Highly efficient and general enantioselective synthesis of naturally occurring isoquinoline alkaloids†
Weilong
Lin
a and
Shengming
Ma
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn; Fax: (+86) 21-6260-9305
bShanghai Key Laboratory of Green Chemistry and Chemical Process, Department of Chemistry East, China Normal University, 3663 North Zhongshan Road, Shanghai 200062, P. R. China
First published on 21st March 2014
Abstract
The highly efficient asymmetric synthesis of (+)-dysoxyline (three steps) and (+)-crispine A (four steps) from readily available different terminal alkynes, benzaldehyde, and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline has been achieved, providing efficient general approaches for achieving libraries of naturally occurring chiral isoquinoline alkaloids.
Introduction
Naturally occurring isoquinoline alkaloids have received broad interest from the scientific community because of their important biological activities (Fig. 1).1–5 For example, (+)-dysoxyline,2a isolated from Dysoxylum lenticellare, displays cardiac activity,6 and this type of 1-phenethyltetrahydroisoquinoline compounds also serves as a biosynthetic precursor for the homoaporphine alkaloids3 and colchicines.7 In addition, (+)-crispine A, isolated from Carduus crispus, shows cytotoxic activity and antitumor activity in vitro.4a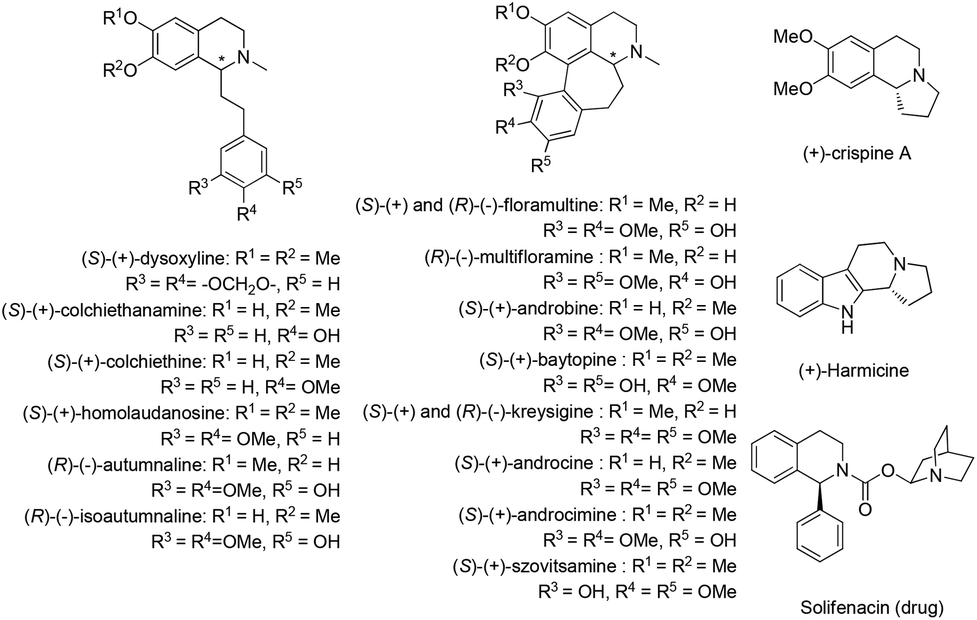 | ||
| Fig. 1 Representative isoquinoline natural products and drugs. | ||
The reported synthesis of optically active (+)-dysoxyline takes 14 steps from a relatively complicated non-commercially available starting material, i.e., 2-bromo-4-methoxy-4-hydroxybenzyl bromide, with 88% ee.8 Herein, we wish to demonstrate a general approach for highly asymmetric synthesis of (+)-dysoxyline in 98% ee, in just three steps from all commercially available starting materials (Scheme 1), which may also be applied to other such targets listed in Fig. 1.
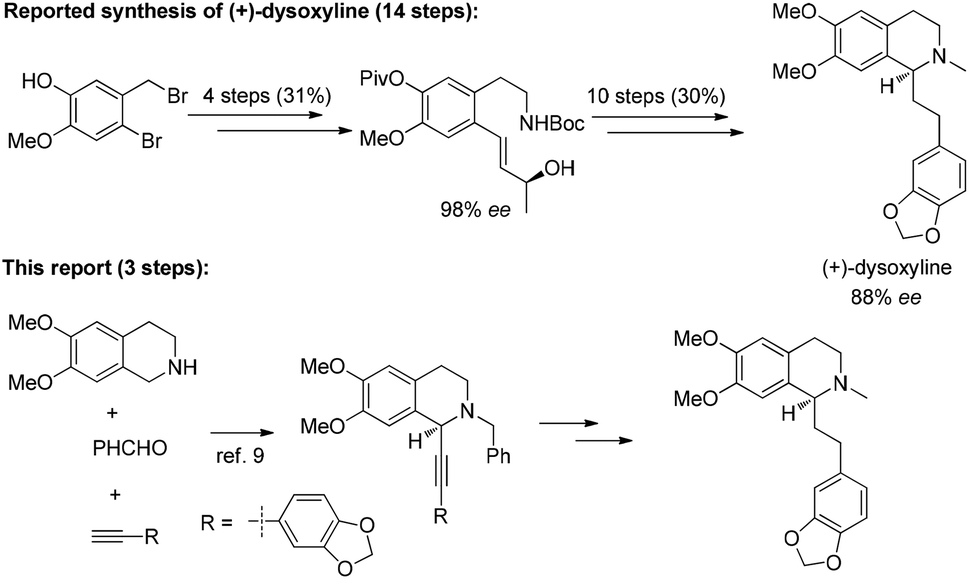 | ||
| Scheme 1 Synthetic routes to (+)-dysoxyline. | ||
Results and discussion
Recently, we reported the highly enantioselective synthesis of C1 substituted chiral tetrahydroisoquinolines with readily available materials terminal alkynes and aldehydes under Cu(I) catalysis.9,10 Thus, we envisioned that 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline would be the starting material for the newly designed synthesis of (+)-dysoxyline (Scheme 2). There are three challenges: (1) the scope of differently structured terminal alkynes in terms of enantioselectivity and yield for the asymmetric abnormal A3 coupling alkynylation reaction;9 (2) in the step of debenzylation, due to the fact that the nitrogen is connected to two arylmethyl groups; thus, racemization to the chiral center is anticipated; (3) such an issue may also occur in the step of hydrogenation.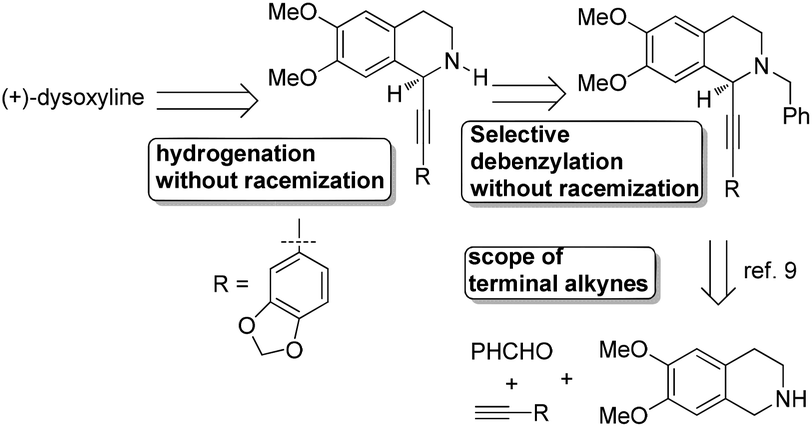 | ||
