Palladium-catalyzed direct ortho-C–H ethoxycarboxylation of anilides at room temperature†
Yumin
Huang
,
Gaocan
Li
,
Jingsheng
Huang
and
Jingsong
You
*
Key Laboratory of Green Chemistry and Technology of Ministry of Education, College of Chemistry, and State Key Laboratory of Biotherapy, West China Medical School, Sichuan University, 29 Wangjiang Road, Chengdu 610064, PR China. E-mail: jsyou@scu.edu.cn; Fax: (+86) 28-85412203; Tel: (+86) 28-85412203
First published on 11th March 2014
Abstract
A Pd-catalyzed direct ortho-C–H ethoxycarboxylation of anilides has been established to form various anthranilic acid derivatives at room temperature. This protocol provides a concise and efficient strategy for diversifying the preparation of anthranilic acid derivatives from readily available starting materials under mild conditions.
Transition metal-catalyzed direct C–H functionalization has emerged as a promising tool for the formation of carbon–carbon and carbon–heteroatom bonds, which effectively avoids time-consuming prefunctionalization of substrates.1 Although a large number of efficient catalyst systems have been reported over the past few years, most of them require the use of harsh reaction conditions, which may result in incompatibilities of functional groups and further limit substrate scope. Therefore, a highly attractive, yet challenging objective is the development of C–H functionalizations with high efficiency and selectivity under mild conditions.2
Direct introduction of a carboxyl or ester group into the aromatic ring via C–H functionalization has been an area of intensive research. In 1980, Fujiwara reported the first example of direct carboxylation reaction of arenes with CO to synthesize aromatic acids.3 After this breakthrough, much effort has been devoted to developing efficient C–H carboxylation reactions by utilizing CO and CO2 as the carboxylation reagent.4 Yu developed the Pd-catalyzed chelation-assisted C–H ortho-carbonylation reaction under a CO atmosphere.4a,c Iwasawa presented the Rh-catalyzed direct carboxylation of the aryl C–H bond with CO2.4d Besides CO and CO2, other carboxylation regents such as carbamoyl chlorides,5 diaziridinone,6 diethyl azodicarboxylate7 and glyoxylates8 have also been established for the direct C–H carboxylation in the past few years. In recent years, the direct carboxylation reaction has become an attractive synthetic method to prepare aromatic acid derivatives.
Anthranilic acid derivatives are important precursors for the synthesis of heterocyclic natural products, pharmacologically important molecules, and biologically active compounds. In particular, they present a privileged architectural unit in heterocyclic frameworks such as quinazoline, quinoline, acridinone and benzoxazinone.9 Although a number of examples have been presented to prepare anthranilic acid derivatives,10 the general procedures have been less developed, which would most likely greatly restrict rapid diversification of significant molecules. Recently, the Pd-catalyzed C–H ortho-carbonylation of anilides with CO was well established for the synthesis of anthranilic acids (eqn (1)).4b,c However, to a certain extent, the handling of the hazardous gas would cause difficulties in the further application of this carbonylation reaction in research laboratories. The Tan group developed a route to anthranilic esters via Pd-catalyzed dehydrogenative/decarbonylative coupling between anilides and glyoxylates with the assistance of the phosphine ligand at 120 °C (eqn (2)).8 In spite of the significant progress made, it is still critically important to develop new efficient methods to synthesize anthranilic acid derivatives under mild conditions from both a scientific and a practical standpoint.
