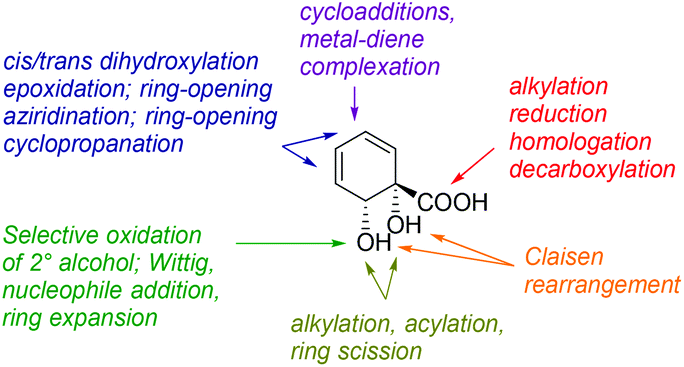 | ||
| Fig. 1 Possible transformations of diol acid 4. | ||
Results and discussion
Ring-saturated derivatives
The diene in 4 readily undergoes hydrogenation over palladium on carbon to give saturated cyclohexane diol acid 5 (Scheme 2). Perhaps surprisingly, this compound has not been reported previously, although the diastereoisomeric trans-diol is known.18 Saturation of ortho,meta arene cis-diols of type 2 to give 3-substituted cyclohexane-1,2-diols and applications of these in catalysis have been reported;19 the analogous approach has not previously been applied to ipso,ortho- arene cis-diols of type 4, however. We wished to target derivatives of 5 with the diol protected, but direct acetonide introduction was surprisingly unsuccessful. To circumvent this, esterification of 5 was carried out prior to ketalisation. Acetonide protection of 6 was successful, albeit with traces of 8 being formed through competing transesterification. Subsequent hydrolysis of ester 7 did indeed give desired acetonide acid 9, but appreciable acetonide migration and deprotection were also observed.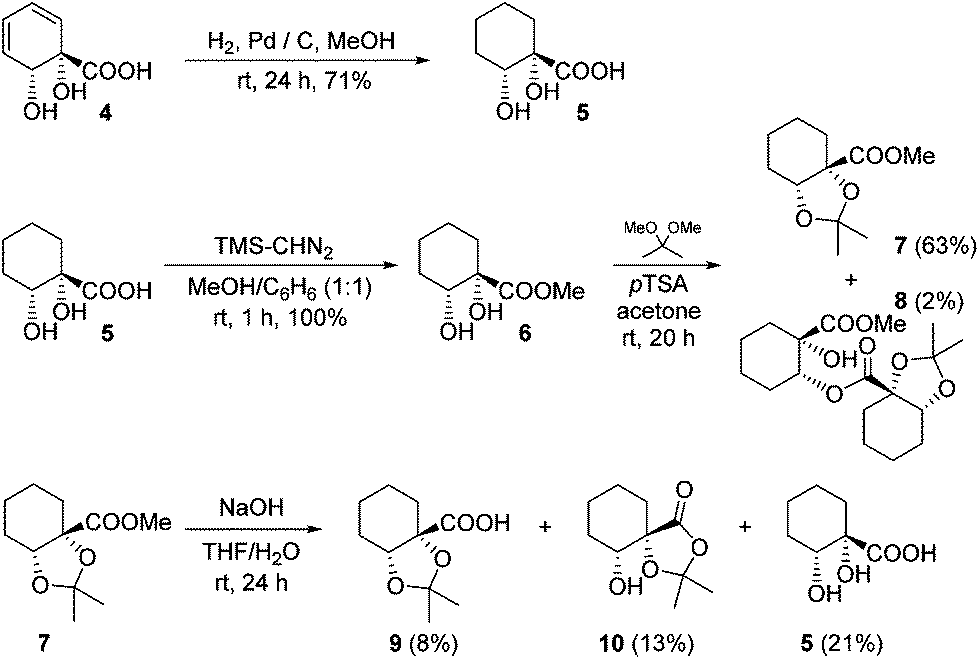 | ||
| Scheme 2 Acetonide acid derivatives of 4. | ||
The structure of 10 was assigned on the basis of its polarity and of the 13C resonance for the ketal carbon (δ = 110.1 ppm, consistent with a five-membered cyclic ketal as opposed to six-membered20), as well as 2D NMR spectroscopic data. In view of the difficulties associated with accessing 9 by means of base-mediated ester cleavage, we instead implemented an approach employing a benzyl ester (Scheme 3). It was found that reliable production of 9 in good yield was best achieved through purification of the final product by dry column chromatography.21
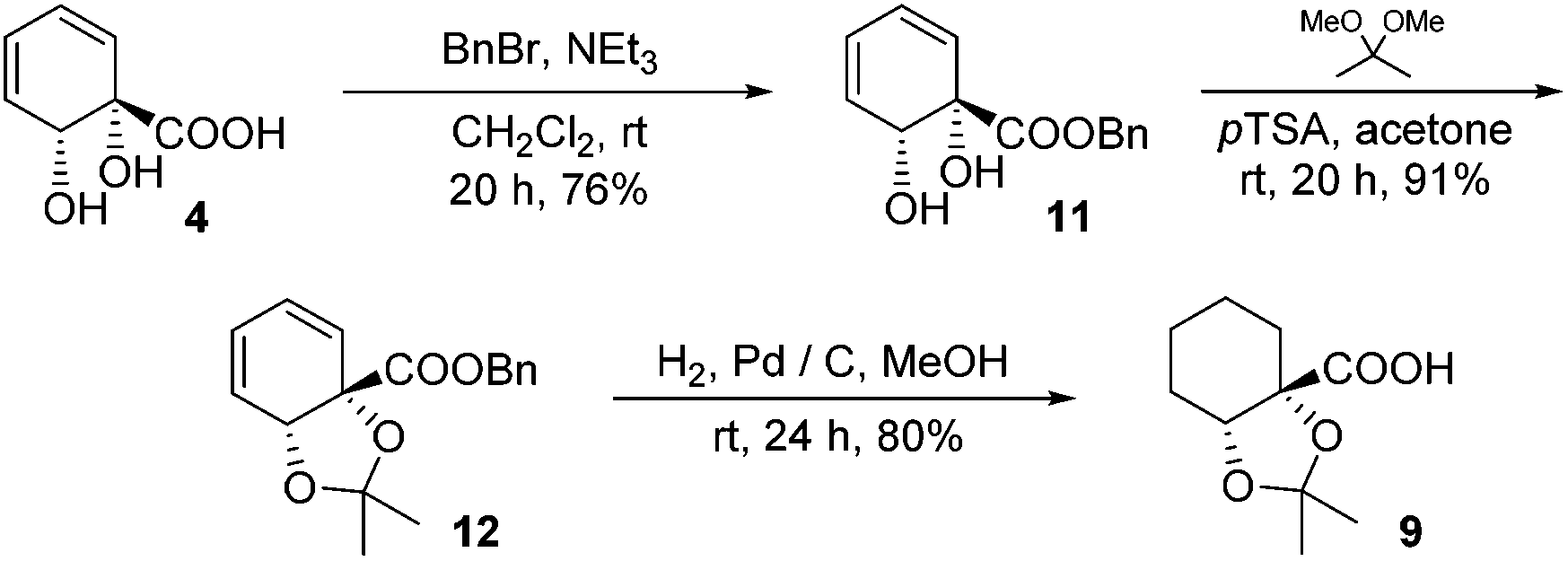 | ||
| Scheme 3 Benzyl ester route to 9. | ||
Other chiral acid building blocks were also targeted; to this end, the diol in benzyl ester 11 was protected as dioxasilole 13 prior to hydrogenation to give 14 (Scheme 4). This last step proved capricious, however, with undesired desilylation also occurring to a varying extent. A bis(ether) derivative was also accessed by permethylation of 5. With a slight excess of alkylating agent incomplete ether formation led to an inseparable mixture of bis(ether) 15 and monoether 16, which required silylation to allow separation to be effected. However, a greater excess of alkylating agent led to clean formation of 15 in good yield and hydrolysis gave bis(ether) acid 18.
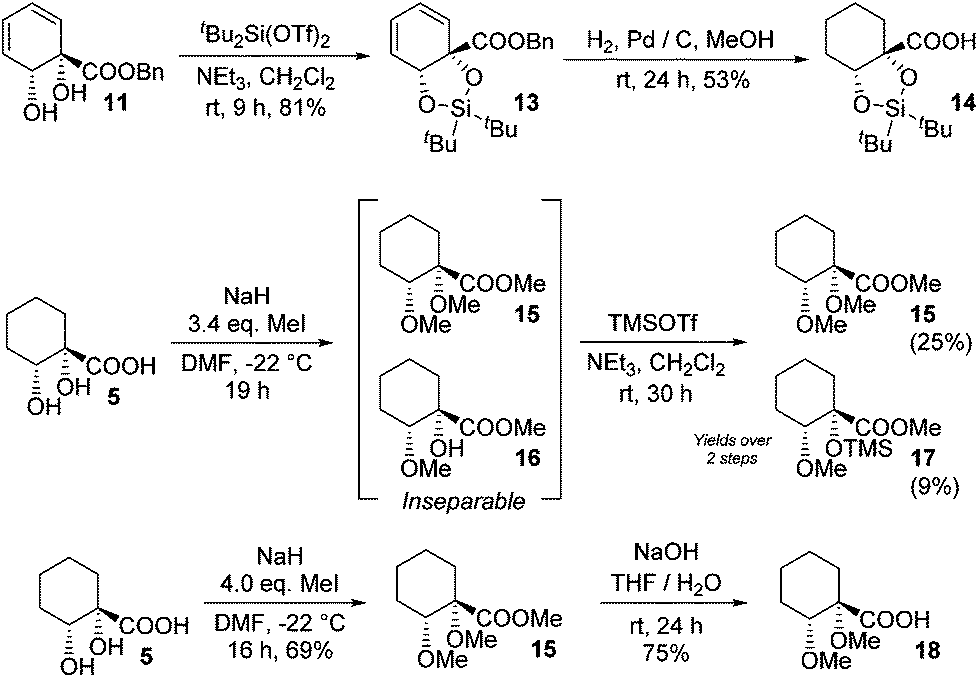 | ||
| Scheme 4 Other protected acid building blocks. | ||
Diol protection as a benzylidene acetal was explored and a moderate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) diastereoselectivity was observed for formation of 19 over 20. Structures were assigned on the basis of NOESY correlations (see ESI†); careful chromatography allowed for isolation of both diastereoisomers in pure form. We next sought to access novel ketones bearing an adjacent quaternary centre. One such target, 21, was available simply by oxidation of byproduct 10. A second such ketone was accessed by a multistep procedure involving silyl protection of the secondary alcohol in 4. Thus, ester 6 was silylated and reduced to give monoprotected triol 23. Choice of reductant proved crucial, since with LiAlH4, yields of 23 were low, with appreciable silyl migration observed, giving rise to 24. In contrast, NaBH4 cleanly effected reduction to 23 only, in good yield. Ketalisation, desilylation and oxidation then gave target ketone 27, a reduced analogue of 21 (Scheme 5).
:2) diastereoselectivity was observed for formation of 19 over 20. Structures were assigned on the basis of NOESY correlations (see ESI†); careful chromatography allowed for isolation of both diastereoisomers in pure form. We next sought to access novel ketones bearing an adjacent quaternary centre. One such target, 21, was available simply by oxidation of byproduct 10. A second such ketone was accessed by a multistep procedure involving silyl protection of the secondary alcohol in 4. Thus, ester 6 was silylated and reduced to give monoprotected triol 23. Choice of reductant proved crucial, since with LiAlH4, yields of 23 were low, with appreciable silyl migration observed, giving rise to 24. In contrast, NaBH4 cleanly effected reduction to 23 only, in good yield. Ketalisation, desilylation and oxidation then gave target ketone 27, a reduced analogue of 21 (Scheme 5).