| Scheme 2 Retrosynthetic analysis of (+)-dysoxyline. | ||
Synthesis of racemic dysoxyline
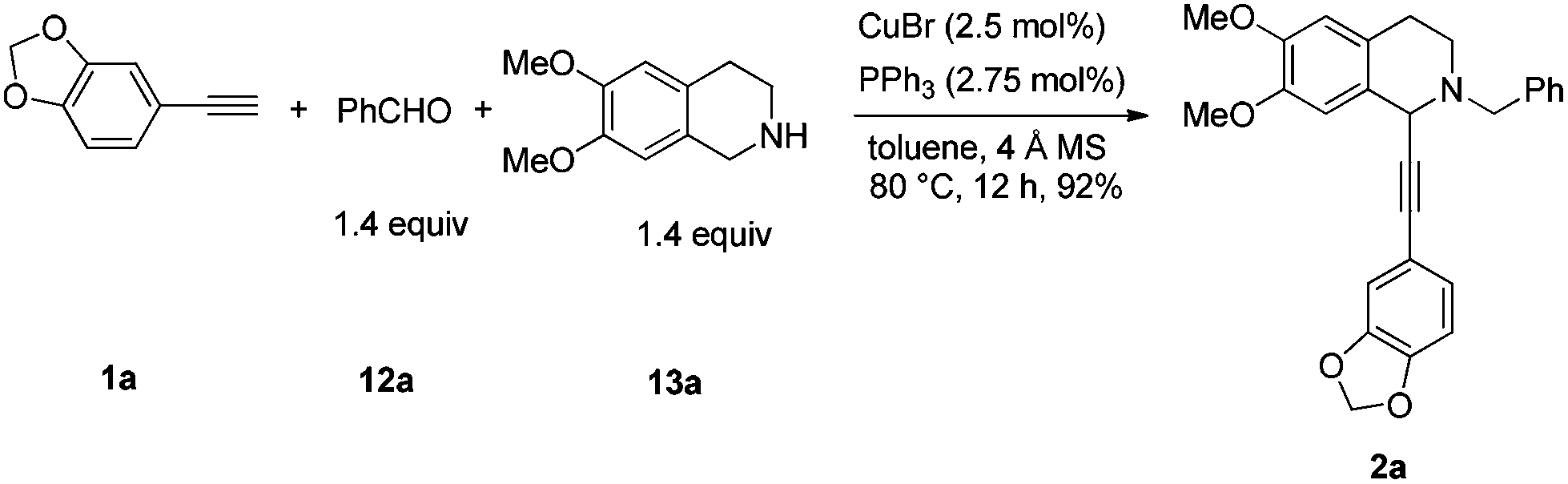
We started this study by developing the synthetic route to the racemic dysoxyline. Delightedly, it was smooth to achieve the total synthesis of racemic dysoxyline in 3 steps from 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline with 73% combined yield, following the idea presented in Scheme 2 (Scheme 3).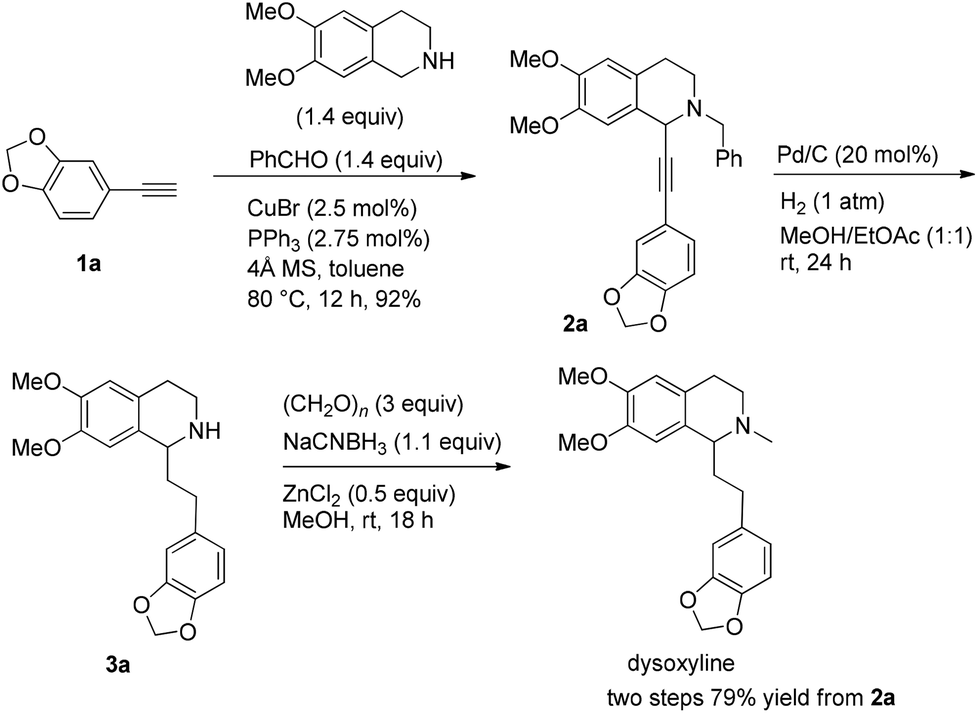 | ||
| Scheme 3 Synthesis of racemic dysoxyline. | ||
Asymmetric synthesis of (+)-dysoxyline
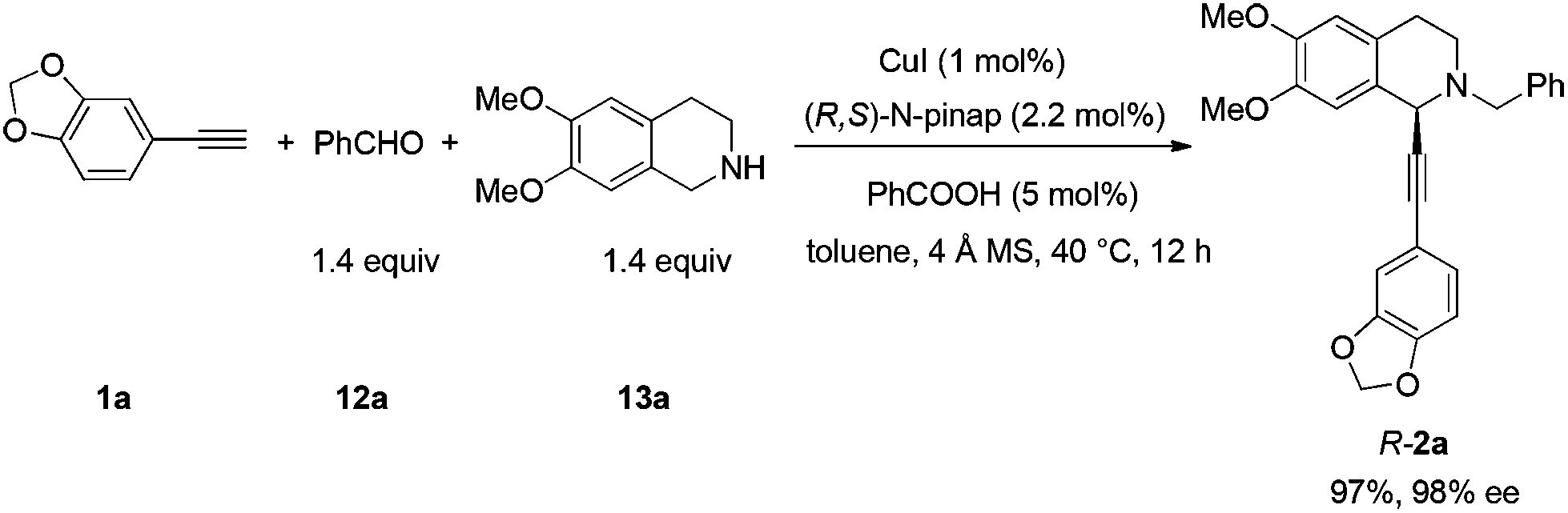
Based on these results, all the commercially available materials, i.e., 5-ethynyl-1,3-benzodioxole,11 benzaldehyde, and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline, were mixed and treated with CuI and (R,S)-N-pinap as the chiral catalyst. It is exciting to observe that the expected propargylic amine 2a was obtained in 97% yield together with 98% ee under the standard conditions.9 As expected, upon exposing 2a to 1 atm of H2 under the catalysis of Pd/C, although the benzyl group was removed and the C–C triple bond was fully hydrogenated, serious racemization occurred. After methylation, disappointedly, (+)-dysoxyline was afforded in only 68% ee, although the yield is still high, indicating a serious racemization in the final two steps (Scheme 4).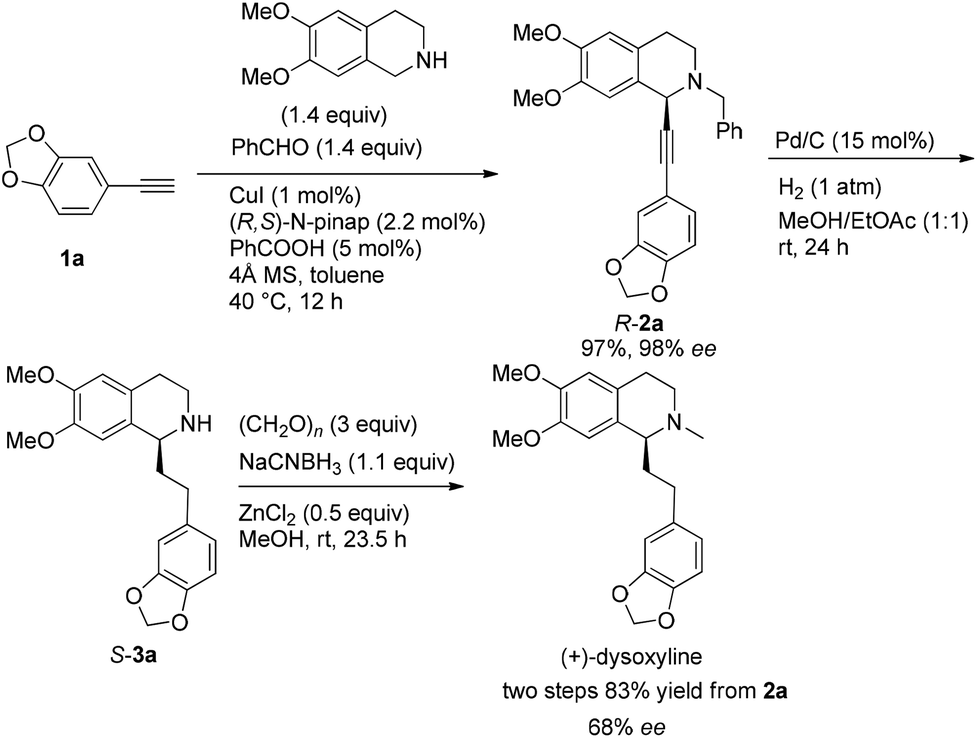 | ||
| Scheme 4 Initial synthesis of (+)-dysoxyline in 68% ee. | ||
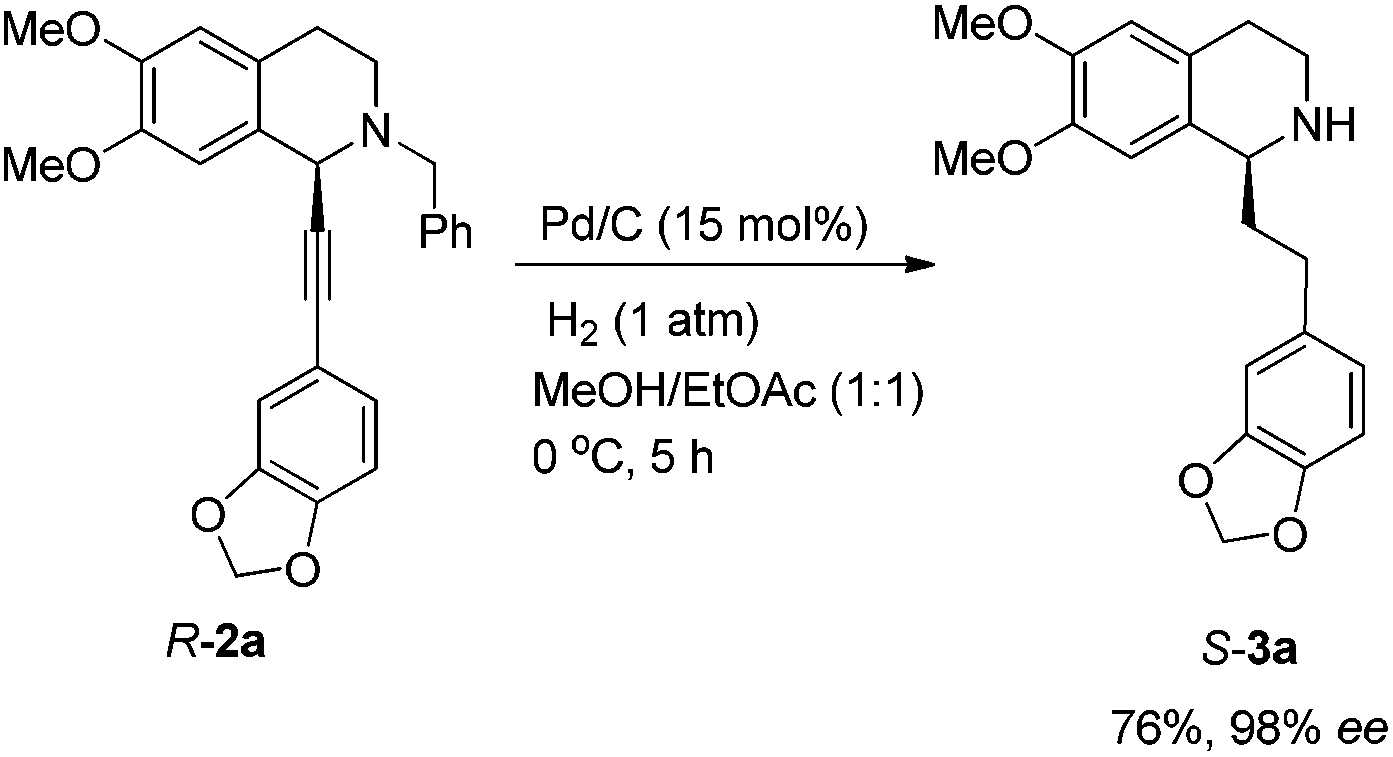
To achieve a highly enantioselective synthesis of (+)-dysoxyline, the optimization of reaction conditions for the hydrogenation under the catalysis of Pd/C was then studied. Firstly, it was observed that 3a was isolated in 79% yield and 78% ee within 24 h (Scheme 5, entry 1). To our delight, the enantioselectivity of 3a was improved to 95% ee by reducing the reaction time to 1 h (Scheme 5, entries 2 and 3) with incomplete conversion. Luckily, 3a was obtained in 76% yield with retained high enantiopurity (98% ee) by reducing the reaction temperature to 0 °C with a reaction time of 5 h (Scheme 5, entry 4).
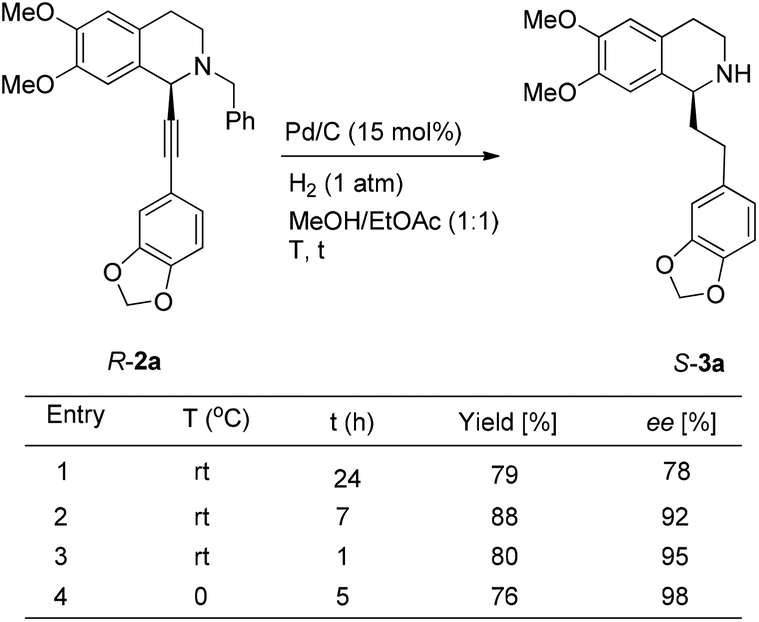 | ||
| Scheme 5 Tackling the issue of selective debenzylation along with hydrogenation. | ||
We next turned to address the racemization in the step of methylation. As shown in Scheme 6, there was a slight racemization when the reaction was conducted at room temperature, probably due to a possible iminium isomerization process before the reduction.9,10 Gladly, methylation could be achieved with retained high enantiopurity (98% ee) at 0 °C (Scheme 6).
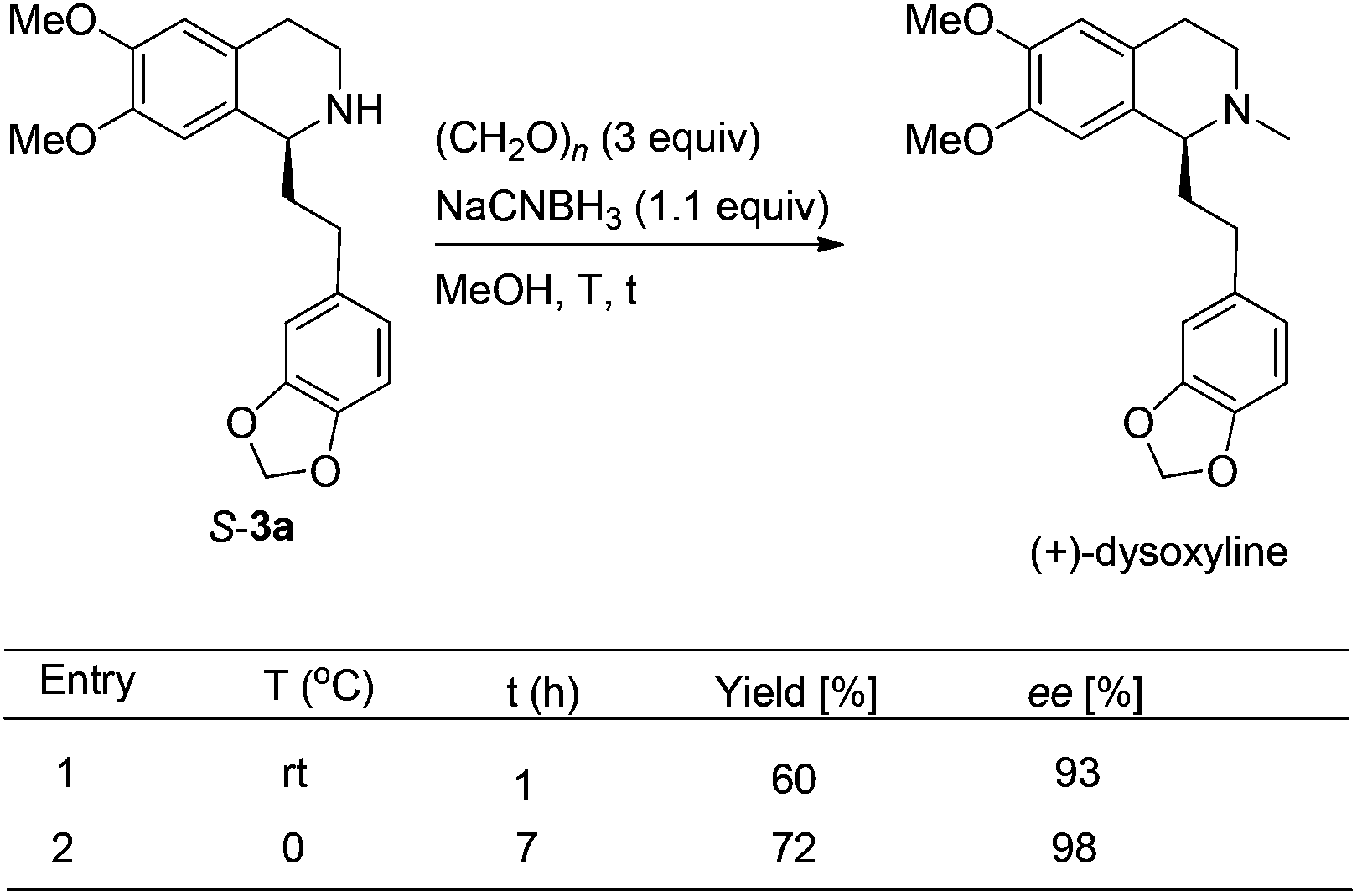 | ||
| Scheme 6 Optimization of the methylation. | ||
After addressing these issues, (+)-dysoxyline was synthesized in 53% overall yield in three steps from 1a (Scheme 7).