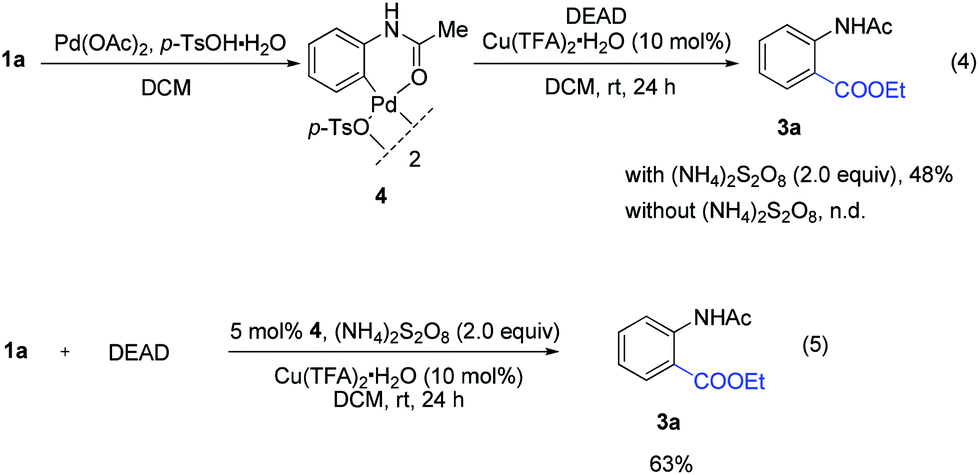
We initially tried to optimize the reaction conditions by using acetanilide 1a and DEAD (Table 1; for details, see ESI†). After screening a number of oxidants (Table 1, entries 1–6), we found that (NH4)2S2O8 could promote this reaction in 28% yield. The yield could be improved slightly by increasing the amount of 2 (Table 1, entry 7). It is worth noting that p-toluenesulfonic acid (p-TsOH) played a critical role in the coupling (Table 1, entry 8). We believe that the TsO− anion is likely attached to Pd(II) to form Pd(OTs)X (X = anion), which might facilitate the ortho-C–H bond cleavage of acetanilide.4c,13 Other acids led to lower yields or did not afford the target product (Table 1, entries 9 and 10). An improved yield was obtained by using Pd(TFA)2 instead of Pd(OAc)2 (Table 1, entry 14). To our delight, addition of Cu(TFA)2·H2O could improve the yield to 68% (Table 1, entry 16). Although the role of Cu(II) salt is unclear at this stage, we speculate that Cu(II) salt may serve as an additive to promote the single electron transfer process in the reaction cycle.14 Finally, the best result was obtained when the reaction was performed in the presence of Pd(TFA)2 (10 mol%), Cu(TFA)2·H2O (10 mol%), (NH4)2S2O8 (2.0 equiv.) and p-TsOH·H2O (0.5 equiv.) in DCM at ambient temperature for 24 h.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Pd | Oxidant | Additive | Solvent | Yieldb (%) |
| a Reactions were carried out using acetanilide (0.3 mmol), DEAD (3.0 equiv.), [Pd] (10 mol%), oxidant (2.0 equiv.), additive (0.5 equiv.) in solvent (1 mL) at rt. b Isolated yield. c DEAD (2.5 equiv.). d Cu(TFA)2·H2O (10 mol%). DCM = CH2Cl2, BQ = benzoquinone, TBHP = tert-butylhydroperoxide, Oxone = potassium monopersulfate. | |||||
| 1c | Pd(OAc)2 | Na2S2O8 | p-TsOH·H2O | DCM | n.d. |
| 2c | Pd(OAc)2 | K2S2O8 | p-TsOH·H2O | DCM | n.d. |
| 3c | Pd(OAc)2 | Oxone | p-TsOH·H2O | DCM | n.d. |
| 4c | Pd(OAc)2 | TBHP | p-TsOH·H2O | DCM | n.d. |
| 5c | Pd(OAc)2 | BQ | p-TsOH·H2O | DCM | n.d. |
| 6c | Pd(OAc)2 | (NH4)2S2O8 | p-TsOH·H2O | DCM | 28% |
| 7 | Pd(OAc)2 | (NH4)2S2O8 | p-TsOH·H2O | DCM | 34% |
| 8 | Pd(OAc)2 | (NH4)2S2O8 | — | DCM | Trace |
| 9 | Pd(OAc)2 | (NH4)2S2O8 | CH3SO3H | DCM | 13% |
| 10 | Pd(OAc)2 | (NH4)2S2O8 | TfOH | DCM | 27% |
| 11 | Pd(OAc)2 | (NH4)2S2O8 | p-TsOH·H2O | MeCN | n.d. |
| 12 | Pd(OAc)2 | (NH4)2S2O8 | p-TsOH·H2O | THF | 21% |
| 13 | Pd(OAc)2 | (NH4)2S2O8 | p-TsOH·H2O | DCE | 16% |
| 14 | Pd(TFA)2 | (NH4)2S2O8 | p-TsOH·H2O | DCM | 48% |