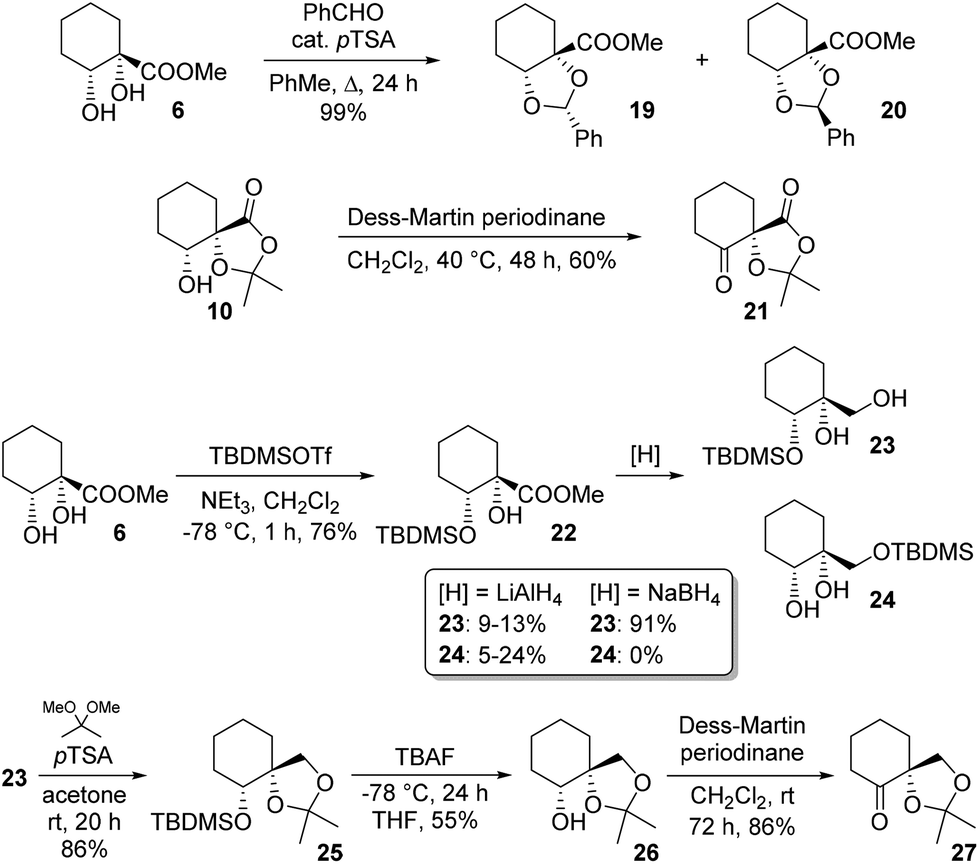 | ||
| Scheme 5 Additional cyclohexyl chirons accessible from 5. | ||
These comparatively minimally functionalised cyclohexyl chirons that all bear a quaternary stereocentre are synthetically valuable insofar as it is difficult to conceive of other means of accessing them as easily, in enantiopure form. We anticipate their finding diverse uses in synthesis and catalysis.
Highly oxygenated derivatives
The structures of arene cis-diols such as 2 and 4 are highly suggestive of applications in the synthesis of cyclitols such as inositols.22 A particular advantage of their use for synthesis of novel inositol derivatives is that they provide ready access to C-substituted derivatives.6d,23 In contrast, use of natural inositols or other carbohydrates as starting materials lends itself to synthesis of O-substituted derivatives, but C-substituted derivatives are accessible only by means of more involved synthetic sequences.The ortho,meta diols of type 2 have been extensively exploited in this context6i,j,p,r,7a,24–27 but no inositol derivatives have been synthesised to date from 4. To access rapidly such a species from 4, we opted to introduce oxygenation into the diene by means of a singlet oxygen photocycloaddition. Use of singlet oxygen to access novel cyclitols from starting materials other than arene cis-diols has previously proven to be a very successful strategy.28 Thus, known silyl ether 28 was transformed to endoperoxide 29 and reduced to protected pentaol 30 by our reported procedure.4i Protection of free hydroxyl groups as methoxymethyl ethers gave 31, which underwent osmium-catalysed dihydroxylation to afford 32 as a single isomer; regiochemistry of osmylation in such systems is well precedented.6y,24d,f,l,25c,l,27d,29 Global deprotection with aqueous acid (and an organic wash to remove silanol) furnished the desired product, but in impure form; attempts using acidic resins also gave impure product. Its purification necessitated exhaustive acetylation to 33, chromatography and ammonolysis to give 34, a C-hydroxymethyl-muco-inositol. C-Hydroxymethyl derivatives of other isomeric inositols are extremely rare30 and those of muco-inositol are wholly unknown (Scheme 6).
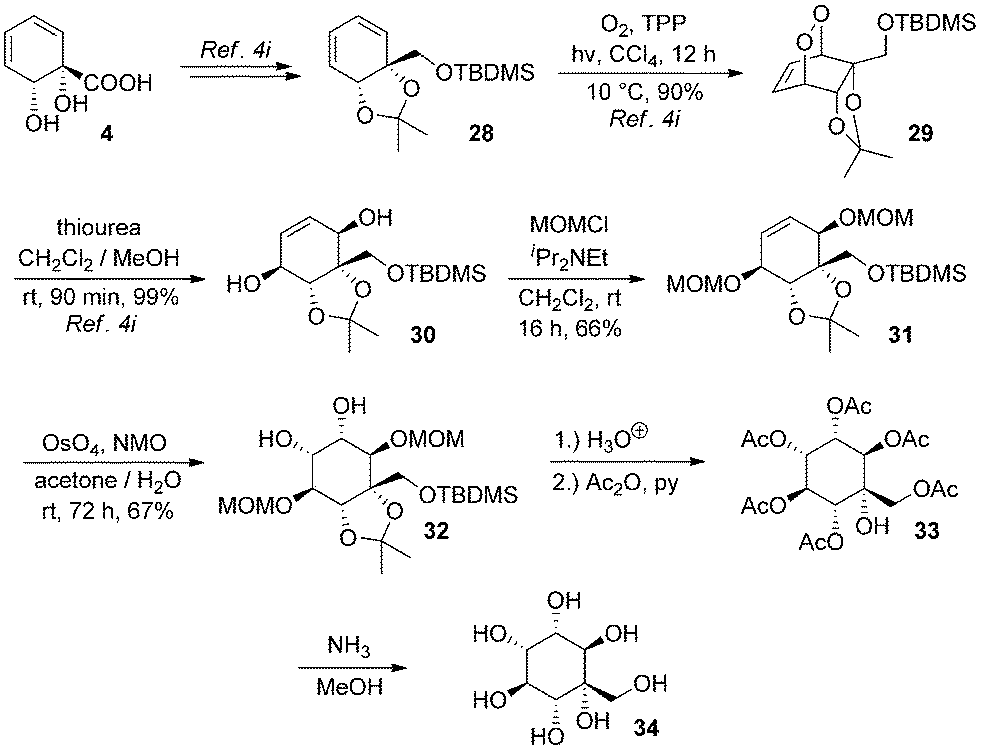 | ||
| Scheme 6 Synthesis of a C-hydroxymethyl inositol. | ||
Endoperoxides derived from diene–1O2 photocycloaddition are versatile intermediates and in addition to reductive cleavage, they are capable of undergoing several other synthetically useful transformations. For example, they undergo base-mediated Kornblum–DeLaMare fragmentation to afford γ-hydroxyenones; we have previously demonstrated such transformations for endoperoxides derived from 4.4i In addition, they may be isomerised to the corresponding bis(epoxides) upon treatment with cobalt–tetraphenylporphine complex.31 This latter transformation has not previously been applied to an endoperoxide derived from 4. To this end, known alcohol 35 was acetylated and subjected to the photocycloaddition. In addition to the expected endoperoxide 37, epoxide 38 was also isolated in small amounts. The structure of 38 was assigned on the basis of its spectroscopic data in comparison with those for the previously reported 39.15 As regards the mechanism of formation of 38, analogous epoxide byproducts of singlet oxygen photocycloaddition have been described previously for other substrates and are believed to arise via a radical pathway.32 Upon treatment with CoTPP, 37 underwent facile isomerisation to bis(epoxide) 40. These epoxides proved resistant to opening with ammonia, with ammonolysis instead cleanly removing the acetate to give 41 (Scheme 7).
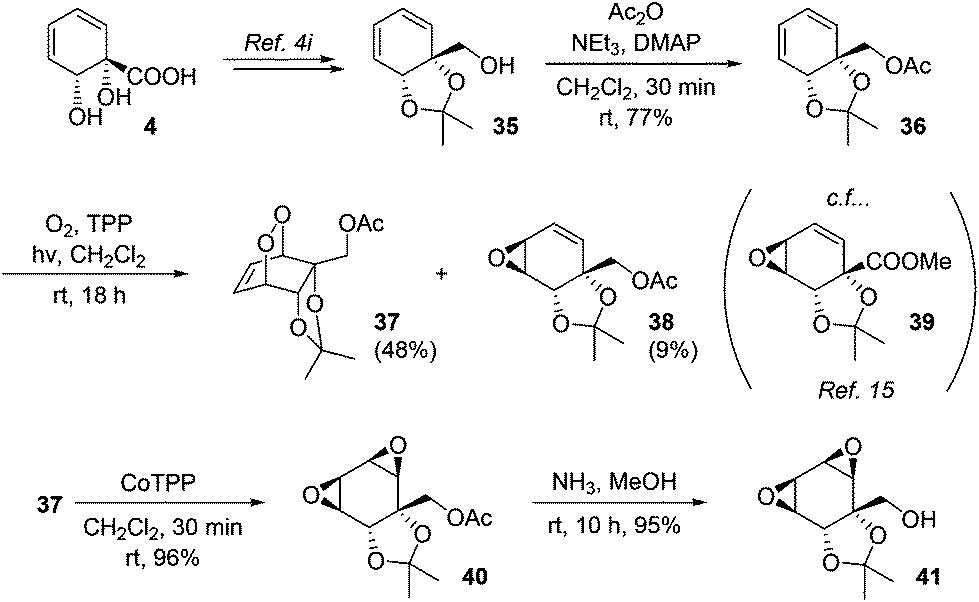 | ||
| Scheme 7 Synthesis of a bis(epoxide). | ||
Conclusions
We have described synthetic sequences which allow for the functionalization of every position on the cyclohexadiene ring of 4. As the stereocentres in 4 are in close proximity to the diene, it has proven possible to introduce additional stereocentres in a highly selective fashion under substrate control. We anticipate that the novel chirons described here may find use in the synthesis of more complex targets.Experimental
General procedures
Reactions were carried out under an atmosphere of nitrogen. In most cases, solvents were obtained by passing through anhydrous alumina columns using an Innovative Technology Inc. PS-400-7 solvent purification system. All other solvents were purchased as “anhydrous” grade from Fisher Scientific. “Petrol” refers to petroleum spirit b.pt 40–60 °C. TLC was performed using aluminium backed plates precoated with Alugram® SIL G/UV 254 nm. Visualization was accomplished by UV light and/or KMnO4 followed by gentle warming. Organic layers were routinely dried with anhydrous MgSO4 and evaporated using a Büchi rotary evaporator. When necessary, further drying was facilitated by high vacuum. Flash column chromatography was carried out using Davisil LC 60 Å silica gel (35–70 micron) purchased from Fisher Scientific. IR spectra were recorded on a Perkin-Elmer 1600 FT IR spectrometer with only selected absorbances quoted as ν in cm−1. NMR spectra were run on Bruker Avance 250, 300, 400 or 500 MHz instruments at 298 K. A micrOTOF electrospray time-of-flight (ESI-TOF) mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) was used; this was coupled to an Agilent 1200 LC system (Agilent Technologies, Waldbronn, Germany). The LC system was used as an autosampler only. 10 μL of sample was injected into a 30:70 flow of water–acetonitrile at 0.6 mL min−1 to the mass spectrometer. For each acquisition 10 μL of calibrant of 5 mM sodium formate was injected after the sample. The observed mass and isotope pattern matched the corresponding theoretical values as calculated from the expected elemental formula.