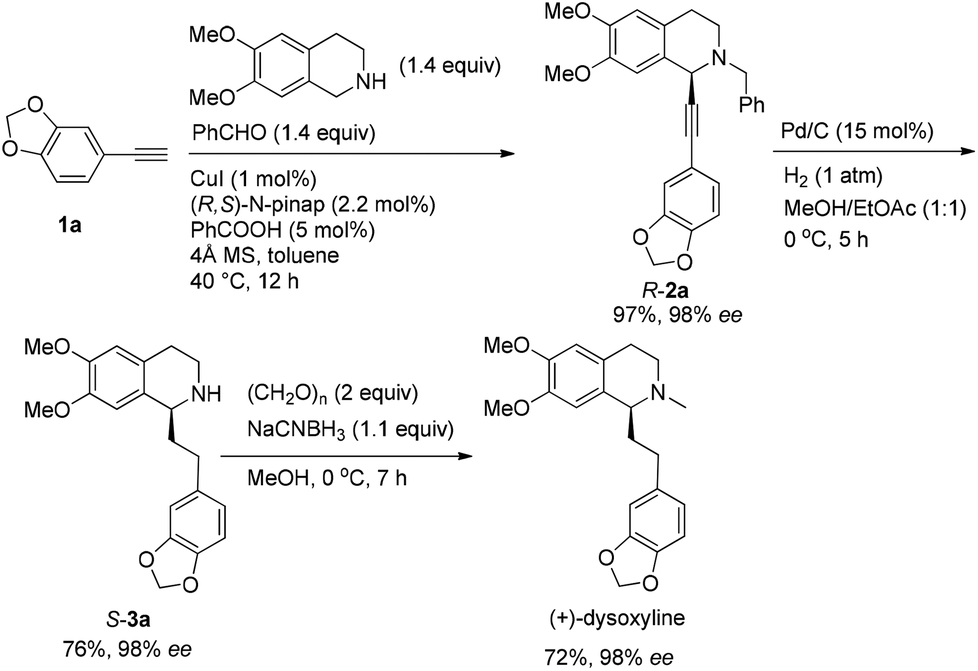 | ||
| Scheme 7 Asymmetric synthesis of (+)-dysoxyline. | ||
Synthesis of racemic crispine A
The total synthesis of racemic crispine A may also be realized in a similar way in 4 steps from 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline with 34% combined yield (Scheme 8).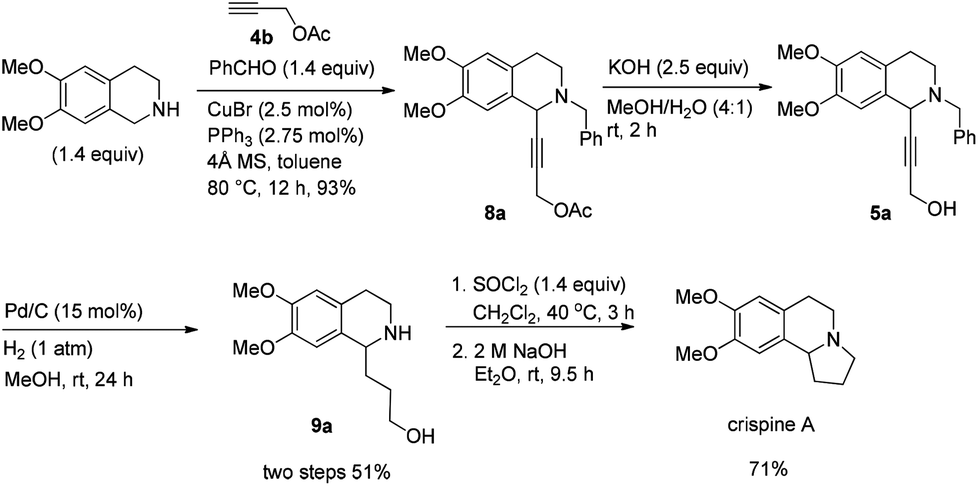 | ||
| Scheme 8 Synthesis of racemic crispine A. | ||
Asymmetric synthesis of (+)-crispine A12
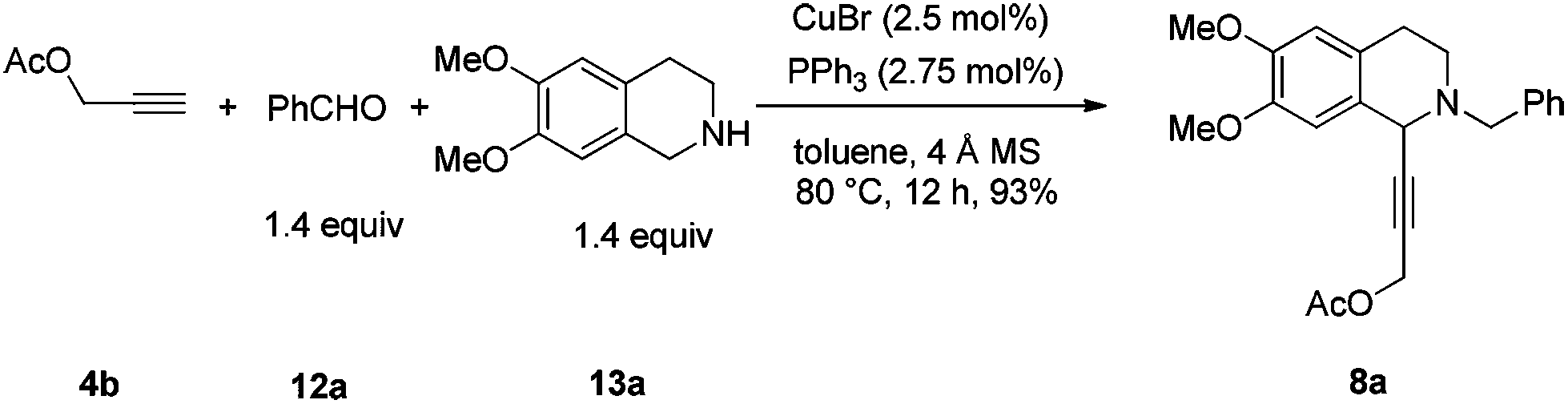
In this case, 2-propyn-1-ol 4a was tried in the first place. Disappointedly, only 36% yield of the desired 5a was afforded together with 36% yield of the normal A3-coupling product 6a. Interestingly, the use of propargyl acetate afforded 8a in 98% yield and 98% ee (Scheme 9).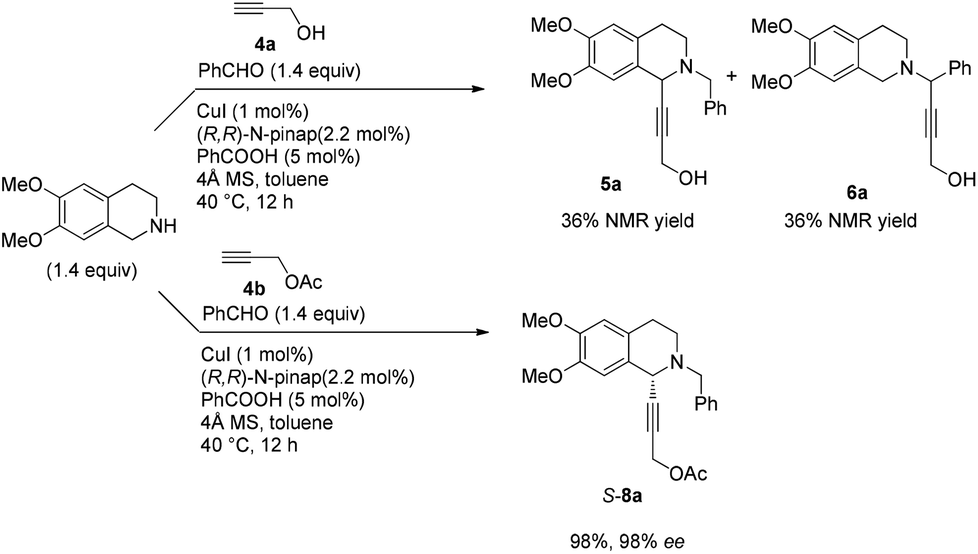 | ||
| Scheme 9 The effect of the protecting group on propargylic alcohol. | ||
Then propargylic alcohol 5a was easily obtained in 96% yield and 98% ee via hydrolysis with potassium hydroxide. However, the debenzylation in 5a was very slow even in methanol, affording 9a in 91% ee with a low yield (Scheme 10).
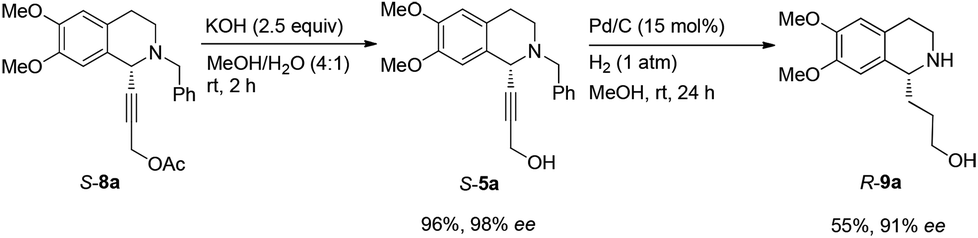 | ||
| Scheme 10 Initial synthesis of 9a. | ||
To achieve a highly enantioselective synthesis of (+)-crispine A, the hydrogenation of 5a was then conducted under a hydrogen pressure of 10 bar. Disappointedly, 9a was obtained in 51% yield and 84% ee. Excitedly, we found that the enantiopurity of 9a was improved to 94% ee when the reaction was conducted in a mixture of methanol–ethyl acetate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); further screening on the solvent effect led to the observation that the hydrogenation in ethyl acetate afforded alcohol 9a in 98% ee and 65% yield (Scheme 11).
:1); further screening on the solvent effect led to the observation that the hydrogenation in ethyl acetate afforded alcohol 9a in 98% ee and 65% yield (Scheme 11).
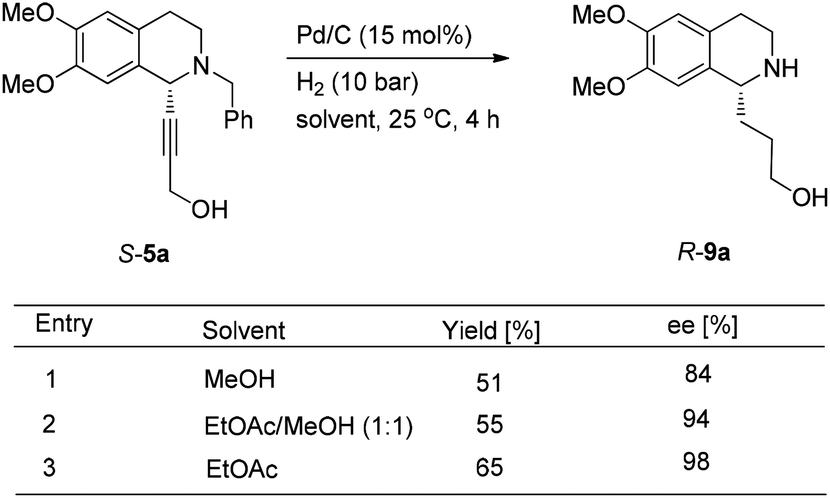 | ||
| Scheme 11 The solvent effect on the hydrogenation of the triple bond. | ||
Gladly, the chlorination at room temperature followed by cyclization afforded (+)-crispine A in 63% yield with retained enantiopurity (98% ee) (Scheme 12).