| 15 | — | (NH4)2S2O8 | p-TsOH·H2O | DCM | n.d. |
| 16d | Pd(TFA)2 | (NH4)2S2O8 | p-TsOH·H2O/Cu(TFA)2·H2O | DCM | 68% |
| 17d | Pd(TFA)2 | Selectfluor | p-TsOH·H2O/Cu(TFA)2·H2O | DCM | 67% |
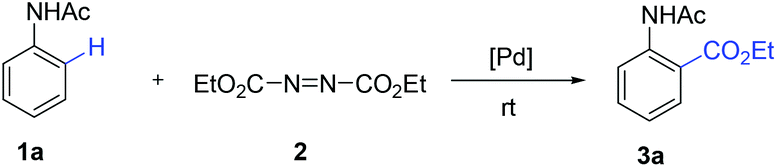
Under the optimized reaction conditions, the scope of substituted acetanilides was tested. To our delight, acetanilides bearing both electron-donating and electron-withdrawing groups could afford the desired products (Table 2, 3b–o). Acetanilides with methyl and t-butyl groups at the para-position gave 3c and 3d in 83% and 73% yields, respectively. A variety of electron-donating groups such as MeO-, nBuO- and BnO-groups could be tolerated, and moderate yields were obtained (Table 2, 3e–g, 3m). The protocol was compatible with the halogen atoms such as fluoro, chloro and bromo, which might be useful for further synthetic transformations (Table 2, 3i–k, 3m–n). It was noted that meta-substituted acetanilides exhibited excellent regioselectivities (Table 2, 3b, 3j–n). The disubstituted acetanilides could smoothly furnish the desired products with excellent regioselectivities (Table 2, 3l–n). In addition, anilides with other directing groups such as pivaloyl, benzoyl and propionyl groups could also undergo the ethoxycarboxylation under the optimized reaction conditions (Table 2, 3p–r).
To get insights into the mechanism, the following controlled experiments were carried out. First, radical scavengers such as hydroquinone, TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy) and BHT (2,6-di-tert-butyl-4-methylphenol) had a dramatic effect on the coupling reaction (Table S8†). Although the radical intermediate could not be captured, we believed that a radical pathway might be involved.7 Subsequently, we found that the stoichiometric reaction of the palladium complex 44c with DEAD could afford the desired product 3a (eqn (4)). However, no reaction occurred in the absence of (NH4)2S2O8. When the reaction was performed using 4 as the catalyst, 3a was obtained in 63% yield (eqn (5)). These observations implied that the ethoxycarboxylation might undergo a Pd(II)/Pd(IV) catalytic cycle.12,15
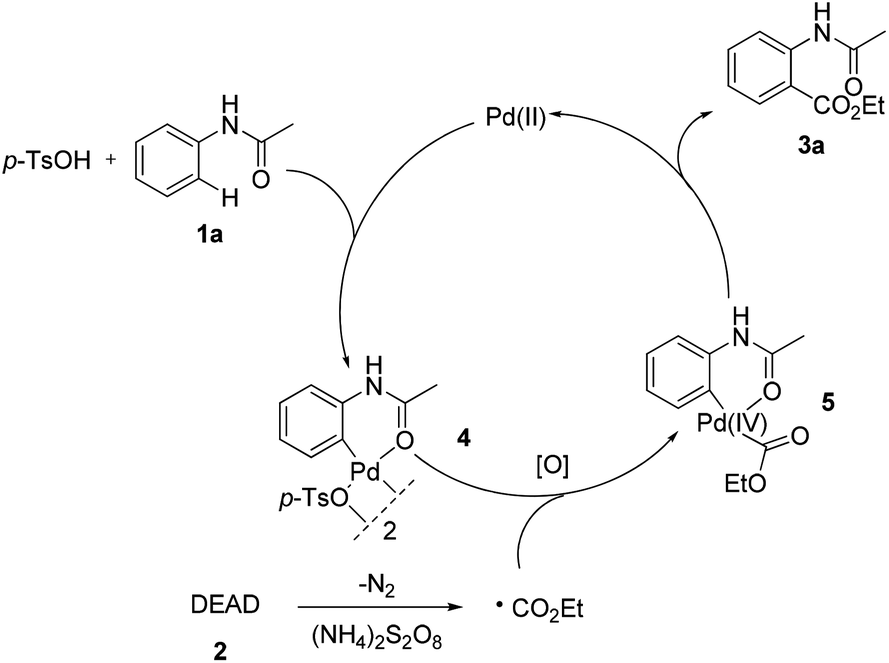 | ||
| Scheme 1 A possible reaction mechanism. | ||