:35:2.5:2.5 EtOAc–petrol–H2O–AcOH) to yield pure 5 (625 mg, 71%) as a white crystalline solid. m.pt = 112–115 °C; Rf = 0.10 (60:35:2.5:2.5 EtOAc–petrol–H2O–AcOH); [α]25D +13.3 (c 0.3, H2O); 1H NMR (300 MHz, D2O) δ = 3.77 (1H, dd, J = 11.5, 3.5 Hz, CH(OH)), 1.60–1.56 (4H, m, CH), 1.40–1.08 (4H, m, CH); 13C NMR (125 MHz, MeOH-d4) δ = 180.1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 79.5 (C(OH)CO2H), 73.7 (CH(OH)), 35.0, 30.8, 25.3, 21.1; vmax (film) 3445, 3086, 2924, 2858, 1736, 1439, 1199, 1138, 1023, 948, 821 cm−1; HRMS (ESI−) m/z calcd for (C7H11O4)−, 159.0663; found 159.0672.
:1), was added dropwise TMS-CHN2 (0.200 mL, 2.0 M solution in THF, 0.400 mmol, 1.4 equiv.) until gas evolution ceased and a yellow colour persisted. The reaction mixture was concentrated under reduced pressure (caution: TMS-CHN2 toxic by inhalation). Pure 6 was obtained as a yellow oil (52 mg, 100%). Rf = 0.05 (30% EtOAc–petrol); [α]D −10.0 (c 0.3, CHCl3); 1H NMR (250 MHz, CDCl3) δ = 3.82–3.74 (1H, m, CH(OH)), 3.77 (3H, s, CH3), 2.86 (2H, br s, OH), 1.83–1.22 (8H, m, CH); 13C NMR (75 MHz, CDCl3) δ = 176.6 (CO), 76.8 (C(OH)(CO2CH3)), 72.2 (CH(OH)), 52.9 (OCH3), 34.2, 30.1, 24.0, 19.8; vmax (film) 3453, 2938, 2861, 1729, 1438, 1272, 1238, 1207, 1149, 1079, 998, 923 cm−1; HRMS (ESI+) m/z calcd for (C8H15O4)+, 175.0965; found 175.0981. NMR Data are in agreement with those previously reported.33
O), 109.0 (C(CH3)2), 81.0 C(OC(CH3)2)(CO2CH3), 75.0 (CH(OC(CH3)2)), 52.5 (OCH3), 32.3, 28.0, 26.0, 25.9, 20.5, 18.7; vmax (film) 2997, 2941, 2873, 1733, 1450, 1383, 1217, 1160, 1054, 1025, 905, 725 cm−1; HRMS (ESI+) m/z calcd for (C11H18NaO4)+, 237.1097; found 237.1079. Data for 8: Rf = 0.85 (50% EtOAc–petrol); [α]25D −21 (c 1.6, CHCl3); 1H NMR (500 MHz, CDCl3) δ = 5.08 (1H, dd, J = 10.0, 6.0 Hz, CH(OCO)), 4.26 (1H, t, J = 3.2 Hz, CH(O)), 3.73 (3H, s, OCH3), 3.24 (1H, s, OH), 2.06–1.24 (16H, m, CH), 1.50 (3H, s, CH3), 1.35 (3H, s, CH3); 13C NMR (125 MHz, CDCl3) δ = 175.3 (CO), 171.8 (CO), 108.9 (C(CH3)3), 80.7, 75.6, 75.2, 74.9, 53.0 (OCH3), 34.1, 32.2, 27.8, 26.2, 25.7, 25.6, 23.6, 20.2, 19.7, 18.3; vmax (film) 3523, 2987, 2938, 2865, 1733, 1449, 1381, 1370, 1243, 1216, 1152, 1124, 1046, 1003, 874, 735 cm−1; HRMS (ESI+) m/z calcd for (C18H29O7)+, 357.1908; found 357.1929.
O), 109.6 (C(CH3)2), 80.7 C(OC(CH3)2)(CO2H), 74.9 (CH(OC(CH3)2)), 32.2, 28.0, 26.0, 25.7, 20.7, 18.5; vmax (film) 2994, 2939, 2873, 2714, 1450, 1371, 1383, 1217, 1174, 905, 725 cm−1; HRMS (ESI−) m/z calcd for (C10H15O4)−, 199.0976; found 199.0975. Data for 10: Rf = 0.83 (50% EtOAc–petrol); [α]25D +2.0 (c 1.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ = 3.75 (1H, dd, J = 11.5, 4.6 Hz, CH(OH)), 1.95–1.93 (1H, m, CH), 1.92–1.89 (1H, m, CH), 1.88 (1H, br s, OH), 1.77–1.80 (1H, m, CH), 1.73 (1H, td, J = 14.0, 4.5 Hz, CH), 1.66 (3H, s, CH3), 1.63 (3H, s, CH3), 1.49–1.55 (1H, m, CH), 1.44–1.48 (1H, m, CH), 1.38 (1H, tt, J = 13.0, 3.5 Hz, CH); 13C NMR (125 MHz, CDCl3) δ = 174.0 (CO), 110.1 (C(CH3)2), 83.2 (C(O)(CO), 71.0 (C(OH)H), 33.7 (CH2), 30.6 (CH2), 29.1 (C(CH3)2), 27.9 (C(CH3)2), 23.6 (CH2), 20.1 (CH2); vmax (film) 3484, 2991, 2939, 2863, 2774, 1448, 1385, 1291, 1262, 1060, 1036, 908, 860, 626 cm−1; HRMS (ESI+) m/z calcd for (C10H17O4)+, 201.1121; found 201.1113.
CH), 5.94 (1H, dddd, J = 9.5, 5.0, 2.5, 0.5 Hz, CCH), 5.82 (1H, ddd, J = 9.5, 2.0, 1.0 Hz, CCH), 5.76 (1H, ddd, J = 9.5, 2.0, 1.0 Hz, CCH), 5.29 (2H, s, CO2–CH2–Ar), 4.87 (1H, m, C(O)H); 13C NMR (75.5 MHz, CDCl3) δ = 175.2 (CO), 135.1, 132.1, 128.8, 128.7, 128.2, 127.0, 124.8, 122.9, 74.1 (C(OH)CO), 71.1 (C(OH)–H), 68.5 (O–CH2); νmax (film) 3451, 3038, 1731, 1660, 1455, 1378, 1234, 1168, 1077, 1020, 909, 753, 695 cm−1; HRMS (+ve ESI-TOF) m/z calculated for (C14H14NaO4)+, 269.0784; found 269.0863.
CH), 5.86–5.79 (1H, m, CCH), 5.18 (2H, s, CH2), 4.97 (1H, d, J = 4.0 Hz, CH(O)), 1.38 (3H, s, CH3), 1.37 (3H, s, CH3). Data in agreement with those previously reported.6e
CH), 5.75–5.71 (1H, m, CCH), 5.25 (1H, d, J = 12.0 Hz, –CHH–), 5.18 (1H, d, J = 12.0 Hz, –CHH–), 4.97 (1H, d, J = 1.8 Hz, CH(OSi)), 0.99 (9H, s, CH3) 0.98 (9H, s, CH3); 13C NMR (75 MHz, CDCl3) δ = 172.2 (CO), 135.6, 128.6, 128.4, 128.3, 126.3, 125.8, 123.9, 122.3, 78.6 (C(OSi)(CO)), 71.5 (CH(OSi)), 67.4 (CH2), 27.0 (CH3), 26.9 (CH3), 21.2 (C(CH3)3), 20.2 (C(CH3)3); vmax (film) 3045, 2966, 2934, 2891, 2859, 1733, 1473, 1229, 1091, 1031, 1012, 1000, 875, 825, 696 cm−1; HRMS (ESI+) m/z calcd for (C22H31O4Si)+, 387.1986; found 387.1993.
O), 77.9 (C(OSi)(CO)), 73.9 (CH(OSi)), 33.0, 30.1, 27.7 (CH3), 27.5 (CH3), 23.7, 20.5, 20.3, 19.6; vmax (film) 3070, 2935, 2894, 2859, 1717, 1448, 1472, 1094, 827 cm−1; HRMS (ESI+) m/z calcd for (C15H29O4Si)+, 301.1830; found 301.1830.
O), 82.9, 81.7, 57.0, 52.1, 51.8, 29.7, 24.8, 23.5, 20.3; vmax (film) 2938, 2861, 1730, 1446, 1373, 1275, 1227, 1099, 1061, 997 cm−1; HRMS (ESI+) m/z calcd for (C10H19O4)+, 203.1278; found 203.1289. Data for 17: Rf = 0.85 (20% EtOAc–petrol); [α]25D −1.4 (c 0.73, CHCl3); 1H NMR (250 MHz, CDCl3) δ = 3.73 (3H, s, CO2CH3), 3.47 (1H, dd, J = 11.0, 4.5 Hz, CH(OCH3)), 3.29 (3H, s, OCH3), 1.85–0.84 (8H, m, CH), 0.15 (9H, s, Si(CH3)3); 13C NMR (75 MHz, CDCl3) δ = 175.6 (CO), 82.2, 76.1, 56.8, 51.8, 36.2, 25.1, 24.0, 20.3, 2.2; vmax (film) 2949, 2860, 1753, 1446, 1366, 1279, 1244, 1156, 1062, 1041, 837, 760, 734 cm−1; HRMS (ESI+) m/z calcd for (C12H25O4Si)+, 261.1517; found 261.1515.
:45:2.5:2.5 EtOAc–petrol–AcOH–H2O) to give 18 as a colourless oil (108 mg, 75%). Rf = 0.25 (50:45:2.5:2.5 EtOAc–petrol–AcOH–H2O); [α]25D −20.5 (c 0.73, CHCl3); 1H NMR (500 MHz, CDCl3) δ = 3.40–3.37 (1H, m, CH(OCH3)), 3.38 (3H, s, OCH3), 3.35 (3H, s, OCH3), 2.18 (1H, dd, J = 15.0, 2.0 Hz, CH), 1.96 (1H, m, CH), 1.82–1.81 (1H, m, CH), 1.68–1.53 (3H, m, CH), 1.37–1.25 (2H, m, CH); 13C NMR (125 MHz, CDCl3) δ = 175.3 (CO), 82.7, 81.2, 57.1, 52.0, 28.1, 24.8, 23.7, 20.0; vmax (film) 3143, 2940, 2860, 2838, 1721, 1464, 1448, 1308, 1196, 1099, 1077, 973, 939 cm−1; HRMS (ESI–) m/z calcd for (C9H15O4)−, 187.0976; found 187.0972.
:1 mixture of 19 and 20 by NMR. Data for 19: Rf = 0.49 (10% EtOAc–petrol); [α]25D −31 (c 0.7, CHCl3); 1H NMR (300 MHz, CDCl3) δ = 7.56–7.52 (2H, m, Ar–H), 7.40–7.37 (3H, m, Ar–H), 5.97 (1H, s, CH(O)(O)), 4.42 (1H, t, J = 4.0 Hz, CH(O)), 3.82 (3H, s, OCH3), 2.13–1.81 (4H, m, CH), 1.76–1.45 (4H, m, CH); 13C NMR (75 MHz, CDCl3) δ = 173.4 (CO), 137.2, 129.3, 128.3, 126.7 (Ar–C), 103.3 (CH(O)(O)(Ar)), 81.5 (C(O)(CO)), 77.3 (C(O)H), 52.5 (OCH3), 31.1, 26.1, 19.4, 18.5; vmax (film) 2951, 2869, 1734, 1451, 1247, 1163, 1088, 1024, 697 cm−1; HRMS (ESI+) m/z calcd for (C15H19O4)+, 263.1278; found 263.1267. Data for 20: Rf = 0.53 (10% EtOAc–petrol); [α]25D −1.6 (c 0.08, CHCl3); 1H NMR (300 MHz, CDCl3) δ = 7.50–7.46 (2H, m, Ar–H), 7.39–7.32 (3H, m, Ar–H), 6.21 (1H, s, CH(O)(O)), 4.53 (1H, t, J = 5.5 Hz, CH(O)), 3.70 (3H, s, OCH3), 2.03–1.89 (4H, m, CH), 1.75–1.39 (4H, m, CH); 13C NMR (75 MHz, CDCl3) δ = 173.0 (CO), 138.9, 129.0, 128.3, 126.4 (Ar–C), 102.8 (CH(O)(O)(Ar)), 81.9 (C(O)(CO)), 75.9 (C(O)H), 52.3 (OCH3), 30.7, 26.2, 20.9, 20.4; vmax (film) 2938, 2863, 1735, 1451, 1247, 1162, 1093, 698 cm−1; HRMS (ESI+) m/z calcd for (C15H19O4)+, 263.1278; found 263.1268.