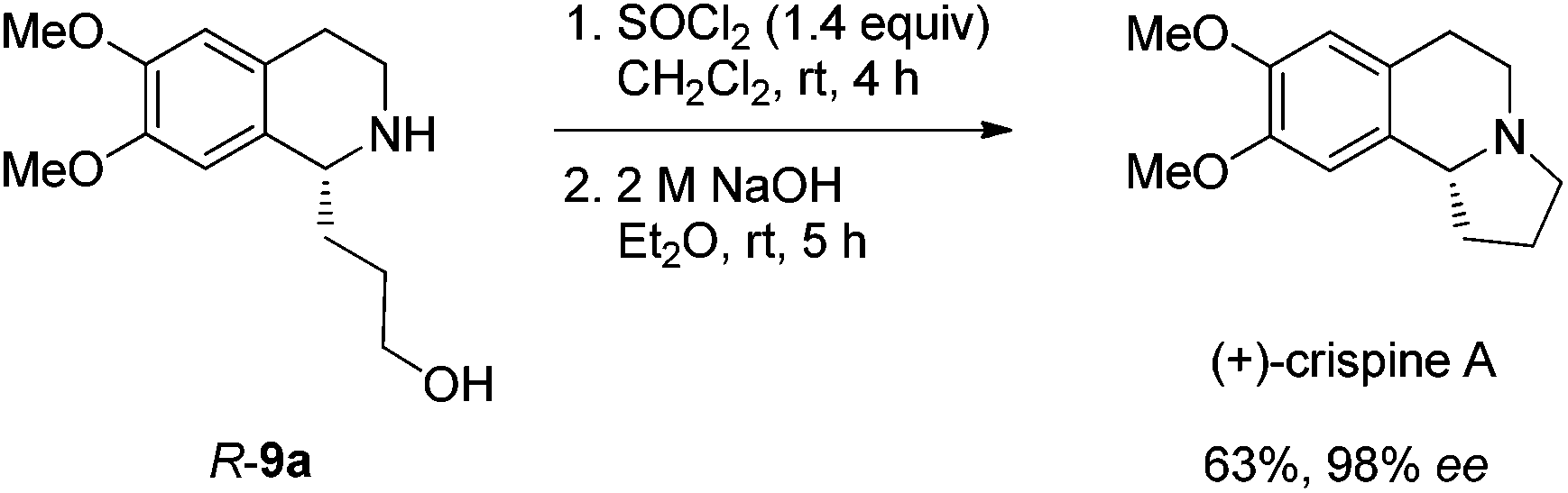 | ||
| Scheme 12 The synthesis of (+)-crispine A via the cyclization of 9a. | ||
Thus, (+)-crispine A was afforded in 39% overall yield in four steps from the commercially available 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (Scheme 13).
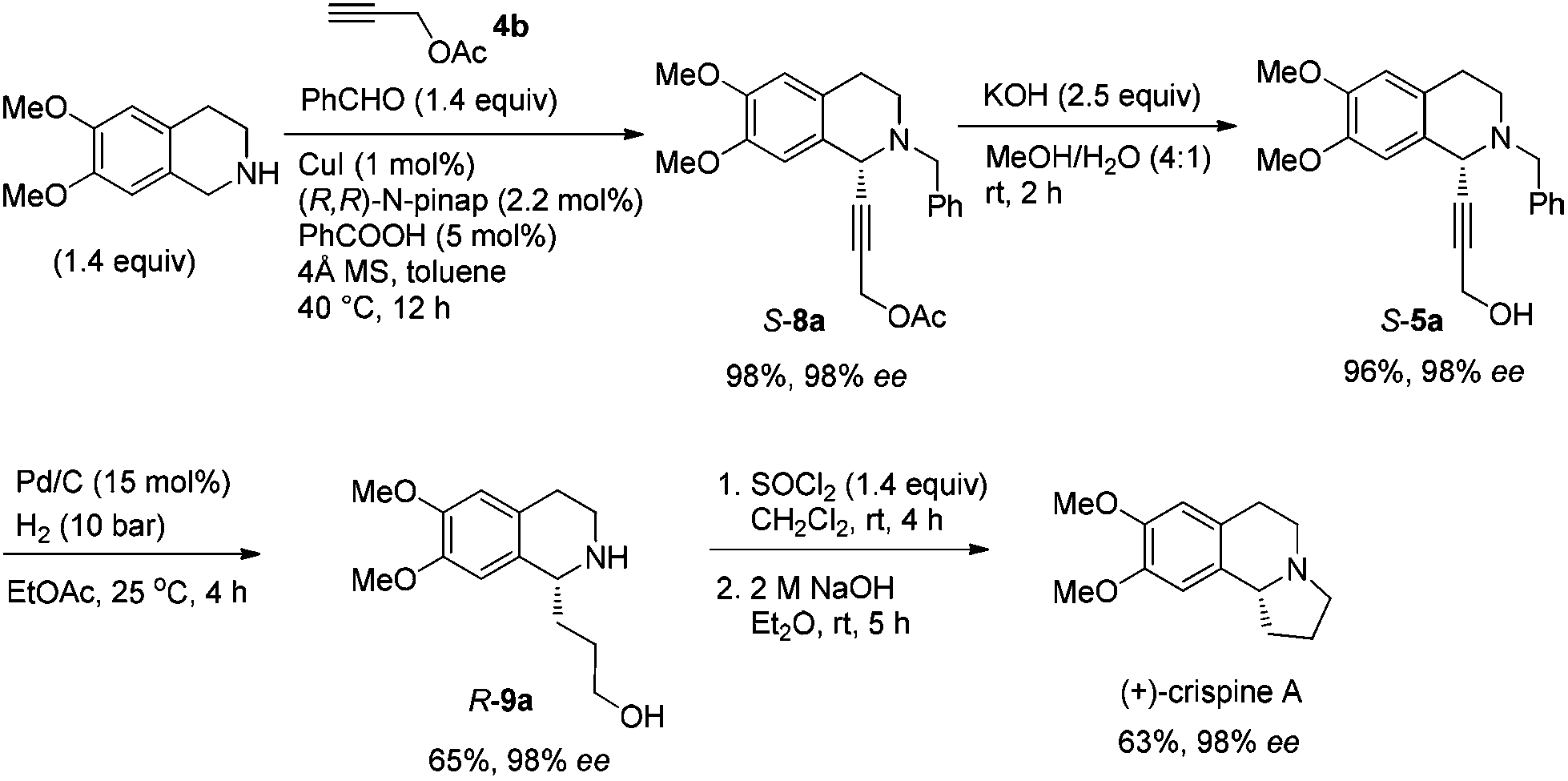 | ||
| Scheme 13 Asymmetric synthesis of (+)-crispine A. | ||
Conclusions
We have achieved the highly efficient asymmetric synthesis of (+)-dysoxyline (three steps) and (+)-crispine A (four steps) from readily available terminal alkynes, aldehydes, and tetrahydroisoquinoline with very high enantiopurity. Due to the easily available nature of the aryl-substituted terminal alkynes/propargylic alcohols and different THIQ derivatives, excellent enantioselectivity, and short synthetic routes, these routes may serve as a general model, providing efficient approaches for achieving libraries of naturally occurring isoquinoline alkaloids. This study will open a new direction for the study of the biological potential of this type of isoquinoline alkaloids. Further studies for achieving biologically active libraries of such natural isoquinoline alkaloids-inspiring compounds and for synthesis of other such natural products listed in Fig. 1 are being actively pursued in this laboratory.Experimental section
General information
CuI (99.5%) and CuBr (99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd; (R,R)-N-pinap (97%) and (R,S)-N-pinap (97%) were purchased from Strem Chemicals and kept in a glove box; 4 Å molecular sieves were purchased from Alfa Aesar and kept in a glove box after activation; Pd/C (10% in mass) was purchased from Sigma-Aldrich. Aldehydes were distilled right before use. Toluene was dried over sodium wire and distilled freshly before use with benzophenone as an indicator. Other reagents were used without further treatment. All the temperatures are referred to the oil baths used.To a Schlenk tube were added PdCl2(PPh3)2 (0.1396 g, 0.2 mmol), PPh3 (0.1315 g, 0.5 mmol), 10a (4.019 g, 20 mmol), Et3N (15 mL), 2-methyl-3-butyn-2-ol (2.530 g, 30 mmol), Et3N (15 mL), and CuI (0.0375 g, 0.2 mmol) sequentially under an Ar atmosphere. The mixture was heated at 90 °C until the completion of the reaction as monitored by TLC (12.5 h). After cooling, the solid was filtered and washed with diethyl ether (50 mL). The solvent was evaporated and the residue was purified by chromatography on silica gel (eluent: petroleum ether–ethyl acetate = 15/1) to afford 11a (3.8126 g, 93%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 6.92 (dd, J = 8.0, 1.6 Hz, 1H, Ar–H), 6.85 (d, J = 1.2 Hz, 1H, Ar–H), 6.72 (d, J = 8.4 Hz, 1H, Ar–H), 5.95 (s, 2H, OCH2O), 2.53 (s, 1H, OH), 1.60 (s, 6H, CH3 × 2); 13C NMR (100 MHz, CDCl3) δ 147.7, 147.2, 126.1, 115.9, 111.5, 108.3, 101.2, 92.1, 81.8, 65.5, 31.4; MS (EI) m/z 205 (M+ + 1, 8.33), 204 (M+, 61.74), 189 (100); IR (neat): ν = 3357, 2980, 2895, 1604, 1502, 1486, 1439, 1222, 1161, 1096, 1036 cm−1.
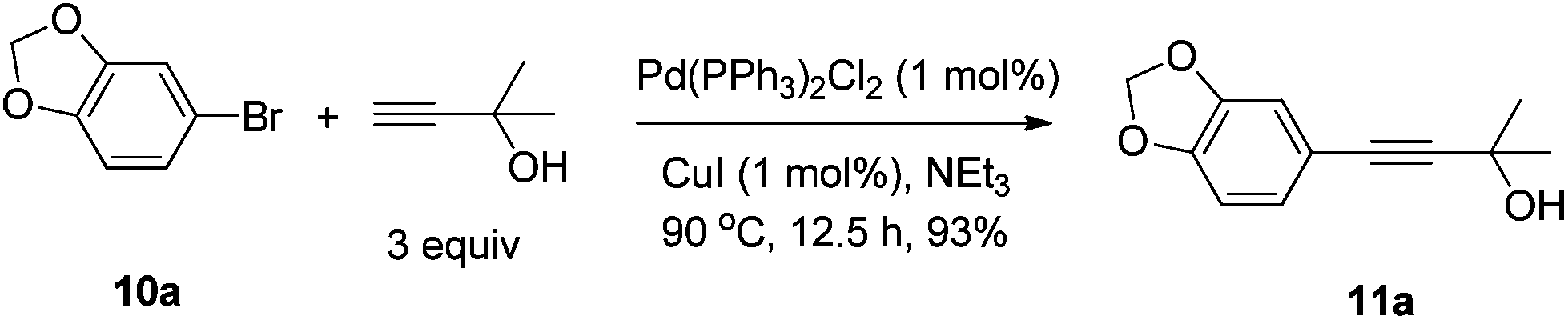
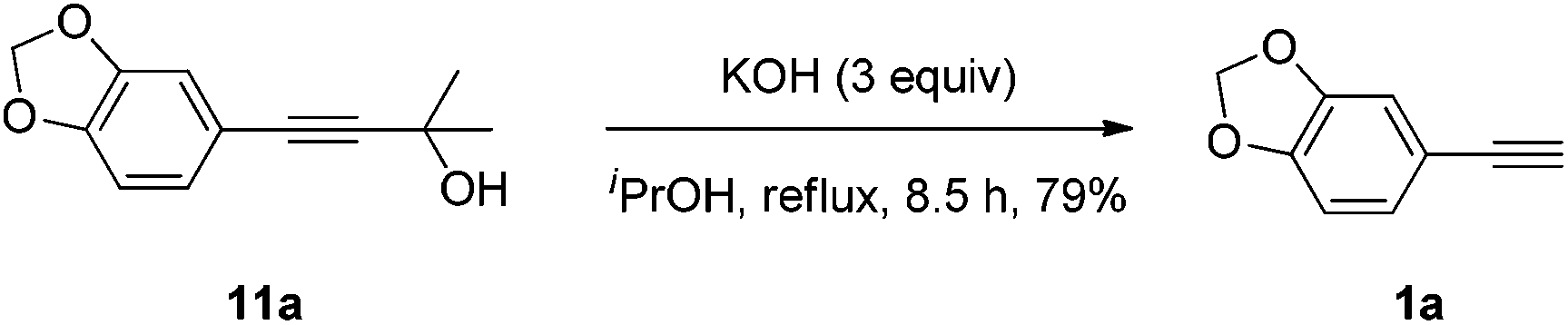
Potassium hydroxide (2.97 g, 52.8 mmol) and isopropanol (45 mL) were added to a round bottomed flask and heated at 50 °C for 10 min. Then, 11a (3.60 g, 17.6 mmol) and isopropanol (55 mL) were added. The mixture was heated under reflux for 8.5 h. After most of the solvent was removed by rotary evaporation, the residue was treated in a mixture of water (50 mL) and diethyl ether (50 mL). After the organic layer was separated, the water was extracted with diethyl ether (50 mL × 3). The solvent was evaporated and the residue was purified by chromatography on silica gel (eluent: petroleum ether (30–60 °C)) to afford 1a (2.0335 g, 79%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.02 (dd, J = 8.0, 1.6 Hz, 1H, Ar–H), 6.93 (d, J = 1.6 Hz, 1H, Ar–H), 6.75 (d, J = 8.4 Hz, 1H, Ar–H), 5.98 (s, 2H, OCH2O), 2.97 (s, 1H, CH); 13C NMR (100 MHz, CDCl3) δ 148.2, 147.3, 126.8, 115.2, 111.9, 108.4, 101.3, 83.5, 75.6; MS (EI) m/z 147 (M+ + 1, 10.17), 146 (M+, 100); IR (neat): ν = 3287, 2897, 2102, 1604, 1502, 1478, 1435, 1331, 1242, 1188, 1092, 1035 cm−1.