In summary, we have developed an efficient Pd(II)-catalyzed ortho-C–H esterification of anilides with a commercially available and easily handled azodicarboxylate agent at ambient temperature. This selective and mild oxidative coupling reaction could be a complementary and practical protocol for the rapid synthesis of anthranilic acid derivatives from the corresponding anilines because of concise manipulation and non-protection against air or moisture.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (973 Program, 2011CB808601), and the National NSF of China (no. 21025205, 21272160, 21321061, J1310008, and J1103315J0104).Notes and references
- For selected reviews, see: (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (c) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (d) B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949 CAS; (e) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (f) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (g) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed.
- For a review, see: (a) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; For selected examples, see: (b) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS; (c) B. Xiao, Y. Fu, J. Xu, T.-J. Gong, J.-J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468 CrossRef CAS PubMed; (d) T. Nishikata, A. R. Abela, S. Huang and B. H. Lipshutz, J. Am. Chem. Soc., 2010, 132, 4978 CrossRef CAS PubMed; (e) P. Fang, M. Li and H. Ge, J. Am. Chem. Soc., 2010, 132, 11898 CrossRef CAS PubMed; (f) T. Nishikata, A. R. Abela and B. H. Lipshutz, Angew. Chem., Int. Ed., 2010, 49, 781 CrossRef CAS PubMed; (g) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed; (h) C. Tang and N. Jiao, J. Am. Chem. Soc., 2012, 134, 18924 CrossRef CAS PubMed; (i) R. Zeng, S. Wu, C. Fu and S. Ma, J. Am. Chem. Soc., 2013, 135, 18284 CrossRef CAS PubMed.
- Y. Fujiwara, T. Kawauchi and H. Taniguchi, J. Chem. Soc., Chem. Commun., 1980, 220 RSC.
- For selected examples, see: (a) R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14082 CrossRef CAS PubMed; (b) C. E. Houlden, M. Hutchby, C. D. Bailey, J. G. Ford, S. N. G. Tyler, M. R. Gagné, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 1830 CrossRef CAS PubMed; (c) R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686 CrossRef CAS PubMed; (d) H. Mizuno, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 1251 CrossRef CAS PubMed.
- T. Kochi, S. Urano, H. Seki, E. Mizushima, M. Sato and F. Kakiuchi, J. Am. Chem. Soc., 2009, 131, 2792 CrossRef CAS PubMed.
- X. Peng, Y. Zhu, T. A. Ramirez, B. Zhao and Y. Shi, Org. Lett., 2011, 13, 5244 CrossRef CAS PubMed.
- W.-Y. Yu, W. N. Sit, K.-M. Lai, Z. Zhou and A. S. C. Chan, J. Am. Chem. Soc., 2008, 130, 3304 CrossRef CAS PubMed.
- S. Wang, Z. Yang, J. Liu, K. Xie, A. Wang, X. Chen and Z. Tan, Chem. Commun., 2012, 48, 9924 RSC.