O), 169.3 (CO), 111.4 (C(CH3)2), 85.2, 39.5, 38.7, 28.4, 27.5, 26.6, 21.0; vmax (film) 2996, 2941, 2921, 2872, 2851, 1784, 1732, 1394, 1380, 1283, 1254, 1130, 1077, 1045, 930 cm−1; HRMS (ESI+) m/z calcd for (C10H14NaO4)+, 221.0784; found 221.0787.
O), 77.5 (C(OH)), 73.8 (CH(OSi)), 52.2, 33.1, 30.1 (CH), 25.6 (SiC(CH3)3), 23.8, 19.6, 17.8 (SiC(CH3)3), −4.1, −5.2; vmax (film) 3555, 3027, 2928, 1705, 1494, 1453, 1281, 908, 732, 699 cm−1; HRMS (ESI+) m/z calcd for (C14H29O4Si)+, 289.1830; found 289.1831.
CH), 4.86 (1H, d, J = 6.5 Hz, OCH2O), 4.85 (1H, d, J = 6.5 Hz, OCH2O), 4.72 (1H, d, J = 6.5 Hz, OCH2O), 4.70 (1H, d, J = 6.5 Hz, OCH2O), 4.40 (1H, d, J = 3.0 Hz, CHO), 4.29 (1H, br s, CHOMOM), 4.22 (1H, br s, CHOMOM), 3.96 (1H, d, J = 11.0 Hz, CHHOTBDMS), 3.49 (1H, d, J = 11.0 Hz, CHHOTBDMS), 3.41 (3H, s, CH2OCH3), 3.39 (3H, s, CH2OCH3), 1.47 (3H, s, O–C–CH3), 1.43 (3H, s, O–C–CH3), 0.89 (9H, s, SiC(CH3)3), 0.04 (6H, s, Si(CH3)2); 13C NMR (75 MHz, CDCl3) δ = 130.3 (HCCH), 127.9 (HCCH), 108.9 (C(CH3)2), 96.3 (OCH2O), 95.5 (OCH2O), 85.3 (4°CCH2O), 78.4 (CHO), 76.8 (CHO), 75.0 (CHO), 61.9 (CH2OTBDMS), 55.5 (OCH3), 55.4 (OCH3), 28.4 (CCH3), 27.0 (CCH3), 25.9 (SiC(CH3)3), 18.3 (SiC(CH3)3), −5.3 (SiCH3), −5.7 (SiCH3); vmax (film) 2954, 1252, 1043, 907, 837, 732, 649 cm−1; HRMS (ESI+) m/z calcd for (C20H38NaO7Si)+, 441.2279; found 441.2285.
:1, 8.0 mL) was added NMO (43.4 mg, 0.370 mmol, 2.0 equiv.) followed dropwise by OsO4 (40 μL, 2.5% w/v in tert-BuOH, 3.7 μmol). The resulting solution was stirred at room temperature for 72 h. The reaction mixture was diluted with EtOAc (30 mL) and extracted with saturated Na2S2O3(aq) (2 × 30 mL). The organic phase was washed with brine (30 mL) and dried over MgSO4. Removal of the solvent under reduced pressure and purification by flash column chromatography (50% EtOAc–petrol) gave 32 as a colourless oil (56 mg, 67%). Rf = 0.43 (50% EtOAc–petrol); [α]25D −10.0 (c 0.1, CHCl3); 1H NMR (300 MHz, CDCl3) δ = 4.82 (2H, s, OCH2O), 4.74 (1H, d, J = 6.5 Hz, OCHHO), 4.67 (1H, d, J = 6.5 Hz, OCHHO), 4.47 (1H, d, J = 2.5 Hz, CHO), 4.18 (1H, m, CHO), 4.06 (1H, br s, CHO), 3.93 (1H, d, J = 11.0 Hz, CHHOTBDMS), 3.94–3.89 (1H, m, CHO) 3.79 (1H, dd, J = 10.0, 4.0 Hz, CHO) 3.64 (1H, d, J = 11.0 Hz, CHHOTBDMS), 3.45 (3H, s, CH2OCH3), 3.40 (3H, s, CH2OCH3), 3.05 (1H, br s, OH), 1.51 (3H, s, C(CH3)2), 1.37 (3H, s, C(CH3)2), 0.89 (9H, s, SiC(CH3)3), 0.07 (3H, s, SiCH3), 0.06 (3H, s, SiCH3); 13C NMR (75 MHz, CDCl3) δ = 108.6 (C(CH3)2), 98.1 (OCH2OCH3), 95.9 (OCH2OCH3), 85.3 (4°CCH2O), 83.0 (CHO), 76.4 (CHO), 73.4 (CHO), 70.9 (CHO), 69.5 (CHO), 61.2 (CH2OTBDMS), 55.9 (CH2OCH3), 55.8 (CH2OCH3), 28.2 (C(CH3)2), 26.3 (C(CH3)2), 25.9 (SiC(CH3)3), 18.3 (SiC(CH3)3), −5.4 (SiCH3), −5.7 (SiCH3); vmax (film) 3455, 2930, 2857, 1463, 1370, 1252, 1217, 1152, 1101, 1033, 999, 948, 919, 859, 837, 814, 777, 675 cm−1; HRMS (ESI+) m/z calcd for (C20H40NaO9Si)+, 475.2334; found 475.2344.
O), 169.7 (2 × CO), 169.6 (CO), 169.0 (CO), 168.3 (CO), 75.0, 71.4, 68.9, 68.8, 68.1, 68.0, 65.0, 20.8 (CH3), 20.7 (CH3), 20.7 (CH3), 20.6 (CH3), 20.5 (CH3), 20.5 (CH3); vmax (film) 3479, 2965, 1745, 1431, 1369, 1218, 1039, 899, 821, 731 cm−1; HRMS (ESI+) m/z calcd for (C19H26NaO13)+, 485.1266; found 485.1318.
CH), 6.03–5.98 (2H, m, CCH), 5.72 (1H, d, J = 10.0 Hz, CCH), 4.41 (1H, d, J = 4.5 Hz, CHOC(CH3)2), 4.15 (1H, d, J = 11.5 Hz, –CHH–), 3.94 (1H, d, J = 11.5 Hz –CHH–), 2.07 (3H, s, OAc), 1.44 (3H, s, CH3), 1.37 (3H, s, CH3); 13C NMR (75 MHz, CDCl3) δ = 170.6 (CO), 128.1, 125.4, 124.3, 123.1 (CC), 106.6 (C(CH3)2), 78.3 (CO(CH2)), 71.8 (C(H)OC(CH3)2), 66.1 (CH2), 27.1, 26.4 (C(CH3)2), 20.8 (COCH3); vmax (film) 2991, 2937, 1741, 1415, 1372, 1239, 1172, 1043, 906, 728, 648 cm−1; HRMS (ESI+) m/z calcd for (C12H16NaO4)+, 247.0941; found 247.0931.
CH), 6.55 (1H, t, J = 8.0 Hz, CCH), 4.88–4.91 (2H, m, C(H)O), 4.55 (1H, d, J = 12.0 Hz –CHH–), 4.31 (1H, d, J = 12.0 Hz –CHH–), 4.25 (1H, d, J = 4.8 Hz, C(H)OC(CH2)3), 2.13 (3H, s, COCH3), 1.39 (3H, s, CH3), 1.30 (3H, s, CH3); 13C NMR (100 MHz, CDCl3) δ = 170.4 (CO), 131.5 (CC), 130.3 (CC), 112.3 (C(CH3)2), 79.9 (CO(CH2), 74.6 (C(H)OC(CH3)2), 72.1 (CHO–O), 71.8 (CHO–O), 65.9 (CH2), 27.8 (C(CH3)2), 26.9 (C(CH3)2), 20.8 (COCH3); vmax (film) 2995, 2988, 2928, 1736, 1348, 1372, 1450, 1244, 1204, 1144, 1039, 919, 743, 712, 644 cm−1; HRMS (ESI+) m/z calcd for (C12H16NaO6)+, 279.0839; found 279.0829. Data for 38: Rf = 0.40 (15% EtOAc–petrol); [α]25D −14.3 (c 0.28, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ = 6.09 (1H, dd, J = 10.0, 4.0 Hz, HCCHCO), 5.70 (1H, dt, J = 10.0 1.5 Hz, HCCHCO), 4.65 (1H, t, J = 1.0 Hz, CHOC(CH3)2), 4.33 (1H, J = 11.0 Hz, –CHH–), 3.83 (1H, J = 11.0 Hz, –CHH–), 3.64 (1H, dd, J = 3.5, 2.5 Hz, CH(O)), 3.37 (1H, m, CH(O)), 2.09 (3H, s), 1.41 (3H, s), 1.37 (3H, s); 13C NMR (75 MHz, CDCl3) 170.4 (CO), 132.5 (CCC(O)CH2, 124.1 (CCCH(O)), 110.5 (C(CH3)2), 79.0 (CO(CH2), 71.3 (C(H)OC(CH3)2), 66.9 (CH2), 50.3 (CH(Oepox)CH(O)), 46.5 (CH(Oepox)CHCH), 27.8 (C–CH3), 26.6 (C–CH3), 20.9 (COCH3); vmax (film) 2989, 2943, 1745, 1455, 1379, 1236, 1181, 1162, 1089, 1060, 1042, 989, 829, 721 cm−1; HRMS (ESI+) m/z calcd for (C12H16NaO5)+, 263.0890; found 263.0903.
O), 110.4 (C(CH3)2), 78.2 (C(O)CH2OAc), 71.9 (C(H)OC(CH3)2), 65.5 (CH2), 51.5 (C(H)O), 50.9 (C(H)O), 47.6 (C(H)O), 47.6 (C(H)O), 28.0 (C(CH3)2), 26.2 (C(CH3)2), 20.8 (COCH3); vmax (film) 2991, 2938, 1742, 1455, 1435, 1380, 1231, 1175, 1063, 1043, 992, 964, 803, 630 cm−1; HRMS (ESI+) m/z calcd for (C12H16NaO6)+, 279.0839; found 279.0861.
O), 79.5 (C(OH)CO2H), 73.7 (CH(OH)), 35.0, 30.8, 25.3, 21.1; vmax (film) 3445, 3086, 2924, 2858, 1736, 1439, 1199, 1138, 1023, 948, 821 cm−1; HRMS (ESI−) m/z calcd for (C7H11O4)−, 159.0663; found 159.0672.
:1), was added dropwise TMS-CHN2 (0.200 mL, 2.0 M solution in THF, 0.400 mmol, 1.4 equiv.) until gas evolution ceased and a yellow colour persisted. The reaction mixture was concentrated under reduced pressure (caution: TMS-CHN2 toxic by inhalation). Pure 6 was obtained as a yellow oil (52 mg, 100%). Rf = 0.05 (30% EtOAc–petrol); [α]D −10.0 (c 0.3, CHCl3); 1H NMR (250 MHz, CDCl3) δ = 3.82–3.74 (1H, m, CH(OH)), 3.77 (3H, s, CH3), 2.86 (2H, br s, OH), 1.83–1.22 (8H, m, CH); 13C NMR (75 MHz, CDCl3) δ = 176.6 (CO), 76.8 (C(OH)(CO2CH3)), 72.2 (CH(OH)), 52.9 (OCH3), 34.2, 30.1, 24.0, 19.8; vmax (film) 3453, 2938, 2861, 1729, 1438, 1272, 1238, 1207, 1149, 1079, 998, 923 cm−1; HRMS (ESI+) m/z calcd for (C8H15O4)+, 175.0965; found 175.0981. NMR Data are in agreement with those previously reported.33
O), 109.0 (C(CH3)2), 81.0 C(OC(CH3)2)(CO2CH3), 75.0 (CH(OC(CH3)2)), 52.5 (OCH3), 32.3, 28.0, 26.0, 25.9, 20.5, 18.7; vmax (film) 2997, 2941, 2873, 1733, 1450, 1383, 1217, 1160, 1054, 1025, 905, 725 cm−1; HRMS (ESI+) m/z calcd for (C11H18NaO4)+, 237.1097; found 237.1079. Data for 8: Rf = 0.85 (50% EtOAc–petrol); [α]25D −21 (c 1.6, CHCl3); 1H NMR (500 MHz, CDCl3) δ = 5.08 (1H, dd, J = 10.0, 6.0 Hz, CH(OCO)), 4.26 (1H, t, J = 3.2 Hz, CH(O)), 3.73 (3H, s, OCH3), 3.24 (1H, s, OH), 2.06–1.24 (16H, m, CH), 1.50 (3H, s, CH3), 1.35 (3H, s, CH3); 13C NMR (125 MHz, CDCl3) δ = 175.3 (CO), 171.8 (CO), 108.9 (C(CH3)3), 80.7, 75.6, 75.2, 74.9, 53.0 (OCH3), 34.1, 32.2, 27.8, 26.2, 25.7, 25.6, 23.6, 20.2, 19.7, 18.3; vmax (film) 3523, 2987, 2938, 2865, 1733, 1449, 1381, 1370, 1243, 1216, 1152, 1124, 1046, 1003, 874, 735 cm−1; HRMS (ESI+) m/z calcd for (C18H29O7)+, 357.1908; found 357.1929.