To a flame-dried Schlenk tube were added CuBr (3.5 mg, 0.025 mmol), PPh3 (7.3 mg, 0.0275 mmol), 4 Å molecular sieves (302.1 mg), and toluene (2 mL) sequentially under an Ar atmosphere. The resulting mixture was then stirred at rt for 30 min. 12a (146.4 mg, 1.4 mmol)/toluene (1 mL), 13a (270.0 mg, 1.4 mmol)/toluene (1 mL), and 1a (145.2 mg, 1.0 mmol)/toluene (2 mL) were then added sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 80 °C with stirring for 12 h as monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtrated through a short column of silica gel eluted with ethyl acetate (50 mL). After evaporation, the residue was purified by chromatography on silica gel (eluent: dichloromethane–ethyl acetate = 80/1) to afford 2a (391.2 mg, 92%) as solid: m.p. 132–133 °C (petroleum ether–ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 7.2 Hz, 2H, Ar–H), 7.34 (t, J = 7.2 Hz, 2H, Ar–H), 7.31–7.24 (m, 1H, Ar–H), 6.98 (dd, J = 8.0, 1.6 Hz, 1H, Ar–H), 6.90 (d, J = 1.6 Hz, 1H, Ar–H), 6.77–6.70 (m, 2H, Ar–H), 6.60 (s, 1H, Ar–H), 5.95 (s, 2H, OCH2O), 4.65 (s, 1H, NCH), 3.92 (d, J = 13.2, 1H, one proton of NCH2Ar), 3.86 (d, J = 16.4, 1H, one proton of NCH2Ar), 3.84 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.08–2.90 (m, 2H, CH2), 2.83–2.76 (m, 1H, one proton of CH2), 2.74–2.66 (m, 1H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 147.9, 147.5, 147.2, 147.1, 138.2, 129.1, 128.1, 127.2, 127.0, 126.1, 125.9, 116.3, 111.6, 111.1, 110.2, 108.2, 101.1, 86.3, 85.7, 59.4, 55.8, 55.7, 53.7, 45.7, 28.5; MS (EI) m/z 428 (M+ + 1, 5.56), 427 (M+, 21.14), 91 (100); IR (KBr): v = 3061, 3027, 2999, 2908, 2830, 1611, 1518, 1504, 1488, 1441, 1408, 1360, 1332, 1248, 1225, 1208, 1134, 1098, 1038 cm−1; Anal. calcd for C27H25NO4 (%): C 75.86, H 5.89, N 3.28; found: C 75.59, H 5.70, N 3.26.
To a Schlenk tube were added 2a (128.7 mg, 0.3 mmol), ethyl acetate (6 mL), methanol (6 mL), and Pd/C (63.7 mg, 0.06 mmol) sequentially, which was purged three times with hydrogen. The suspension was stirred for 24 h at room temperature under a hydrogen pressure of 1 atm, and was filtered through a short column of Celite eluted with methanol (5 mL × 6). After evaporation, the crude product was then submitted to the next step without further purification and characterization.
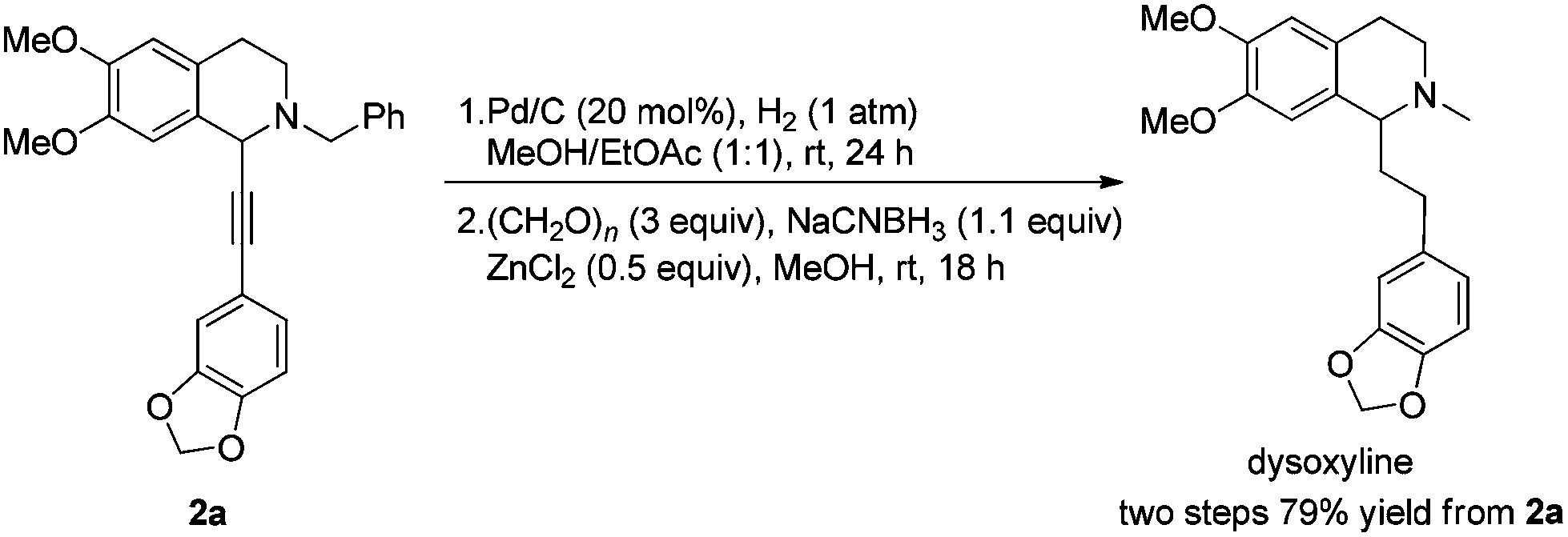
To another Schlenk tube were added paraformaldehyde (27.2 mg, 0.9 mmol), ZnCl2 (21.4 mg, 0.15 mmol), the crude product (prepared in the previous step), MeOH (3 mL), NaCNBH3 (20.9 mg, 0.33 mmol), and MeOH (3 mL) sequentially. The mixture was stirred for 18 h at room temperature. After evaporation, water (10 mL) was added and the mixture was extracted with ethyl acetate (10 mL × 4). The extract was dried over Na2SO4, filtered, and condensed. The residual product was purified by silica gel column chromatography (eluent: dichloromethane–methanol = 40/1) to provide dysoxyline (84.5 mg, 79%) as an oil: 1H NMR (400 MHz, CDCl3) δ 6.72 (d, J = 8.0 Hz, 1H, Ar–H), 6.68 (d, J = 1.2 Hz, 1H, Ar–H), 6.63 (d, J = 7.6 Hz, 1H, Ar–H), 6.57 (s, 1H, Ar–H), 6.53 (s, 1H, Ar–H), 5.90 (s, 2H, OCH2O), 3.85 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.42 (t, J = 5.4 Hz, 1H, NCH), 3.20–3.10 (m, 1H, one proton of CH2), 2.82–2.61 (m, 4H, CH2 × 2), 2.54–2.44 (m, 4H, NCH3 and one proton of CH2), 2.06–1.97 (m, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ 147.3, 147.1, 147.0, 145.3, 136.5, 129.4, 126.4, 120.9, 111.0, 109.7, 108.8, 108.0, 100.6, 62.4, 55.8, 55.6, 47.9, 42.5, 37.0, 31.2, 25.2; MS (EI) m/z 192 (100); IR (neat): v = 2932, 1609, 1514, 1503, 1487, 1463, 1440, 1243, 1224, 1186, 1123, 1102, 1068, 1035 cm−1.
To a flame-dried Schlenk tube was added (R,S)-N-pinap (0.0739 g, 0.132 mmol) inside a glove box. CuI (0.0115 g, 0.06 mmol), 4 Å molecular sieves (1.8021 g), and toluene (12 mL) were sequentially added under an Ar atmosphere. The Schlenk tube was then stirred at rt for 30 min. PhCOOH (0.0365 g, 0.3 mmol), 12a (0.8916 g, 8.4 mmol)/toluene (6 mL), 13a (1.6297 g, 8.4 mmol)/toluene (6 mL), and 1a (0.8763 g, 6.0 mmol)/toluene (12 mL) were then added sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 40 °C with stirring for 12 h as monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtrated through a short column of silica gel eluted with ethyl acetate (100 mL). After evaporation, the residue was purified by chromatography on silica gel to afford R-2a (2.4880 g, 97%) (eluent: dichloromethane–ethyl acetate = 80/1) as a solid: 98% ee (HPLC conditions: Chiralcel OD-H column, hexane–i-PrOH = 95/5, 1.0 mL min−1, λ = 214 nm, tR(major) = 28.7 min, tR(minor) = 21.1 min); [α]25D = +112.6 (c = 0.99, CHCl3); m.p. 132–133 °C (petroleum ether–ethyl acetate); 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 7.2 Hz, 2H, Ar–H), 7.34 (t, J = 7.4 Hz, 2H, Ar–H), 7.31–7.24 (m, 1H, Ar–H), 6.98 (dd, J = 8.0, 1.6 Hz, 1H, Ar–H), 6.90 (d, J = 1.2 Hz, 1H, Ar–H), 6.76–6.70 (m, 2H, Ar–H), 6.60 (s, 1H, Ar–H), 5.96 (s, 2H, OCH2O), 4.65 (s, 1H, NCH), 3.92 (d, J = 12.8, 1H, one proton of NCH2Ar), 3.87 (d, J = 15.6, 1H, one proton of NCH2Ar), 3.85 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.07–2.90 (m, 2H, CH2), 2.84–2.76 (m, 1H, one proton of CH2), 2.74–2.66 (m, 1H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 147.9, 147.5, 147.2, 147.1, 138.2, 129.1, 128.1, 127.2, 127.0, 126.1, 125.9, 116.3, 111.6, 111.1, 110.2, 108.2, 101.1, 86.3, 85.7, 59.4, 55.8, 55.7, 53.7, 45.7, 28.5; MS (EI) m/z 428 (M+ + 1, 4.09), 427 (M+, 18.87), 91 (100); IR (KBr): v = 3060, 3027, 2999, 2907, 2831, 1611, 1518, 1504, 1488, 1441, 1360, 1332, 1248, 1225, 1208, 1134, 1098, 1038 cm−1; Anal. calcd for C27H25NO4 (%): C 75.86, H 5.89, N 3.28; found: C 75.77, H 5.82, N 3.21.
To a Schlenk tube were added R-2a (426.9 mg, 1.0 mmol), ethyl acetate (10 mL), methanol (10 mL), and Pd/C (160.0 mg, 0.15 mmol, 10% in mass) sequentially, which was purged three times with hydrogen. The suspension was stirred for 5 h at 0 °C under a hydrogen pressure of 1 atm, and was filtered through a short column of Celite eluted with methanol (10 mL × 6). After evaporation, the residual product was purified by silica gel column chromatography (eluent: dichloromethane–methanol–ammonia solution = 800/10/4) to provide S-3a (283.4 mg, 76%, purity = 92% based on the 1H NMR analysis with CH2Br2 as the internal standard) as an oil: 98% ee (HPLC conditions: Chiralcel OD-H column, hexane–i-PrOH = 60/40, 1.0 mL min−1, λ = 214 nm, tR(major) = 16.1 min, tR(minor) = 34.6 min); [α]24D = −12.7 (c = 0.61, CHCl3) (lit.8 [α]20D = −2.7 (c = 0.56, CHCl3)); 1H NMR (400 MHz, CDCl3) δ 6.75–6.65 (m, 3H, Ar–H), 6.57 (s, 2H, Ar–H), 5.91 (s, 2H, OCH2O), 3.93 (dd, J = 9.0, 3.4 Hz, 1H, Ar–H), 3.85 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.27–3.19 (m, 1H, one proton of CH2), 3.03–2.95 (m, 1H, one proton of CH2), 2.80–2.61 (m, 4H, CH2 × 2), 2.12–1.91 (m, 2H, CH2), 1.64 (s, 1H, NH); 13C NMR (100 MHz, CDCl3) δ 147.4, 147.1, 147.0, 145.4, 136.1, 131.1, 127.1, 120.9, 111.6, 108.9, 108.7, 108.0, 100.6, 55.9, 55.7, 54.8, 40.9, 38.4, 32.0, 29.4; MS (EI) m/z 342 (M+ + 1, 0.23), 341 (M+, 0.7), 192 (100); IR (neat): v = 2931, 2909, 1608, 1503, 1487, 1463, 1440, 1242, 1221, 1186, 1110, 1035, 1001 cm−1.
To a Schlenk tube were added S-3a (170.6 mg, 0.46 mmol, 92% purity) and MeOH (5 mL), and the solution was cooled to 0 °C. Then to the solution was added paraformaldehyde (45.1 mg, 1.5 mmol), NaCNBH3 (35.1 mg, 0.55 mmol), and MeOH (5 mL) sequentially. The mixture was stirred for 7 h at 0 °C. After concentration, water (10 mL) was added and the mixture was extracted with ethyl acetate (10 mL × 6). The extract was dried over Na2SO4, filtered, and evaporated. The residual product was purified by silica gel column chromatography (eluent: ethyl acetate–methanol–ammonia solution = 200/10/1) to provide (+)-dysoxyline (117.3 mg, 72%) as an oil: 98% ee (HPLC conditions: Chiralcel IF column, hexane–i-PrOH = 80/20, 0.5 mL min−1, λ = 214 nm, tR(major) = 19.6 min, tR(minor) = 27.8 min); [α]25D = +21.3 (c = 0.37, EtOH) (lit.2a [α]25D = +22 (c = 0.34, EtOH)); 1H NMR (400 MHz, CDCl3) δ 6.71 (d, J = 8.0 Hz, 1H, Ar–H), 6.68 (s, 1H, Ar–H), 6.63 (d, J = 8.0 Hz, 1H, Ar–H), 6.57 (s, 1H, Ar–H), 6.53 (s, 1H, Ar–H), 5.89 (s, 2H, OCH2O), 3.85 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.42 (t, J = 5.2 Hz, 1H, NCH), 3.20–3.10 (m, 1H, one proton of CH2), 2.82–2.61 (m, 4H, CH2 × 2), 2.54–2.44 (m, 4H, NCH3 and one proton of CH2), 2.09–1.96 (m, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ 147.3, 147.1, 147.0, 145.3, 136.5, 129.4, 126.4, 120.9, 111.0, 109.7, 108.8, 108.0, 100.6, 62.4, 55.8, 55.6, 47.9, 42.5, 37.0, 31.2, 25.2; MS (EI) m/z 192 (100); IR (neat): v = 2932, 2908, 2831, 1608, 1503, 1487, 1463, 1440, 1405, 1242, 1222, 1186, 1110, 1034, 1001 cm−1.