- (a) A. Krantz, R. W. Spencer, T. F. Tam, T. J. Liak, L. J. Copp, E. M. Thomas and S. P. Rafferty, J. Med. Chem., 1990, 33, 464 CrossRef CAS; (b) R. J. Griffin, S. Srinivasan, K. Bowman, A. H. Calvert, N. J. Curtin, D. R. Newell, L. C. Pemberton and B. T. Golding, J. Med. Chem., 1998, 41, 5247 CrossRef CAS PubMed; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (d) P.-W. Hsieh, F.-R. Chang, C.-H. Chang, P.-W. Cheng, L.-C. Chiang, F.-L. Zeng, K.-H. Lin and Y.-C. Wu, Bioorg. Med. Chem. Lett., 2004, 14, 4751 CrossRef CAS PubMed; (e) C. Gil and S. Bräse, Chem.–Eur. J., 2005, 11, 2680 CrossRef CAS PubMed; (f) M. de F. Pereira, V. Thiéry and T. Besson, Tetrahedron, 2007, 63, 847 CrossRef PubMed; (g) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC.
- (a) M. T. Bogert and V. J. Chambers, J. Am. Chem. Soc., 1905, 27, 649 CrossRef CAS; (b) W. C. Sumpter and W. F. Jones, J. Am. Chem. Soc., 1943, 65, 1802 CrossRef CAS; (c) S. H. Merrill, J. Org. Chem., 1960, 25, 1882 CrossRef CAS; (d) Z.-L. Zhou, S. M. Kher, S. X. Cai, E. R. Whittemore, S. A. Espitia, J. E. Hawkinson, M. Tran, R. M. Woodward, E. Weber and J. F. W. Keana, Bioorg. Med. Chem., 2003, 11, 1769 CrossRef CAS; (e) A. Kamal, N. Shankaraiah, N. Markandeya and C. S. Reddy, Synlett, 2008, 1297 CrossRef CAS PubMed.
- For reviews, see: (a) S. Dandapani and D. P. Curran, Chem.–Eur. J., 2004, 10, 3130 CrossRef CAS PubMed; (b) T. Y. S. But and P. H. Toy, Chem.–Asian J., 2007, 2, 1340 CrossRef CAS PubMed; (c) X. Xu, G. Cheng and X. Li, Chin. J. Org. Chem., 2012, 32, 1024 CrossRef CAS; For selected examples, see: (d) K. Muñiz and A. Iglesias, Angew. Chem., Int. Ed., 2007, 46, 6350 CrossRef PubMed; (e) X. Xu, X. Li, L. Ma, N. Ye and B. Weng, J. Am. Chem. Soc., 2008, 130, 14048 CrossRef CAS PubMed; (f) X. Xu and X. Li, Org. Lett., 2009, 11, 1027 CrossRef CAS PubMed; (g) B. Han, X.-L. Yang, R. Fang, W. Yu, C. Wang, X.-Y. Duan and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 8816 CrossRef CAS PubMed; (h) Y. Amaoka, S. Kamijo, T. Hoshikawa and M. Inoue, J. Org. Chem., 2012, 77, 9959 CrossRef CAS PubMed.
- F. Yang, F. Song, W. Li, J. Lan and J. You, RSC Adv., 2013, 3, 9649 RSC.
- (a) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS PubMed; (b) C. E. Houlden, C. D. Bailey, J. G. Ford, M. R. Gagné, G. C. Lloyd-Jones and K. I. Booker-Milburn, J. Am. Chem. Soc., 2008, 130, 10066 CrossRef CAS PubMed.
- C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC.
- For a review on Pd(IV) chemistry, see: (a) A. J. Canty, Acc. Chem. Res., 1992, 25, 83 CrossRef CAS; For selected examples, see: (b) C. S. Yeung, X. Zhao, N. Borduas and V. M. Dong, Chem. Sci., 2010, 1, 331 RSC; (c) J. Karthikeyan and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 9880 CrossRef CAS PubMed; (d) T.-S. Jiang and G.-W. Wang, J. Org. Chem., 2012, 77, 9504 CrossRef CAS PubMed; (e) X. Sun, G. Shan, Y. Sun and Y. Rao, Angew. Chem., Int. Ed., 2013, 52, 4440 CrossRef CAS PubMed.
- (a) T.-S. Mei, X. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806 CrossRef CAS PubMed; (b) K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data of NMR and MS spectra for all new compounds, copies of NMR spectra, and experimental procedures. See DOI: 10.1039/c3qo00069a |
| This journal is © the Partner Organisations 2014 |