O), 109.6 (C(CH3)2), 80.7 C(OC(CH3)2)(CO2H), 74.9 (CH(OC(CH3)2)), 32.2, 28.0, 26.0, 25.7, 20.7, 18.5; vmax (film) 2994, 2939, 2873, 2714, 1450, 1371, 1383, 1217, 1174, 905, 725 cm−1; HRMS (ESI−) m/z calcd for (C10H15O4)−, 199.0976; found 199.0975. Data for 10: Rf = 0.83 (50% EtOAc–petrol); [α]25D +2.0 (c 1.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ = 3.75 (1H, dd, J = 11.5, 4.6 Hz, CH(OH)), 1.95–1.93 (1H, m, CH), 1.92–1.89 (1H, m, CH), 1.88 (1H, br s, OH), 1.77–1.80 (1H, m, CH), 1.73 (1H, td, J = 14.0, 4.5 Hz, CH), 1.66 (3H, s, CH3), 1.63 (3H, s, CH3), 1.49–1.55 (1H, m, CH), 1.44–1.48 (1H, m, CH), 1.38 (1H, tt, J = 13.0, 3.5 Hz, CH); 13C NMR (125 MHz, CDCl3) δ = 174.0 (CO), 110.1 (C(CH3)2), 83.2 (C(O)(CO), 71.0 (C(OH)H), 33.7 (CH2), 30.6 (CH2), 29.1 (C(CH3)2), 27.9 (C(CH3)2), 23.6 (CH2), 20.1 (CH2); vmax (film) 3484, 2991, 2939, 2863, 2774, 1448, 1385, 1291, 1262, 1060, 1036, 908, 860, 626 cm−1; HRMS (ESI+) m/z calcd for (C10H17O4)+, 201.1121; found 201.1113.
CH), 5.94 (1H, dddd, J = 9.5, 5.0, 2.5, 0.5 Hz, CCH), 5.82 (1H, ddd, J = 9.5, 2.0, 1.0 Hz, CCH), 5.76 (1H, ddd, J = 9.5, 2.0, 1.0 Hz, CCH), 5.29 (2H, s, CO2–CH2–Ar), 4.87 (1H, m, C(O)H); 13C NMR (75.5 MHz, CDCl3) δ = 175.2 (CO), 135.1, 132.1, 128.8, 128.7, 128.2, 127.0, 124.8, 122.9, 74.1 (C(OH)CO), 71.1 (C(OH)–H), 68.5 (O–CH2); νmax (film) 3451, 3038, 1731, 1660, 1455, 1378, 1234, 1168, 1077, 1020, 909, 753, 695 cm−1; HRMS (+ve ESI-TOF) m/z calculated for (C14H14NaO4)+, 269.0784; found 269.0863.
CH), 5.86–5.79 (1H, m, CCH), 5.18 (2H, s, CH2), 4.97 (1H, d, J = 4.0 Hz, CH(O)), 1.38 (3H, s, CH3), 1.37 (3H, s, CH3). Data in agreement with those previously reported.6e
CH), 5.75–5.71 (1H, m, CCH), 5.25 (1H, d, J = 12.0 Hz, –CHH–), 5.18 (1H, d, J = 12.0 Hz, –CHH–), 4.97 (1H, d, J = 1.8 Hz, CH(OSi)), 0.99 (9H, s, CH3) 0.98 (9H, s, CH3); 13C NMR (75 MHz, CDCl3) δ = 172.2 (CO), 135.6, 128.6, 128.4, 128.3, 126.3, 125.8, 123.9, 122.3, 78.6 (C(OSi)(CO)), 71.5 (CH(OSi)), 67.4 (CH2), 27.0 (CH3), 26.9 (CH3), 21.2 (C(CH3)3), 20.2 (C(CH3)3); vmax (film) 3045, 2966, 2934, 2891, 2859, 1733, 1473, 1229, 1091, 1031, 1012, 1000, 875, 825, 696 cm−1; HRMS (ESI+) m/z calcd for (C22H31O4Si)+, 387.1986; found 387.1993.
O), 77.9 (C(OSi)(CO)), 73.9 (CH(OSi)), 33.0, 30.1, 27.7 (CH3), 27.5 (CH3), 23.7, 20.5, 20.3, 19.6; vmax (film) 3070, 2935, 2894, 2859, 1717, 1448, 1472, 1094, 827 cm−1; HRMS (ESI+) m/z calcd for (C15H29O4Si)+, 301.1830; found 301.1830.
O), 82.9, 81.7, 57.0, 52.1, 51.8, 29.7, 24.8, 23.5, 20.3; vmax (film) 2938, 2861, 1730, 1446, 1373, 1275, 1227, 1099, 1061, 997 cm−1; HRMS (ESI+) m/z calcd for (C10H19O4)+, 203.1278; found 203.1289. Data for 17: Rf = 0.85 (20% EtOAc–petrol); [α]25D −1.4 (c 0.73, CHCl3); 1H NMR (250 MHz, CDCl3) δ = 3.73 (3H, s, CO2CH3), 3.47 (1H, dd, J = 11.0, 4.5 Hz, CH(OCH3)), 3.29 (3H, s, OCH3), 1.85–0.84 (8H, m, CH), 0.15 (9H, s, Si(CH3)3); 13C NMR (75 MHz, CDCl3) δ = 175.6 (CO), 82.2, 76.1, 56.8, 51.8, 36.2, 25.1, 24.0, 20.3, 2.2; vmax (film) 2949, 2860, 1753, 1446, 1366, 1279, 1244, 1156, 1062, 1041, 837, 760, 734 cm−1; HRMS (ESI+) m/z calcd for (C12H25O4Si)+, 261.1517; found 261.1515.
:45:2.5:2.5 EtOAc–petrol–AcOH–H2O) to give 18 as a colourless oil (108 mg, 75%). Rf = 0.25 (50:45:2.5:2.5 EtOAc–petrol–AcOH–H2O); [α]25D −20.5 (c 0.73, CHCl3); 1H NMR (500 MHz, CDCl3) δ = 3.40–3.37 (1H, m, CH(OCH3)), 3.38 (3H, s, OCH3), 3.35 (3H, s, OCH3), 2.18 (1H, dd, J = 15.0, 2.0 Hz, CH), 1.96 (1H, m, CH), 1.82–1.81 (1H, m, CH), 1.68–1.53 (3H, m, CH), 1.37–1.25 (2H, m, CH); 13C NMR (125 MHz, CDCl3) δ = 175.3 (CO), 82.7, 81.2, 57.1, 52.0, 28.1, 24.8, 23.7, 20.0; vmax (film) 3143, 2940, 2860, 2838, 1721, 1464, 1448, 1308, 1196, 1099, 1077, 973, 939 cm−1; HRMS (ESI–) m/z calcd for (C9H15O4)−, 187.0976; found 187.0972.
:1 mixture of 19 and 20 by NMR. Data for 19: Rf = 0.49 (10% EtOAc–petrol); [α]25D −31 (c 0.7, CHCl3); 1H NMR (300 MHz, CDCl3) δ = 7.56–7.52 (2H, m, Ar–H), 7.40–7.37 (3H, m, Ar–H), 5.97 (1H, s, CH(O)(O)), 4.42 (1H, t, J = 4.0 Hz, CH(O)), 3.82 (3H, s, OCH3), 2.13–1.81 (4H, m, CH), 1.76–1.45 (4H, m, CH); 13C NMR (75 MHz, CDCl3) δ = 173.4 (CO), 137.2, 129.3, 128.3, 126.7 (Ar–C), 103.3 (CH(O)(O)(Ar)), 81.5 (C(O)(CO)), 77.3 (C(O)H), 52.5 (OCH3), 31.1, 26.1, 19.4, 18.5; vmax (film) 2951, 2869, 1734, 1451, 1247, 1163, 1088, 1024, 697 cm−1; HRMS (ESI+) m/z calcd for (C15H19O4)+, 263.1278; found 263.1267. Data for 20: Rf = 0.53 (10% EtOAc–petrol); [α]25D −1.6 (c 0.08, CHCl3); 1H NMR (300 MHz, CDCl3) δ = 7.50–7.46 (2H, m, Ar–H), 7.39–7.32 (3H, m, Ar–H), 6.21 (1H, s, CH(O)(O)), 4.53 (1H, t, J = 5.5 Hz, CH(O)), 3.70 (3H, s, OCH3), 2.03–1.89 (4H, m, CH), 1.75–1.39 (4H, m, CH); 13C NMR (75 MHz, CDCl3) δ = 173.0 (CO), 138.9, 129.0, 128.3, 126.4 (Ar–C), 102.8 (CH(O)(O)(Ar)), 81.9 (C(O)(CO)), 75.9 (C(O)H), 52.3 (OCH3), 30.7, 26.2, 20.9, 20.4; vmax (film) 2938, 2863, 1735, 1451, 1247, 1162, 1093, 698 cm−1; HRMS (ESI+) m/z calcd for (C15H19O4)+, 263.1278; found 263.1268.
O), 169.3 (CO), 111.4 (C(CH3)2), 85.2, 39.5, 38.7, 28.4, 27.5, 26.6, 21.0; vmax (film) 2996, 2941, 2921, 2872, 2851, 1784, 1732, 1394, 1380, 1283, 1254, 1130, 1077, 1045, 930 cm−1; HRMS (ESI+) m/z calcd for (C10H14NaO4)+, 221.0784; found 221.0787.
O), 77.5 (C(OH)), 73.8 (CH(OSi)), 52.2, 33.1, 30.1 (CH), 25.6 (SiC(CH3)3), 23.8, 19.6, 17.8 (SiC(CH3)3), −4.1, −5.2; vmax (film) 3555, 3027, 2928, 1705, 1494, 1453, 1281, 908, 732, 699 cm−1; HRMS (ESI+) m/z calcd for (C14H29O4Si)+, 289.1830; found 289.1831.
CH), 4.86 (1H, d, J = 6.5 Hz, OCH2O), 4.85 (1H, d, J = 6.5 Hz, OCH2O), 4.72 (1H, d, J = 6.5 Hz, OCH2O), 4.70 (1H, d, J = 6.5 Hz, OCH2O), 4.40 (1H, d, J = 3.0 Hz, CHO), 4.29 (1H, br s, CHOMOM), 4.22 (1H, br s, CHOMOM), 3.96 (1H, d, J = 11.0 Hz, CHHOTBDMS), 3.49 (1H, d, J = 11.0 Hz, CHHOTBDMS), 3.41 (3H, s, CH2OCH3), 3.39 (3H, s, CH2OCH3), 1.47 (3H, s, O–C–CH3), 1.43 (3H, s, O–C–CH3), 0.89 (9H, s, SiC(CH3)3), 0.04 (6H, s, Si(CH3)2); 13C NMR (75 MHz, CDCl3) δ = 130.3 (HCCH), 127.9 (HCCH), 108.9 (C(CH3)2), 96.3 (OCH2O), 95.5 (OCH2O), 85.3 (4°CCH2O), 78.4 (CHO), 76.8 (CHO), 75.0 (CHO), 61.9 (CH2OTBDMS), 55.5 (OCH3), 55.4 (OCH3), 28.4 (CCH3), 27.0 (CCH3), 25.9 (SiC(CH3)3), 18.3 (SiC(CH3)3), −5.3 (SiCH3), −5.7 (SiCH3); vmax (film) 2954, 1252, 1043, 907, 837, 732, 649 cm−1; HRMS (ESI+) m/z calcd for (C20H38NaO7Si)+, 441.2279; found 441.2285.