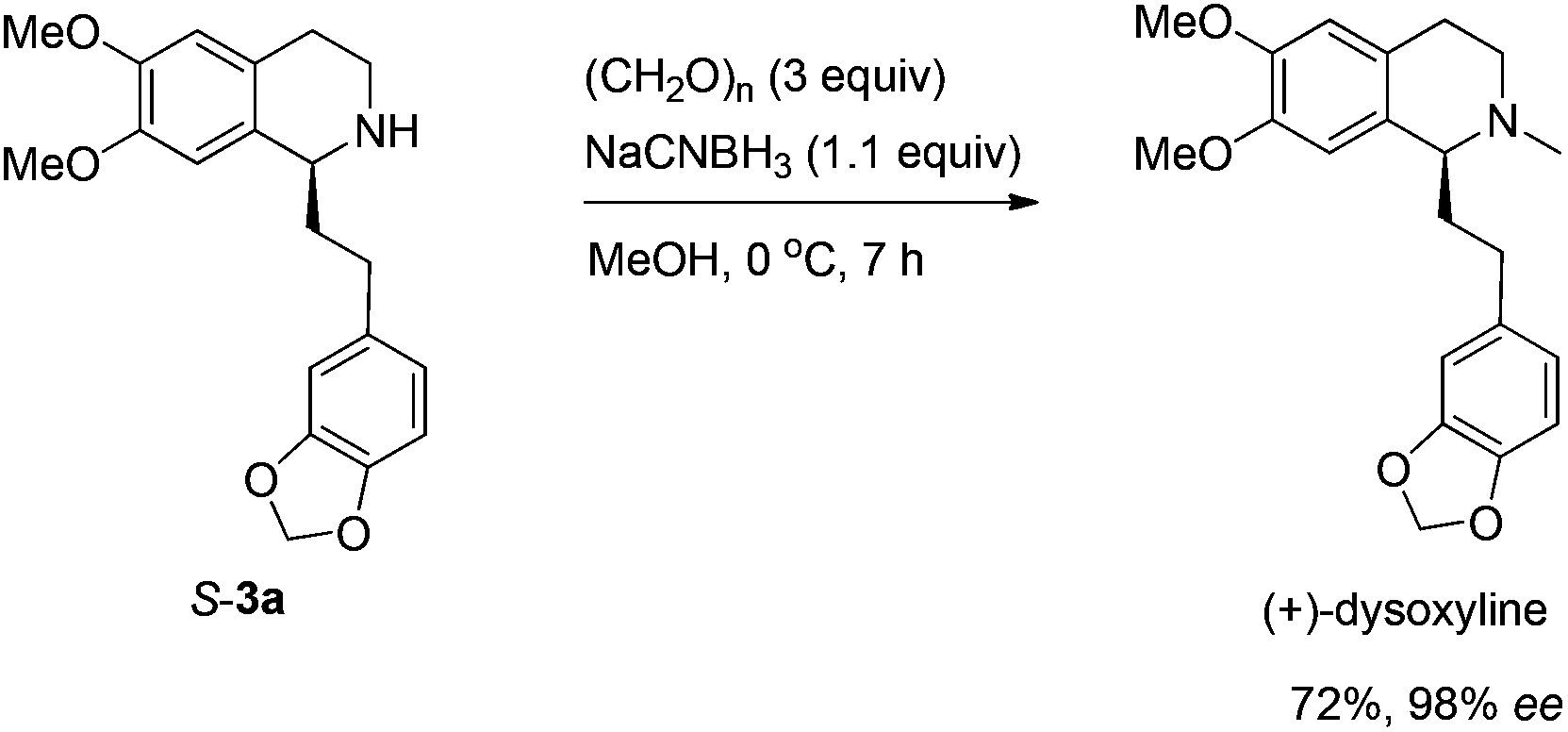
To a flame-dried Schlenk tube were added CuBr (3.6 mg, 0.025 mmol), PPh3 (7.4 mg, 0.0275 mmol), 4 Å molecular sieves (302.1 mg), and toluene (2 mL) sequentially under an Ar atmosphere. The mixture was then stirred at rt for 30 min. 12a (149.0 mg, 1.4 mmol)/toluene (1 mL), 13a (271.0 mg, 1.4 mmol)/toluene (1 mL), and 4b (98.2 mg, 1.0 mmol)/toluene (2 mL) were then added sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 80 °C with stirring for 12 h as monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtrated through a short column of silica gel eluted with ethyl acetate (50 mL). After evaporation, the residue was purified by chromatography on silica gel (eluent: petroleum ether–ethyl acetate = 6/1) to afford 8a (353.1 mg, 93%) as an oil: 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 6.8 Hz, 2H, Ar–H), 7.33 (t, J = 7.2 Hz, 2H, Ar–H), 7.27 (t, J = 7.2 Hz, 1H, Ar–H), 6.65 (s, 1H, Ar–H), 6.58 (s, 1H, Ar–H), 4.75 (s, 2H, OCH2), 4.52 (s, 1H, NCH), 3.85 (d, J = 13.6, 1H, one proton of NCH2Ar), 3.79 (d, J = 13.2, 1H, one proton of NCH2Ar), 3.84 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 2.98–2.86 (m, 2H, ArCH2), 2.82–2.74 (m, 1H, one proton of CH2), 2.71–2.61 (m, 1H, one proton of CH2), 2.11 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 170.0, 148.0, 147.1, 138.0, 129.0, 128.1, 127.0, 126.5, 125.9, 111.1, 110.1, 84.6, 80.4, 59.2, 55.7, 55.6, 53.1, 52.4, 45.6, 28.3, 20.6; MS (ESI) m/z 380 ([M + H]+); IR (neat): v = 2936, 2913, 2831, 1739, 1611, 1517, 1463, 1452, 1375, 1358, 1254, 1219, 1131, 1090, 1019 cm−1; HRMS calcd for C23H26NO4 ([M + H]+): 380.1856. Found: 380.1861.
To a Schlenk tube were added KOH (29.1 mg, 0.5 mmol), mixed solvent (1 mL, MeOH–H2O = 4/1 by volume), and 8a (76.0 mg, 0.2 mmol)/mixed solvent (1 mL, MeOH–H2O = 4/1 by volume) sequentially. The mixture was then stirred at room temperature. After 2 h as monitored by TLC, the reaction was complete, and MeOH was removed by rotary evaporation. Water (5 mL) was added and the solution was extracted with ethyl acetate (10 mL × 3). The organic phase was dried over anhydrous Na2SO4. After filtration and evaporation, the crude product was then submitted to the next step without further purification and characterization.
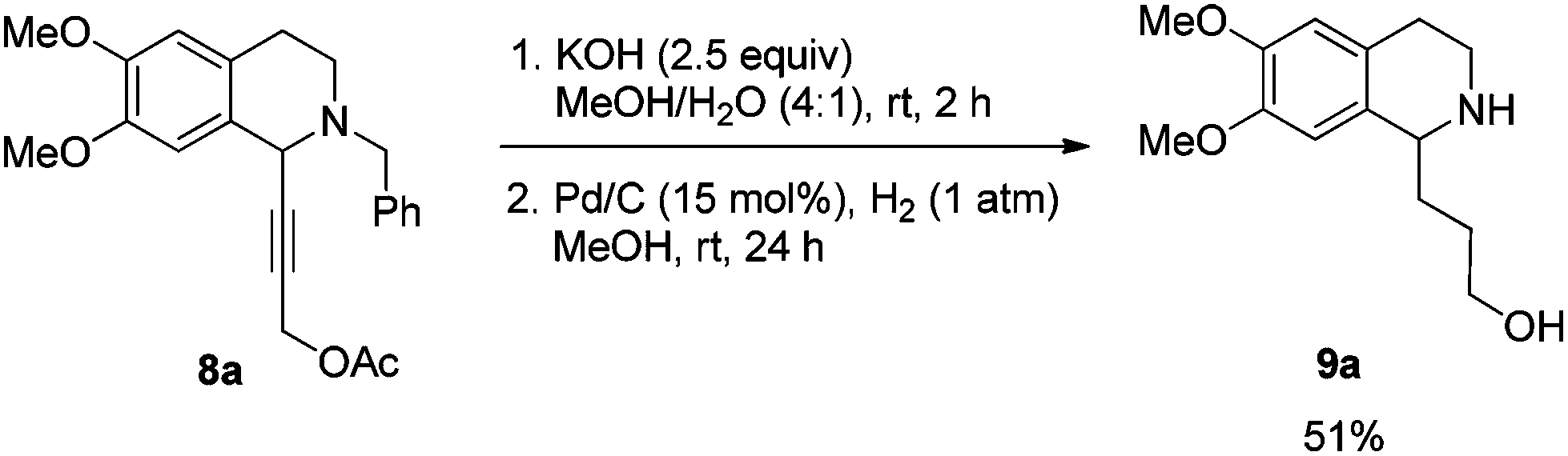
To another Schlenk tube was added the crude product, methanol (4 mL), and Pd/C (32.1 mg, 0.03 mmol, 10% in mass) sequentially, which was purged three times with hydrogen. The suspension was stirred for 24 h at room temperature under a hydrogen pressure of 1 atm, and was filtered over a short column of Celite eluted with methanol (5 mL × 6). After evaporation, the residual product was purified by silica gel column chromatography (eluent: dichloromethane–methanol = 5/1) to provide 9a (25.7 mg, 51%) as an oil: 1H NMR (400 MHz, CDCl3) δ 6.60 (s, 1H, Ar–H), 6.56 (s, 1H, Ar–H), 4.38 (s, 2H, NH and OH), 3.98–3.93 (m, 1H, NCH), 3.85 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.70–3.62 (m, 1H, one proton of CH2), 3.60–3.51 (m, 1H, one proton of CH2), 3.25–3.15 (m, 1H, one proton of CH2), 3.10–3.00 (m, 1H, one proton of CH2), 2.83–2.61 (m, 2H, CH2), 2.05–1.90 (m, 2H, CH2), 1.85–1.65 (m, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ 147.4, 147.3, 130.1, 126.5, 111.5, 109.2, 62.7, 55.9, 55.7, 55.3, 39.6, 35.4, 30.5, 28.5; MS (EI) m/z 251 (M+, 0.2), 192 (100); IR (neat): v = 3302, 2931, 2832, 1610, 1512, 1463, 1354, 1254, 1221, 1112, 1011 cm−1.
To a Schlenk tube were added SOCl2 (13.1 mg, 0.095 mmol) and dichloromethane (0.5 mL). The solution was heated at 40 °C, and 0.5 mL of dichloromethane containing 9a (17.0 mg, 0.068 mmol) was added dropwise. The resulting mixture was heated at 40 °C for 3 h. After evaporation, the crude product was then submitted to the next step without further purification and characterization.
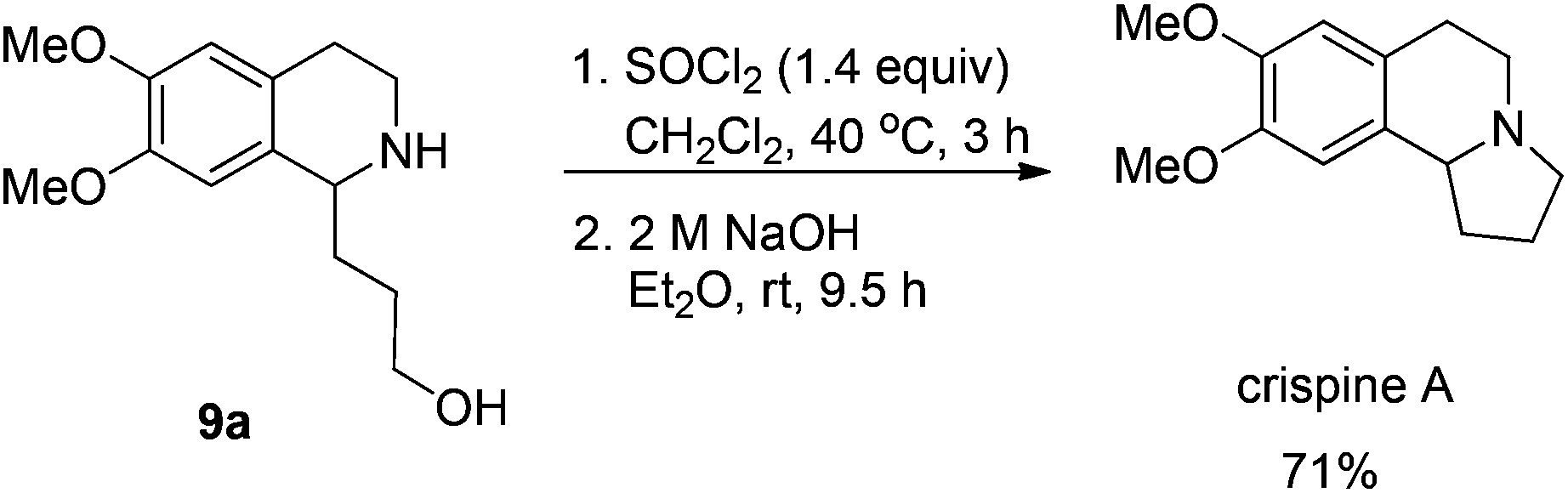
To the Schlenk tube containing the crude product were added an aqueous 2 M NaOH solution (0.5 mL, 1.0 mmol) and diethyl ether (1 mL) sequentially. The suspension was stirred for 9.5 h at room temperature, the organic layer was separated and the water was extracted with diethyl ether (5 mL × 3). The organic layer was dried over Na2SO4, filtered, and evaporated. The residual product was purified by silica gel column chromatography (eluent: dichloromethane–methanol = 15/1–5/1) to provide crispine A (11.2 mg, 71%) as a solid: m.p. 84–86 °C (petroleum ether–diethyl ether) (lit.15 m.p. 88–89 °C); 1H NMR (400 MHz, CDCl3) δ 6.61 (s, 1H, Ar–H), 6.57 (s, 1H, Ar–H), 3.850 (s, 3H, OCH3), 3.845 (s, 3H, OCH3), 3.43 (t, J = 8.0 Hz, 1H, NCH), 3.22–3.15 (m, 1H, one proton of CH2), 3.11–2.96 (m, 2H, CH2), 2.79–2.69 (m, 1H, one proton of CH2), 2.69–2.52 (m, 2H, CH2), 2.38–2.27 (m, 1H, one proton of CH2), 2.00–1.80 (m, 2H, CH2), 1.79–1.66 (m, 1H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 147.2, 147.1, 130.8, 126.1, 111.2, 108.7, 62.9, 55.9, 55.8, 53.1, 48.3, 30.4, 27.9, 22.1; MS (EI) m/z 234 (M+ + 1, 6.25), 233 (M+, 43.47), 232 (100); IR (KBr): v = 2924, 2853, 2794, 1611, 1512, 1464, 1376, 1357, 1321, 1263, 1229, 1214, 1138, 1015 cm−1.
To a flame-dried Schlenk tube was added (R,R)-N-pinap (247.0 mg, 0.44 mmol) inside a glove box. CuI (38.4 mg, 0.2 mmol), 4 Å molecular sieves (6.0213 g), and toluene (40 mL) were sequentially added under an Ar atmosphere. The Schlenk tube was then stirred at rt for 30 min. PhCOOH (122.6 mg, 1.0 mmol), 12a (2.9901 g, 28 mmol)/toluene (20 mL), 13a (5.4213 g, 28 mmol)/toluene (20 mL), and 4b (2.0251 g, 20.0 mmol)/toluene (40 mL) were then added sequentially under an Ar atmosphere. The Schlenk tube was then placed in a pre-heated oil bath at 40 °C with stirring for 12 h as monitored by TLC. After cooling to room temperature, the crude reaction mixture was filtrated through a short column of silica gel eluted with ethyl acetate (100 mL). After evaporation, the residue was purified by chromatography on silica gel to afford S-8a (7.6763 g, 98%) (eluent: petroleum ether–ethyl acetate = 6/1) as an oil: 98% ee (HPLC conditions: Chiralcel AD-H column, hexane–i-PrOH = 85/15, 1.0 mL min−1, λ = 214 nm, tR(major) = 13.2 min, tR(minor) = 10.1 min); [α]25D = −77.4 (c = 1.02, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 6.8 Hz, 2H, Ar–H), 7.32 (t, J = 7.4 Hz, 2H, Ar–H), 7.26 (t, J = 7.0 Hz, 1H, Ar–H), 6.64 (s, 1H, Ar–H), 6.57 (s, 1H, Ar–H), 4.74 (t, J = 1.6 Hz, 2H, OCH2), 4.51 (s, 1H, NCH), 3.85 (d, J = 13.2, 1H, one proton of NCH2Ar), 3.78 (d, J = 13.2, 1H, one proton of NCH2Ar), 3.82 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 2.98–2.86 (m, 2H, ArCH2), 2.81–2.73 (m, 1H, one proton of CH2), 2.70–2.61 (m, 1H, one proton of CH2), 2.09 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 170.0, 148.0, 147.1, 138.0, 129.0, 128.1, 127.0, 126.5, 125.9, 111.1, 110.1, 84.6, 80.4, 59.2, 55.8, 55.6, 53.1, 52.4, 45.6, 28.3, 20.6; MS (ESI) m/z 380 ([M + H]+); IR (neat): v = 2936, 2911, 2831, 1739, 1611, 1516, 1463, 1452, 1375, 1358, 1254, 1219, 1131, 1090, 1019 cm−1; HRMS calcd for C23H26NO4 ([M + H]+): 380.1856. Found: 380.1858.
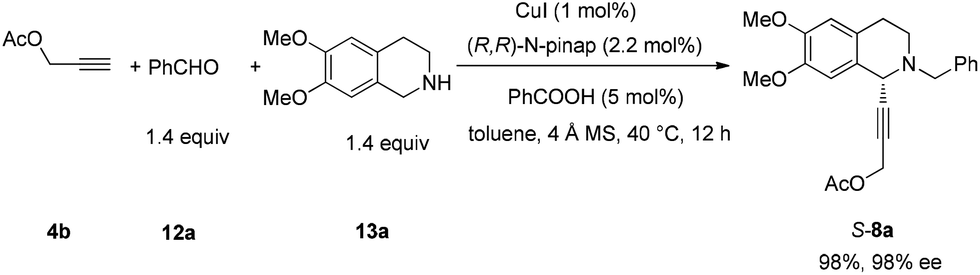
To a Schlenk tube, KOH (2.6668 g, 47.5 mmol), mixed solvent (80 mL, MeOH–H2O = 4/1 by volume), and S-8a (7.2096 g, 19.0 mmol)/mixed solvent (80 mL, MeOH–H2O = 4/1 by volume) were added sequentially. The mixture was then stirred at room temperature. After 2 h as monitored by TLC, the reaction was complete, and then MeOH was removed by rotary evaporation. Water (5 mL) was added and the solution was extracted with ethyl acetate (50 mL × 4) and the organic phase was dried over anhydrous Na2SO4. After filtration and evaporation, the residual product was purified by silica gel column chromatography (eluent: petroleum ether–ethyl acetate = 3/2) to provide S-5a (6.1661 g, 96%) as an oil: 98% ee (HPLC conditions: Chiralcel OD-H column, hexane–i-PrOH = 95/5, 0.4 mL min−1, λ = 214 nm, tR(major) = 41.8 min, tR(minor) = 49.7 min); [α]25D = −72.1 (c = 0.99, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.2 Hz, 2H, Ar–H), 7.35–7.23 (m, 3H, Ar–H), 6.62 (s, 1H, Ar–H), 6.56 (s, 1H, Ar–H), 4.51 (s, 1H, NCH), 4.26 (s, 2H, OCH2), 3.81 (d, J = 6.8, 1H, one proton of NCH2Ar), 3.77 (d, J = 13.2, 1H, one proton of NCH2Ar), 3.82 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 2.98–2.85 (m, 2H, ArCH2), 2.80–2.70 (m, 1H, one proton of CH2), 2.68–2.57 (m, 1H, one proton of CH2), 2.36 (s, 1H, OH); 13C NMR (100 MHz, CDCl3) δ 148.1, 147.2, 138.0, 129.2, 128.2, 127.1, 126.9, 125.9, 111.2, 110.1, 85.1, 83.2, 59.2, 55.9, 55.8, 53.3, 51.0, 45.4, 28.2; MS (ESI) m/z 338 ([M + H]+); IR (neat): v = 3499, 3060, 2914, 2833, 1611, 1516, 1463, 1452, 1358, 1254, 1223, 1128, 1085, 1015 cm−1; HRMS calcd for C21H24NO3 ([M + H]+): 338.1751. Found: 338.1755.
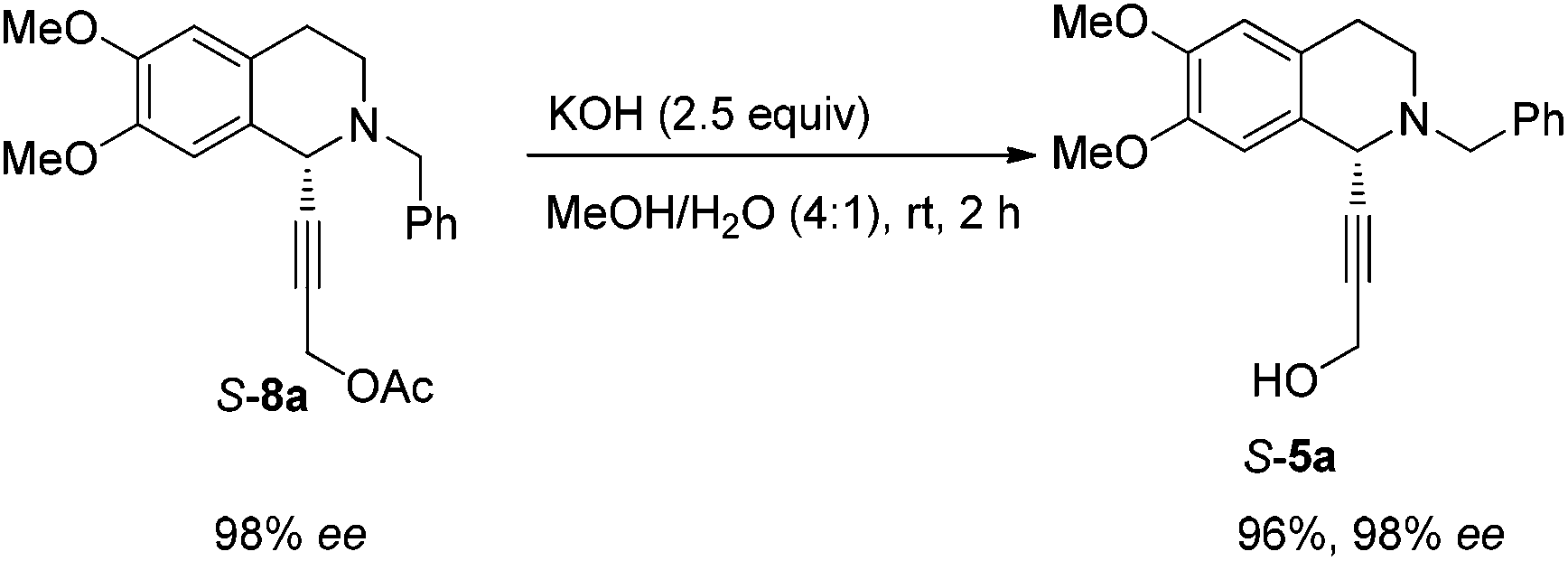
To a vial were added S-5a (337.5 mg, 1.0 mmol), ethyl acetate (10 mL), and Pd/C (160.0 mg, 0.15 mmol, 10% in mass) sequentially. The vial was transferred to a Parr steel autoclave, which was purged three times with hydrogen and finally pressurized to 10 bar. The suspension was stirred for 4 h at 25 °C, and was filtered over a small pad of Celite eluted with methanol (15 mL × 4). After evaporation, the residual product was purified by silica gel column chromatography (eluent: dichloromethane–methanol–ammonia solution = 100/10/0.5) to provide R-9a (170.9 mg, 65%, purity = 95% based on the 1H NMR analysis with CH2Br2 as the internal standard) as an oil: 98% ee (HPLC conditions: Chiralcel OD-H column, hexane–i-PrOH = 80/20, 0.1% triethylamine, 0.4 mL min−1, λ = 214 nm, tR(major) = 24.2 min, tR(minor) = 15.9 min); [α]25D = +34.9 (c = 0.99, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.59 (s, 1H, Ar–H), 6.56 (s, 1H, Ar–H), 4.37 (s, 2H, NH and OH), 3.98–3.93 (m, 1H, NCH), 3.85 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.69–3.61 (m, 1H, one proton of CH2), 3.59–3.51 (m, 1H, one proton of CH2), 3.25–3.15 (m, 1H, one proton of CH2), 3.10–3.00 (m, 1H, one proton of CH2), 2.83–2.61 (m, 2H, CH2), 2.05–1.90 (m, 2H, CH2), 1.85–1.65 (m, 2H, CH2); 13C NMR (100 MHz, CDCl3) δ 147.4, 147.3, 130.1, 126.5, 111.5, 109.2, 62.7, 55.9, 55.7, 55.3, 39.6, 35.4, 30.5, 28.5; MS (EI) m/z 251 (M+, 0.27), 192 (100); IR (neat): v = 3303, 2931, 2832, 1610, 1512, 1463, 1355, 1254, 1221, 1112, 1011 cm−1.