:1, 8.0 mL) was added NMO (43.4 mg, 0.370 mmol, 2.0 equiv.) followed dropwise by OsO4 (40 μL, 2.5% w/v in tert-BuOH, 3.7 μmol). The resulting solution was stirred at room temperature for 72 h. The reaction mixture was diluted with EtOAc (30 mL) and extracted with saturated Na2S2O3(aq) (2 × 30 mL). The organic phase was washed with brine (30 mL) and dried over MgSO4. Removal of the solvent under reduced pressure and purification by flash column chromatography (50% EtOAc–petrol) gave 32 as a colourless oil (56 mg, 67%). Rf = 0.43 (50% EtOAc–petrol); [α]25D −10.0 (c 0.1, CHCl3); 1H NMR (300 MHz, CDCl3) δ = 4.82 (2H, s, OCH2O), 4.74 (1H, d, J = 6.5 Hz, OCHHO), 4.67 (1H, d, J = 6.5 Hz, OCHHO), 4.47 (1H, d, J = 2.5 Hz, CHO), 4.18 (1H, m, CHO), 4.06 (1H, br s, CHO), 3.93 (1H, d, J = 11.0 Hz, CHHOTBDMS), 3.94–3.89 (1H, m, CHO) 3.79 (1H, dd, J = 10.0, 4.0 Hz, CHO) 3.64 (1H, d, J = 11.0 Hz, CHHOTBDMS), 3.45 (3H, s, CH2OCH3), 3.40 (3H, s, CH2OCH3), 3.05 (1H, br s, OH), 1.51 (3H, s, C(CH3)2), 1.37 (3H, s, C(CH3)2), 0.89 (9H, s, SiC(CH3)3), 0.07 (3H, s, SiCH3), 0.06 (3H, s, SiCH3); 13C NMR (75 MHz, CDCl3) δ = 108.6 (C(CH3)2), 98.1 (OCH2OCH3), 95.9 (OCH2OCH3), 85.3 (4°CCH2O), 83.0 (CHO), 76.4 (CHO), 73.4 (CHO), 70.9 (CHO), 69.5 (CHO), 61.2 (CH2OTBDMS), 55.9 (CH2OCH3), 55.8 (CH2OCH3), 28.2 (C(CH3)2), 26.3 (C(CH3)2), 25.9 (SiC(CH3)3), 18.3 (SiC(CH3)3), −5.4 (SiCH3), −5.7 (SiCH3); vmax (film) 3455, 2930, 2857, 1463, 1370, 1252, 1217, 1152, 1101, 1033, 999, 948, 919, 859, 837, 814, 777, 675 cm−1; HRMS (ESI+) m/z calcd for (C20H40NaO9Si)+, 475.2334; found 475.2344.
O), 169.7 (2 × CO), 169.6 (CO), 169.0 (CO), 168.3 (CO), 75.0, 71.4, 68.9, 68.8, 68.1, 68.0, 65.0, 20.8 (CH3), 20.7 (CH3), 20.7 (CH3), 20.6 (CH3), 20.5 (CH3), 20.5 (CH3); vmax (film) 3479, 2965, 1745, 1431, 1369, 1218, 1039, 899, 821, 731 cm−1; HRMS (ESI+) m/z calcd for (C19H26NaO13)+, 485.1266; found 485.1318.
CH), 6.03–5.98 (2H, m, CCH), 5.72 (1H, d, J = 10.0 Hz, CCH), 4.41 (1H, d, J = 4.5 Hz, CHOC(CH3)2), 4.15 (1H, d, J = 11.5 Hz, –CHH–), 3.94 (1H, d, J = 11.5 Hz –CHH–), 2.07 (3H, s, OAc), 1.44 (3H, s, CH3), 1.37 (3H, s, CH3); 13C NMR (75 MHz, CDCl3) δ = 170.6 (CO), 128.1, 125.4, 124.3, 123.1 (CC), 106.6 (C(CH3)2), 78.3 (CO(CH2)), 71.8 (C(H)OC(CH3)2), 66.1 (CH2), 27.1, 26.4 (C(CH3)2), 20.8 (COCH3); vmax (film) 2991, 2937, 1741, 1415, 1372, 1239, 1172, 1043, 906, 728, 648 cm−1; HRMS (ESI+) m/z calcd for (C12H16NaO4)+, 247.0941; found 247.0931.
CH), 6.55 (1H, t, J = 8.0 Hz, CCH), 4.88–4.91 (2H, m, C(H)O), 4.55 (1H, d, J = 12.0 Hz –CHH–), 4.31 (1H, d, J = 12.0 Hz –CHH–), 4.25 (1H, d, J = 4.8 Hz, C(H)OC(CH2)3), 2.13 (3H, s, COCH3), 1.39 (3H, s, CH3), 1.30 (3H, s, CH3); 13C NMR (100 MHz, CDCl3) δ = 170.4 (CO), 131.5 (CC), 130.3 (CC), 112.3 (C(CH3)2), 79.9 (CO(CH2), 74.6 (C(H)OC(CH3)2), 72.1 (CHO–O), 71.8 (CHO–O), 65.9 (CH2), 27.8 (C(CH3)2), 26.9 (C(CH3)2), 20.8 (COCH3); vmax (film) 2995, 2988, 2928, 1736, 1348, 1372, 1450, 1244, 1204, 1144, 1039, 919, 743, 712, 644 cm−1; HRMS (ESI+) m/z calcd for (C12H16NaO6)+, 279.0839; found 279.0829. Data for 38: Rf = 0.40 (15% EtOAc–petrol); [α]25D −14.3 (c 0.28, CH2Cl2); 1H NMR (300 MHz, CDCl3) δ = 6.09 (1H, dd, J = 10.0, 4.0 Hz, HCCHCO), 5.70 (1H, dt, J = 10.0 1.5 Hz, HCCHCO), 4.65 (1H, t, J = 1.0 Hz, CHOC(CH3)2), 4.33 (1H, J = 11.0 Hz, –CHH–), 3.83 (1H, J = 11.0 Hz, –CHH–), 3.64 (1H, dd, J = 3.5, 2.5 Hz, CH(O)), 3.37 (1H, m, CH(O)), 2.09 (3H, s), 1.41 (3H, s), 1.37 (3H, s); 13C NMR (75 MHz, CDCl3) 170.4 (CO), 132.5 (CCC(O)CH2, 124.1 (CCCH(O)), 110.5 (C(CH3)2), 79.0 (CO(CH2), 71.3 (C(H)OC(CH3)2), 66.9 (CH2), 50.3 (CH(Oepox)CH(O)), 46.5 (CH(Oepox)CHCH), 27.8 (C–CH3), 26.6 (C–CH3), 20.9 (COCH3); vmax (film) 2989, 2943, 1745, 1455, 1379, 1236, 1181, 1162, 1089, 1060, 1042, 989, 829, 721 cm−1; HRMS (ESI+) m/z calcd for (C12H16NaO5)+, 263.0890; found 263.0903.
O), 110.4 (C(CH3)2), 78.2 (C(O)CH2OAc), 71.9 (C(H)OC(CH3)2), 65.5 (CH2), 51.5 (C(H)O), 50.9 (C(H)O), 47.6 (C(H)O), 47.6 (C(H)O), 28.0 (C(CH3)2), 26.2 (C(CH3)2), 20.8 (COCH3); vmax (film) 2991, 2938, 1742, 1455, 1435, 1380, 1231, 1175, 1063, 1043, 992, 964, 803, 630 cm−1; HRMS (ESI+) m/z calcd for (C12H16NaO6)+, 279.0839; found 279.0861.
Acknowledgements
We thank EPSRC for funding (Studentship to JAG, Grant EP/G03768X/1; Impact Acceleration Account award to SEL).Notes and references
- For reviews, see: (a) D. J.-Y. D. Bon, B. Lee, M. G. Banwell and I. A. Cade, Chim. Oggi, 2012, 30, 22 CAS; (b) T. Hudlický, Pure Appl. Chem., 2010, 82, 1785 CrossRef; (c) T. Hudlický and J. W. Reed, Synlett, 2009, 685 CrossRef; (d) K. A. B. Austin, M. Matveenko, T. A. Reekie and M. G. Banwell, Chem. Aust., 2008, 75, 3 CAS; (e) D. R. Boyd and T. D. H. Bugg, Org. Biomol. Chem., 2006, 4, 181 RSC; (f) R. A. Johnson, Org. React., 2004, 63, 117 CAS; (g) M. G. Banwell, A. J. Edwards, G. J. Harfoot, K. A. Jolliffe, M. D. McLeod, K. J. McRae, S. G. Stewart and M. Vögtle, Pure Appl. Chem., 2003, 75, 223 CrossRef CAS; (h) T. Hudlický, D. Gonzales and D. T. Gibson, Aldrichimica Acta, 1999, 32, 35 Search PubMed; (i) D. A. Widdowson, D. W. Ribbons and S. D. Thomas, Janssen Chim. Acta, 1990, 8, 3 CAS.
- (a) J. M. Wallis and J. K. Kochi, J. Am. Chem. Soc., 1988, 110, 8207 CrossRef CAS; (b) W. B. Motherwell and A. S. Williams, Angew. Chem., Int. Ed. Engl., 1995, 34, 2031 Search PubMed; (c) P. M. J. Jung, W. B. Motherwell and A. S. Williams, Chem. Commun., 1997, 1283 RSC; (d) Y. Feng, C.-Y. Ke, G. Xue and L. Que Jr., Chem. Commun., 2009, 50 RSC.
- D. T. Gibson, J. R. Koch, C. L. Schuld and R. E. Kallio, Biochem., 1968, 7, 3795 CrossRef CAS.
- For recent (2011 onwards) examples, see: (a) D. K. Winter, M. A. Endoma-Arias, T. Hudlický, J. A. Beutler and J. A. Porco Jr., J. Org. Chem., 2013, 78, 7617 CrossRef CAS PubMed; (b) V. Varghese and T. Hudlický, Synlett, 2013, 369 CAS; (c) X. Ma, M. G. Banwell and A. C. Willis, J. Nat. Prod., 2013, 76, 1514 CrossRef CAS PubMed; (d) B. D. Schwartz, E. Matoušová, R. White, M. G. Banwell and A. C. Willis, Org. Lett., 2013, 15, 1934 CrossRef CAS PubMed; (e) D. J.-Y. D. Bon, M. G. Banwell, J. S. Ward and A. C. Willis, Tetrahedron, 2013, 69, 1363 CrossRef CAS PubMed; (f) D. R. Boyd, N. D. Sharma, J. G. Carroll, P. L. Loke, C. R. O'Dowd and C. C. R. Allen, RSC Adv., 2013, 3, 10944 RSC; (g) J. C. Ramos, M. Brovetto and G. A. Seoane, Org. Lett., 2013, 15, 1982 CrossRef CAS PubMed; (h) M. G. Banwell, N. Y. Gao, X. Ma, L. Petit, L. V. White, B. D. Schwartz, A. C. Willis and I. A. Cade, Pure Appl. Chem., 2012, 84, 1329 CrossRef CAS; (i) M. J. Palframan, G. Kociok-Köhn and S. E. Lewis, Chem.–Eur. J., 2012, 18, 4766 CrossRef CAS PubMed; (j) D. R. Boyd, N. D. Sharma, M. Kaik, P. B. A. McIntyre, P. J. Stevenson and C. C. R. Allen, Org. Biomol. Chem., 2012, 10, 2774 RSC; (k) M. K. Sharma, M. G. Banwell, A. C. Willis and A. D. Rae, Chem. Asian J., 2012, 7, 676 CrossRef CAS PubMed; (l) M. G. Banwell, N. Y. Gao, B. D. Schwartz and L. V. White, Top. Curr. Chem., 2012, 309, 163 CAS; (m) D. J.-Y. D. Bon, M. G. Banwell, I. A. Cade and A. C. Willis, Tetrahedron, 2011, 67, 8348 CrossRef CAS PubMed; (n) M. A. A. Endoma-Arias and T. Hudlický, Tetrahedron Lett., 2011, 52, 6632 CrossRef CAS PubMed; (o) B. D. Schwartz, M. G. Banwell and I. A. Cade, Tetrahedron Lett., 2011, 52, 4526 CrossRef CAS PubMed; (p) L. V. White, B. D. Schwartz, M. G. Banwell and A. C. Willis, J. Org. Chem., 2011, 76, 6250 CrossRef CAS PubMed; (q) M. J. Palframan, G. Kociok-Köhn and S. E. Lewis, Org. Lett., 2011, 13, 3150 CrossRef CAS PubMed; (r) J. Duchek, T. G. Piercy, J. Gilmet and T. Hudlický, Can. J. Chem., 2011, 89, 709 CrossRef CAS; (s) M. G. Banwell, A. L. Lehmann, R. S. Menon and A. C. Willis, Pure Appl. Chem., 2011, 83, 411 CrossRef CAS PubMed; (t) D. R. Adams, C. Aichinger, J. Collins, U. Rinner and T. Hudlický, Synlett, 2011, 725 CAS; (u) M. Labora, V. Schapiro and E. Pandolfi, Tetrahedron: Asymmetry, 2011, 22, 1705 CrossRef CAS PubMed; (v) X. Ma, J. C. Jury and M. G. Banwell, Tetrahedron Lett., 2011, 52, 2192 CrossRef CAS PubMed.
- For selected examples, see: (a) L. Werner, A. Machara, B. Sullivan, I. Carrera, M. Moser, D. R. Adams and T. Hudlický, J. Org. Chem., 2011, 76, 10050 CrossRef CAS PubMed; (b) L. Werner, A. Machara and T. Hudlický, Adv. Synth. Catal., 2010, 352, 195 CrossRef CAS; (c) F. Fabris, J. Collins, B. Sullivan, H. Leisch and T. Hudlický, Org. Biomol. Chem., 2009, 7, 2619 RSC; (d) B. Sullivan, I. Carrera, M. Drouin and T. Hudlický, Angew. Chem., Int. Ed., 2009, 48, 4229 Search PubMed; (e) M. Matveenko, A. C. Willis and M. G. Banwell, Tetrahedron Lett., 2008, 49, 7018 CrossRef CAS PubMed ; erratum, 2009, 50, 2982; ; (f) J.-J. Shie, J.-M. Fang and C.-H. Wong, Angew. Chem., Int. Ed., 2008, 47, 5788 CrossRef CAS PubMed; (g) A. T. Omori, K. J. Finn, H. Leisch, R. J. Carroll and T. Hudlický, Synlett, 2007, 2859 CAS; (h) M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395 CrossRef CAS PubMed; (i) M. G. Charest, D. R. Siegel and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 8292 CrossRef CAS PubMed; (j) I. W. Davies and P. J. Reider, Chem. Ind., 1996, 11, 412 CrossRef; (k) I. W. Davies, C. H. Senanayake, L. Castonguay, R. D. Larsen, T. R. Verhoeven and P. J. Reider, Tetrahedron Lett., 1995, 36, 7619 CrossRef CAS.
- For selected examples, see: (a) J. A. Griffen, J. C. White, G. Kociok-Köhn, M. D. Lloyd, A. Wells, T. C. Arnot and S. E. Lewis, Tetrahedron, 2013, 69, 5989 CrossRef CAS PubMed; (b) R. M. B. Carrilho, V. Heguaburu, V. Schapiro, E. Pandolfi, L. Kollár and M. M. Pereira, Tetrahedron, 2012, 68, 6935 CrossRef CAS PubMed; (c) V. de la Sovera, A. Bellomo, J. M. Pena, D. Gonzalez and H. A. Stefani, Mol. Diversity, 2011, 15, 163 CrossRef PubMed; (d) J. Duchek, D. R. Adams and T. Hudlický, Chem. Rev., 2011, 111, 4223 CrossRef CAS PubMed; (e) S. Pilgrim, G. Kociok-Köhn, M. D. Lloyd and S. E. Lewis, Chem. Commun., 2011, 47, 4799 RSC; (f) J. C. Ramos, P. Bracco, M. Mazzini, J. R. Fernández, D. Gamenara and G. A. Seoane, Tetrahedron: Asymmetry, 2010, 21, 969 CrossRef CAS PubMed; (g) D. R. Boyd, N. D. Sharma, N. I. Bowers, G. B. Coen, J. F. Malone, C. R. O'Dowd, P. J. Stevenson and C. C. R. Allen, Org. Biomol. Chem., 2010, 8, 1415 RSC; (h) A. Bellomo, A. Bertucci, H. Stefanic, Á. Vázquez and D. Gonzalez, Tetrahedron: Asymmetry, 2009, 20, 2673 CrossRef CAS PubMed; (i) A. Bellomo, J. B. Bonilla, J. López-Prados, M. Martín-Lomas and D. Gonzalez, Tetrahedron: Asymmetry, 2009, 20, 2061 CrossRef CAS PubMed; (j) A. Bellomo, S. Camarano, C. Rossini and D. Gonzalez, Carbohydr. Res., 2009, 344, 44 CrossRef CAS PubMed; (k) A. Bellomo, D. Gonzalez and H. A. Stefani, J. Organomet. Chem., 2008, 693, 1136 CrossRef CAS PubMed; (l) A. Bellomo and D. Gonzalez, Tetrahedron Lett., 2007, 48, 3047 CrossRef CAS PubMed; (m) G. Fonseca and G. A. Seoane, Tetrahedron: Asymmetry, 2005, 16, 1393 CrossRef CAS PubMed; (n) D. R. Boyd, N. D. Sharma, N. M. Llamas, J. F. Malone, C. R. O'Dowd and C. C. R. Allen, Org. Biomol. Chem., 2005, 3, 1953 RSC; (o) M. H. Parker, B. E. Maryanoff and A. B. Reitz, Synlett, 2004, 2095 CrossRef CAS PubMed; (p) C. Vitelio, A. Bellomo, M. Brovetto, G. Seoane and D. Gonzalez, Carbohydr. Res., 2004, 339, 1773 CrossRef CAS PubMed; (q) M. G. Banwell, X. Ma, N. Asano, K. Ikeda and J. N. Lambert, Org. Biomol. Chem., 2003, 1, 2035 RSC; (r) B. J. Paul, J. Willis, T. Martinot, I. Ghiviriga, K. A. Abboud and T. Hudlický, J. Am. Chem. Soc., 2002, 124, 10416 CrossRef CAS PubMed; (s) M. G. Banwell, A. M. Bray and D. J. Wong, New J. Chem., 2001, 25, 1351 RSC; (t) S. H. Lee and C. S. Cheong, Bull. Korean Chem. Soc., 2000, 21, 1061 CAS; (u) M. G. Banwell, C. DeSavi, D. C. R. Hockless, S. Pallich and K. G. Watson, Synlett, 1999, S1, 885–888 Search PubMed; (v) M. Banwell, C. DeSavi and K. Watson, J. Chem. Soc., Perkin Trans. 1, 1998, 2251 RSC; (w) M. Banwell, C. DeSavi and K. Watson, Chem. Commun., 1998, 1189 RSC; (x) M. Desjardins, L. E. Brammer Jr. and T. Hudlický, Carbohydr. Res., 1997, 304, 39 CrossRef CAS; (y) T. Hudlický, K. A. Abboud, D. A. Entwistle, R. Fan, R. Maurya, A. J. Thorpe, J. Bolonick and B. Myers, Synthesis, 1996, 897 CrossRef PubMed.
- (a) V. P. Bui and T. Hudlický, Tetrahedron, 2004, 60, 641 CrossRef CAS PubMed; (b) M. G. Williams, P. E. Olson, K. J. Tautvydas, R. M. Bitner, R. A. Mader and L. P. Wackett, Appl. Microbiol. Biotechnol., 1990, 34, 316 CrossRef CAS; (c) D. G. H. Ballard, A. Courtis, I. M. Shirley and S. C. Taylor, Macromolecules, 1988, 21, 294 CrossRef CAS; (d) D. G. H. Ballard, A. Courtis, I. M. Shirley and S. C. Taylor, J. Chem. Soc., Chem. Commun., 1983, 954 RSC.
- (a) J. L. Royo, E. Moreno-Ruiz, A. Cebolla and E. Santero, J. Biotechnol., 2005, 116, 113 CrossRef CAS PubMed; (b) J. Y. Kim, K. Lee, Y. Kim, C.-K. Kim and K. Lee, Lett. Appl. Microbiol., 2003, 36, 343 CrossRef CAS; (c) A. Berry, T. C. Dodge, M. Pepsin and W. Weyler, J. Ind. Microbiol. Biotechnol., 2002, 28, 127 CrossRef CAS; (d) B. D. Ensley, B. J. Ratzkin, T. D. Osslund, M. J. Simon, L. P. Wackett and D. T. Gibson, Science, 1983, 222, 167 CAS.
- (a) T. D. H. Bugg, Tetrahedron, 2003, 59, 7075 CrossRef CAS; (b) D. T. Gibson and R. E. Parales, Curr. Opin. Biotechnol., 2000, 11, 236 CrossRef CAS.
- D. R. Boyd, N. D. Sharma, M. V. Hand, M. R. Groocock, N. A. Kerley, H. Dalton, J. Chima and G. N. Sheldrake, J. Chem. Soc., Chem. Commun., 1993, 974 RSC.
- A. M. Reiner and G. D. Hegeman, Biochemistry, 1971, 10, 2530 CrossRef CAS.
- A. E. G. Cass, D. W. Ribbons, J. T. Rossiter and S. R. Williams, Biochem. Soc. Trans., 1986, 14, 1268 CAS.
- S.-Y. Sun, X. Zhang, Q. Zhou, J.-C. Chen and G.-Q. Chen, Appl. Microbiol. Biotechnol., 2008, 80, 977 CrossRef CAS PubMed.
- G. N. Jenkins, D. W. Ribbons, D. A. Widdowson, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Perkin Trans. 1, 1995, 2647 RSC.
- A. G. Myers, D. R. Siegel, D. J. Buzard and M. G. Charest, Org. Lett., 2001, 3, 2923 CrossRef CAS PubMed.
- (a) M. D. Mihovilovic, H. G. Leisch and K. Mereiter, Tetrahedron Lett., 2004, 45, 7087 CrossRef CAS PubMed; (b) T. C. M. Fischer, H. G. Leisch and M. D. Mihovilovic, Monatsh. Chem., 2010, 141, 699 CrossRef CAS.
- (a) M. Ali Khan, M. F. Mahon, J. P. Lowe, A. J. W. Stewart and S. E. Lewis, Chem.–Eur. J., 2012, 18, 13480 CrossRef CAS PubMed; (b) D. van der Waals, T. Pugh, M. Ali Khan, A. J. W. Stewart, A. L. Johnson and S. E. Lewis, Chem. Central J., 2011, 5, 80 CrossRef CAS PubMed; (c) J. A. Griffen, A. M. Le Coz, G. Kociok-Köhn, M. Ali Khan, A. J. W. Stewart and S. E. Lewis, Org. Biomol. Chem., 2011, 9, 3920 RSC; (d) M. Ali Khan, J. P. Lowe, A. L. Johnson, A. J. W. Stewart and S. E. Lewis, Chem. Commun., 2011, 47, 215 RSC; (e) M. Ali Khan, M. F. Mahon, A. J. W. Stewart and S. E. Lewis, Organometallics, 2010, 29, 199 CrossRef CAS.
- (a) J. P. Guthrie, J. Cossar and J. Lu, Can. J. Chem., 1991, 69, 1904 CrossRef CAS; (b) G. Hesse and G. Krehbiel, Liebigs Ann., 1955, 593, 42 CAS.