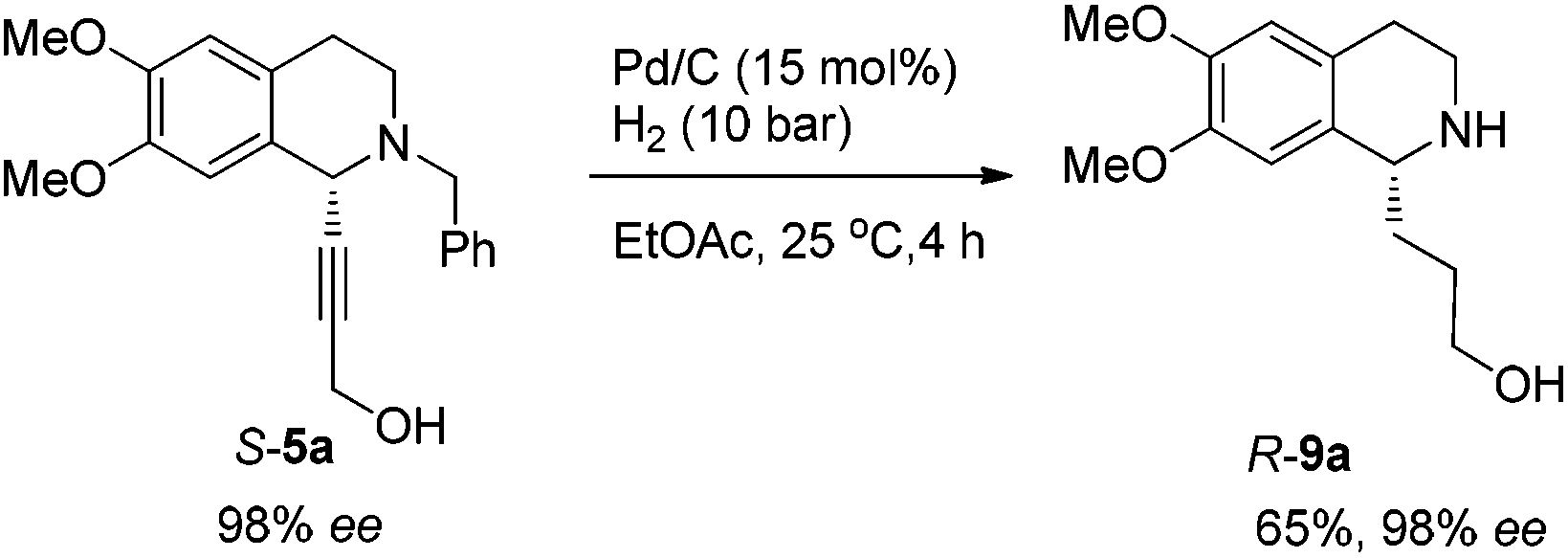
To a flame dried Schlenk tube were added dichloromethane (2.5 mL) and SOCl2 (84.3 mg, 0.7 mmol). Then 2.5 mL of dichloromethane containing R-9a (126.0 mg, 0.48 mmol, 95% purity) were added dropwise. The resulting mixture was stirred for 4 h at room temperature, and the solvent was removed under reduced pressure to afford the crude product.
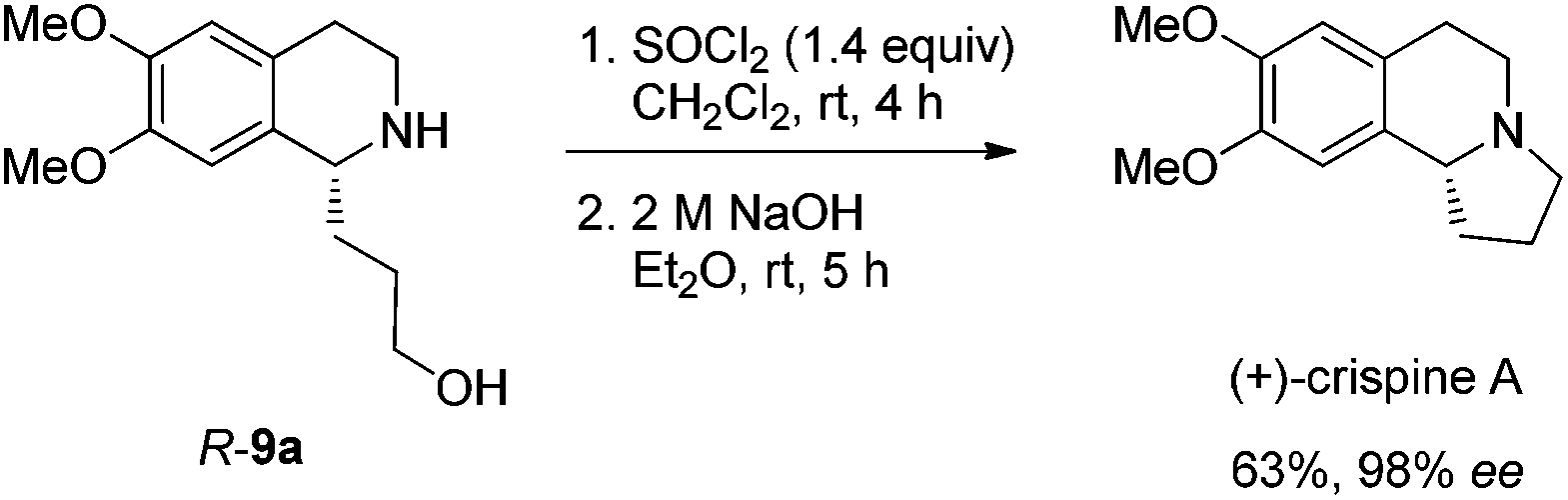
To the Schlenk tube containing the crude product were added an aqueous 2 M NaOH solution (2.5 mL, 5 mmol) and diethyl ether (10 mL) sequentially. The suspension was stirred for 5 h at room temperature, the organic layer was separated and the water was extracted with diethyl ether (10 mL × 4). The extract was dried over Na2SO4, filtered, and evaporated. The residual product was purified by silica gel column chromatography (eluent: ethyl acetate–methanol–ammonia solution = 300/100/4 for the first round to provide 42.1 mg of pure product; the impure part was submitted to the second round under the same eluent to provide 27.8 mg) to provide (+)-crispine A (totally 69.9 mg, 63%) as a solid: 98% ee (HPLC conditions: Chiralcel OD-H column, hexane–i-PrOH = 80/20, 1.0 mL min−1, λ = 214 nm, tR(major) = 12.2 min, tR(minor) = 8.6 min); [α]18D = +105.2 (c = 1.08, CHCl3) (lit.12a [α]25D = +100.4 (c = 1.0, CHCl3); lit.4a [α]25D = +91.0 (MeOH)); m.p. 85–87 °C (petroleum ether–diethyl ether) (lit.4a m.p. 87–89 °C); 1H NMR (300 MHz, CDCl3) δ 6.61 (s, 1H, Ar–H), 6.57 (s, 1H, Ar–H), 3.852 (s, 3H, OCH3), 3.848 (s, 3H, OCH3), 3.43 (t, J = 8.3 Hz, 1H, NCH), 3.23–3.14 (m, 1H, one proton of CH2), 3.13–2.96 (m, 2H, CH2), 2.80–2.50 (m, 3H, CH2 and one proton of CH2), 2.40–2.26 (m, 1H, one proton of CH2), 2.02–1.81 (m, 2H, CH2), 1.80–1.64 (m, 1H, one proton of CH2); 13C NMR (100 MHz, CDCl3) δ 147.2, 147.1, 130.8, 126.1, 111.2, 108.7, 62.9, 55.9, 55.8, 53.1, 48.3, 30.4, 27.9, 22.1; MS (EI) m/z 234 (M+ + 1, 6.1), 233 (M+, 42.84), 232 (100); IR (KBr): v = 2924, 2853, 2792, 1611, 1511, 1464, 1376, 1357, 1321, 1263, 1229, 1214, 1138, 1015 cm−1.
Acknowledgements
Financial support from the National Basic Research Program (2011CB808700) and the National Natural Science Foundation of China (21232006) is greatly appreciated.Notes and references
- (a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed; (b) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444 RSC.
- (a) A. J. Aladesanmi, C. J. Kelly and J. D. Leary, J. Nat. Prod., 1983, 46, 127 CrossRef CAS; (b) E. Tojo, M. A. Onur, A. J. Freyer and M. Shamma, J. Nat. Prod., 1990, 53, 634 CrossRef CAS; (c) A. J. Freyerm, M. H. Abu Zarga and S. Firdous, J. Nat. Prod., 1987, 50, 684 CrossRef.
- E. Tojo, J. Nat. Prod., 1989, 52, 909 CrossRef CAS.
- (a) Q. Zhang, G. Tu, Y. Zhao and T. Cheng, Tetrahedron, 2002, 58, 6795 CrossRef CAS; (b) T. S. Kam and K. M. Sim, Phytochemistry, 1998, 47, 145 CrossRef CAS; (c) T. Itoh, M. Miyazaki, K. Nagata, M. Yokoya, S. Nakamura and A. Ohsawa, Heterocycles, 2002, 58, 115 CrossRef CAS PubMed.
- C. J. Kelleher, L. Cardozo, C. R. Chapple, F. Haab and A. M. Ridder, BJU Int., 2005, 95, 81 CrossRef PubMed.
- A. J. Aladesanmi and O. R. Ilesanmi, J. Nat. Prod., 1987, 50, 1041 CrossRef CAS.
- P. W. Sheldrake, K. E. Suckling, R. N. Woodhouse, A. J. Murtagh, R. B. Herbert, A. C. Barker, J. Staunton and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1998, 3003 RSC.
- R. J. Reddy, N. Kawai and J. Uenishi, J. Org. Chem., 2012, 77, 11101 CrossRef CAS PubMed.
- W. Lin, T. Cao, W. Fan, Y. Han, J. Kuang, H. Luo, B. Miao, X. Tang, Q. Yu, W. Yuan, J. Zhang, C. Zhu and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 277 CrossRef CAS PubMed.
- For the pioneering study on a racemic version of such abnormal A3 coupling reactions, see: D. Das, A. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 3765 CrossRef CAS PubMed . For a recent study on such a racemic version of such a reaction with THIQ, see: Q. Zheng, W. Meng, G. Jiang and Z. Yu, Org. Lett., 2013, 15, 5928 CrossRef PubMed . For other related studies, see: (a) L. Ma, W. Chen and D. Seidel, J. Am. Chem. Soc., 2012, 134, 15305 Search PubMed; (b) D. Das and D. Seidel, Org. Lett., 2013, 15, 4358 Search PubMed; (c) D. Seidel, Org. Chem. Front., 2014 10.1039/C4QO00022F.
- 5-Ethynyl-1,3-benzodioxole 1a is commercially available. In this study it was obtained via Sonogashira cross-coupling followed by removal of the 2-hydroxy-2-propyl group in 73% overall yield.
- (a) J. Szawkalo, A. Zawadzka, K. Wojtasiewicz, A. Leniewski, J. Drabowicz and Z. Czarnocki, Tetrahedron: Asymmetry, 2005, 16, 3619 CrossRef CAS PubMed; (b) T. R. Wu and J. M. Chong, J. Am. Chem. Soc., 2006, 128, 9646 CrossRef CAS PubMed; (c) S. M. Allin, S. N. Gaskell, J. M. R. Towler, P. C. Bulman Page, B. Saha, M. J. McKenzie and W. P. Martin, J. Org. Chem., 2007, 72, 8972 CrossRef CAS PubMed; (d) J. Szawkalo, S. J. Czarnocki, A. Zawadzka, K. Wojtasiewicz, A. Leniewski, J. K. Maurin, Z. Czarnocki and J. Drabowicz, Tetrahedron: Asymmetry, 2007, 18, 406 CrossRef CAS PubMed; (e) K. R. Bailey, A. J. Ellis, R. Reiss, T. J. Snape and N. J. Turner, Chem. Commun., 2007, 3640 RSC; (f) G. Hou, J. Xie, P. Yan and Q. Zhou, J. Am. Chem. Soc., 2009, 131, 1366 CrossRef CAS PubMed; (g) M. Amat, V. Elias, N. Llor, F. Subrizi, E. Molins and J. Bosch, Eur. J. Org. Chem., 2010, 4017 CrossRef CAS PubMed; (h) M. Miyazaki, N. Ando, K. Sugai, Y. Seito, H. Fukuoka, T. Kanemitsu, K. Nagata, Y. Odanaka, K. T. Nakamura and T. Itoh, J. Org. Chem., 2011, 76, 534 CrossRef CAS PubMed; (i) M. Gurram, B. Gyimothy, R. Wang, S. Q. Lam, F. Ahmed and R. J. Herr, J. Org. Chem., 2011, 76, 1605 CrossRef CAS PubMed; (j) G. Barker, J. L. McGrath, A. Klapars, D. Stead, G. Zhou, K. R. Campos and P. O'Brien, J. Org. Chem., 2011, 76, 5936 CrossRef CAS PubMed; (k) F. Louafi, J. Moreau, S. Shahane, S. Golhen, T. Roisnel, S. Sinbandhit and J. Hurvois, J. Org. Chem., 2011, 76, 9720 CrossRef CAS PubMed; (l) N. Kawai, M. Matsuda and J. Uenishi, Tetrahedron, 2011, 67, 8648 CrossRef CAS PubMed; (m) R. Sanchez-Obregon, B. Ortiz, V. M. Mastranzo, F. Yuste and J. L. Garcia Ruano, Tetrahedron Lett., 2013, 54, 1893 CrossRef CAS PubMed; (n) S. C. K. Rotte, A. G. Chittiboyina and I. A. Khan, Eur. J. Org. Chem., 2013, 6355 CrossRef CAS PubMed.
- U. B. Vasconcelos, A. Schrader, G. D. Vilela, A. C. A. Borges and A. A. Merlo, Tetrahedron, 2008, 64, 4619 CrossRef CAS PubMed.
- X. Tang, C. Zhu, T. Cao, J. Kuang, W. Lin, S. Ni, J. Zhang and S. Ma, Nat. Commun., 2013, 4, 2450, DOI:10.1038/ncomms3450.
- F. D. King, Tetrahedron, 2007, 63, 2053 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00058g |
| This journal is © the Partner Organisations 2014 |