- D. R. Boyd, N. D. Sharma, M. V. Berberian, K. S. Dunne, C. Hardacre, M. Kaik, B. Kelly, J. F. Malone, S. T. McGregor and P. J. Stevenson, Adv. Synth. Catal., 2010, 352, 855 CrossRef CAS.
- J. G. Buchanan, A. R. Edgar, D. I. Rawson, P. Shahidi and R. H. Wightman, Carbohydr. Res., 1982, 100, 75 CrossRef CAS.
- (a) J. T. Sharp, I. Gosney and A. G. Rowley, in Practical Organic Chemistry, Chapman and Hall, New York, 1989 Search PubMed; (b) J. M. Bohen, M. M. Joullié, F. A. Kaplan and B. Loev, J. Chem. Educ., 1973, 50, 367 CrossRef CAS.
- For reviews on inositols, see: (a) B. Kilbas and M. Balci, Tetrahedron, 2011, 67, 2355 CrossRef CAS PubMed; (b) S. J. Conway and G. J. Miller, Nat. Prod. Rep., 2007, 24, 687 RSC; (c) B. V. L. Potter and D. Lampe, Angew. Chem., Int. Ed. Engl., 1995, 34, 1933 CrossRef CAS.
- T. Hudlický, D. A. Entwistle, K. K. Pitzer and A. J. Thorpe, Chem. Rev., 1996, 96, 1195 CrossRef PubMed.
- (a) H. A. J. Carless and Y. Dove, Tetrahedron: Asymmetry, 1996, 7, 649 CrossRef CAS; (b) H. A. J. Carless and S. S. Malik, J. Chem. Soc., Chem. Commun., 1995, 2447 RSC; (c) H. A. J. Carless, K. Busia, Y. Dove and S. S. Malik, J. Chem. Soc., Perkin Trans. 1, 1993, 2505 RSC; (d) H. A. J. Carless and K. Busia, Carbohydr. Res., 1992, 234, 207 CrossRef CAS; (e) H. A. J. Carless, Tetrahedron Lett., 1992, 33, 6379 CrossRef CAS; (f) H. A. J. Carless, K. Busia and O. Z. Oak, Synlett, 1993, 672 CrossRef CAS; (g) H. A. J. Carless and S. S. Malik, Tetrahedron: Asymmetry, 1992, 3, 1135 CrossRef CAS; (h) H. A. J. Carless, J. Chem. Soc., Chem. Commun., 1992, 234 RSC; (i) H. A. J. Carless and O. Z. Oak, Tetrahedron Lett., 1991, 32, 1671 CrossRef CAS; (j) H. A. J. Carless and O. Z. Oak, J. Chem. Soc., Chem. Commun., 1991, 61 RSC; (k) H. A. J. Carless and K. Busia, Tetrahedron Lett., 1990, 31, 3449 CrossRef CAS; (l) H. A. J. Carless and K. Busia, Tetrahedron Lett., 1990, 31, 1617 CrossRef CAS; (m) H. A. J. Carless, J. A. Billinge and O. Z. Oak, Tetrahedron Lett., 1989, 30, 3113 CrossRef CAS; (n) H. A. J. Carless and O. Z. Oak, Tetrahedron Lett., 1989, 30, 1719 CrossRef CAS.
- (a) S. Freeman and T. Hudlický, Bioorg. Med. Chem. Lett., 2004, 14, 1209 CrossRef CAS PubMed; (b) B. J. Paul, E. Hobbs, P. Buccino and T. Hudlický, Tetrahedron Lett., 2001, 42, 6433 CrossRef CAS; (c) B. J. Paul, T. A. Martinot, J. Willis and T. Hudlický, Synthesis, 2001, 952 CrossRef CAS PubMed; (d) T. Hudlický, N. Restrepo-Sanchez, P. D. Kary and L. M. Jaramillo-Gomez, Carbohydr. Res., 2000, 324, 200 CrossRef; (e) T. Hudlický, C. D. Claeboe, L. E. Brammer Jr., L. Koroniak, G. Butora and I. Ghiviriga, J. Org. Chem., 1999, 64, 4909 CrossRef PubMed; (f) K. A. Oppong, T. Hudlický, F. Yan, C. York and B. V. Nguyen, Tetrahedron, 1999, 55, 2875 CrossRef CAS; (g) L. E. Brammer Jr. and T. Hudlický, Tetrahedron: Asymmetry, 1998, 9, 2011 CrossRef; (h) M. Desjardins, L. E. Brammer Jr. and T. Hudlický, Carbohydr. Res., 1997, 304, 39 CrossRef CAS; (i) B. V. Nguyen, C. York and T. Hudlický, Tetrahedron, 1997, 53, 8807 CrossRef CAS; (j) T. Hudlický, K. A. Abboud, J. Bolonick, R. Maurya, M. L. Stanton and A. J. Thorpe, Chem. Commun., 1996, 1717 RSC; (k) T. Hudlický, K. A. Abboud, D. A. Entwistle, R. Fan, R. Maurya, A. J. Thorpe, J. Bolonick and B. Myers, Synthesis, 1996, 897 CrossRef PubMed; (l) T. Hudlický and A. J. Thorpe, Synlett, 1994, 899 CrossRef; (m) T. Hudlický, M. Mandel, J. Rouden, R. S. Lee, B. Bachmann, T. Dudding, K. J. Yost and J. S. Merola, J. Chem. Soc., Perkin Trans. 1, 1994, 1553 RSC; (n) M. Mandel and T. Hudlický, J. Chem. Soc., Perkin Trans. 1, 1993, 741 RSC ; erratum, p. 1537; ; (o) M. Mandel, T. Hudlický, L. D. Kwart and G. M. Whited, J. Org. Chem., 1993, 58, 2331 CrossRef CAS.
- T. Tschamber, F. Backenstrass, H. Fritz and J. Streith, Helv. Chim. Acta, 1992, 75, 1052 CrossRef CAS.
- (a) S. V. Ley, A. J. Redgrave and L. L. Yeung, ACS Symp. Ser., 1991, 463, 132 CrossRef CAS PubMed; (b) S. V. Ley, Pure Appl. Chem., 1990, 62, 2031 CrossRef CAS; (c) S. V. Ley, M. Parra, A. J. Redgrave and F. Sternfeld, Tetrahedron, 1990, 46, 4995 CrossRef CAS; (d) S. V. Ley and F. Sternfeld, Tetrahedron, 1989, 45, 3463 CrossRef CAS; (e) S. V. Ley, M. Parra, A. J. Redgrave, F. Sternfeld and A. Vidal, Tetrahedron Lett., 1989, 30, 3557 CrossRef CAS; (f) S. V. Ley and F. Sternfeld, Tetrahedron Lett., 1988, 29, 5305 CrossRef CAS; (g) S. V. Ley, F. Sternfeld and S. Taylor, Tetrahedron Lett., 1987, 28, 225 CrossRef CAS.
- For examples, see: (a) A. Baran, G. Aydin, T. Savran, E. Sahin and M. Balci, Org. Lett., 2013, 15, 4350 CrossRef CAS PubMed; (b) Z. Ekmekci and M. Balci, Eur. J. Org. Chem., 2012, 4988 CrossRef CAS; (c) A. Baran, S. Çambul, M. Nebioglu and M. Balci, J. Org. Chem., 2012, 77, 5086 CrossRef CAS PubMed; (d) N. I. Kurbanoğlu, M. Çelik, H. Kilic, C. Alp, E. Şahin and M. Balci, Tetrahedron, 2010, 66, 3485 CrossRef PubMed; (e) S. Cantekin, A. Baran, R. Çalişkan and M. Balci, Carbohydr. Res., 2009, 344, 426 CrossRef CAS PubMed; (f) A. Baran and M. Balci, J. Org. Chem., 2009, 74, 88 CrossRef CAS PubMed; (g) A. Coşkun, M. Güney, A. Daştan and M. Balci, Tetrahedron, 2007, 63, 4944 CrossRef PubMed; (h) M. Çelik and M. Balci, ARKIVOC, 2007, 150 Search PubMed; (i) E. Salamci, B. Bayram and M. Balci, Turk. J. Chem., 2006, 30, 441 CAS; (j) Ş. D. Yardımcı, N. Kaya and M. Balci, Tetrahedron, 2006, 62, 10633 CrossRef PubMed; (k) L. Kelebekli, M. Çelik, E. Şahin, Y. Kara and M. Balci, Tetrahedron Lett., 2006, 47, 7031 CrossRef CAS PubMed; (l) L. Kelebekli, Y. Kara and M. Balci, Carbohydr. Res., 2005, 340, 1940 CrossRef CAS PubMed; (m) M. S. Gültekin, M. Çelik, E. Turkut, C. Tanyeli and M. Balci, Tetrahedron: Asymmetry, 2004, 15, 453 CrossRef PubMed ; erratum, p. 1959; ; (n) M. S. Gültekin, E. Salamci and M. Balci, Carbohydr. Res., 2003, 338, 1615 CrossRef; (o) Y. Kara and M. Balci, Tetrahedron, 2003, 59, 2063 CrossRef CAS.
- (a) G. Mehta and S. Sen, Tetrahedron Lett., 2010, 51, 503 CrossRef CAS PubMed; (b) G. Luchetti, K. Ding, M. d'Alarcao and A. Kornienko, Synthesis, 2008, 3142 CAS; (c) Y. J. Lee, K. Lee, S. I. Jung, H. B. Jeon and K. S. Kim, Tetrahedron, 2005, 61, 1987 CrossRef CAS PubMed; (d) G. Mehta, R. S. Senaiar and S. Sen, Proc. Indian Acad. Sci. A, 2005, 71, 121 CAS; (e) G. Mehta, R. S. Senaiar and M. K. Bera, Chem.–Eur. J., 2003, 9, 2264 CrossRef CAS PubMed; (f) A. Kornienko and M. d'Alarcao, Tetrahedron: Asymmetry, 1999, 10, 827 CrossRef; (g) K. K. Reddy, J. R. Falck and J. Capdevila, Tetrahedron Lett., 1993, 34, 7869 CrossRef CAS.
- (a) H. Paulsen and F. R. Heiker, Liebigs Ann., 1981, 2180 CAS; (b) H. Paulsen and F. R. Heiker, Angew. Chem., 1980, 92, 930 CrossRef CAS; (c) S. J. Angyal, J. E. Klavins and J. A. Mills, Aust. J. Chem., 1974, 27, 1075 CrossRef CAS; (d) T. Posternak, D. Janjic, E. A. C. Lucken and A. Szente, Helv. Chim. Acta, 1967, 50, 1027 CrossRef CAS; (e) T. Posternak, Proc. 2nd Meet. Fed. Eur. Biochem. Soc., 1966, p. 31; (f) T. Posternak and J.-G. Falbriard, Helv. Chim. Acta, 1960, 43, 2142 CrossRef; (g) T. Posternak and D. Reymond, Helv. Chim. Acta, 1953, 36, 1370 CrossRef CAS; (h) T. Posternak, Helv. Chim. Acta, 1950, 33, 1594 CrossRef CAS; (i) T. Posternak, Helv. Chim. Acta, 1944, 27, 457 CrossRef CAS.
- (a) J. D. Boyd, C. S. Foote and D. K. Imagawa, J. Am. Chem. Soc., 1980, 102, 3641 CrossRef CAS; (b) Y. Sütbeyaz, H. Seçen and M. Balci, J. Org. Chem., 1988, 53, 2312 CrossRef; (c) M. Balci and Y. Sütbeyaz, Tetrahedron Lett., 1983, 24, 311 CrossRef CAS.
- (a) C. W. Jefford, Chem. Soc. Rev., 1993, 22, 59 RSC; (b) A. A. Frimer, Chem. Rev., 1979, 79, 359 CrossRef CAS; (c) C. W. Jefford and C. G. Rimbault, J. Org. Chem., 1978, 43, 1908 CrossRef CAS.
- (a) T. Nagasawa, N. Shimada, M. Torihata and S. Kuwahara, Tetrahedron, 2010, 66, 4965 CrossRef CAS PubMed; (b) J. Huang and E. J. Corey, Org. Lett., 2003, 5, 3455 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all novel compounds, as well as selected 2D NMR spectra. See DOI: 10.1039/c3qo00057e |
| This journal is © the Partner Organisations 2014 |