An efficient synthesis of (NH)-phenanthridinones via ligand-free copper-catalyzed annulation†
Yan-Fu
Chen
,
Yi-Sheng
Wu
,
Yu-Huei
Jhan
and
Jen-Chieh
Hsieh
*
Department of Chemistry, Tamkang University, New Taipei City, 25137, Taiwan, Republic of China. E-mail: jchsieh@mail.tku.edu.tw; Fax: +886-2-26209924; Tel: +886-2-26215656 ext 2545
First published on 26th February 2014
Abstract
An efficient and concise procedure for the ligand-free copper-catalyzed cascade reaction of C–O and C–N bond coupling was developed, which afforded various (NH)-phenanthridinones in moderate to good yields with tolerance of a wide variety of substrates. This method could be useful for the syntheses of natural alkaloids.
Introduction
Phenanthridinone is an important building block for organic synthesis because it has been frequently discovered in a wide variety of natural alkaloids1 and in addition possesses several types of bioactivity such as antilymphoma, antileukemia, antitumor, antiviral and inhibitor of HIV-1 integrase etc.2 Traditional approaches for the syntheses of phenanthridinones are via intramolecular annulation of the corresponding aminocarboxylatebiphenyls3 or the reductive cyclization of nitrocarbonylbiphenyls.4 Other common strategies include radical cyclization,5 Schmidt reaction/electrophilic aromatic substitution of biphenylcarboxylic acids,6 Beckmann rearrangement of fluorenones,7 microwave-assisted anionic ring closure reaction,8 anionic cycloaromatization of 1-aryl-3-hexen-1,5-diynes,9 oxidative coupling reaction of N-arylbenzamide10 and Pd-catalyzed annulation of aryne with o-halobenzamides.11In recent years, palladium-catalyzed C–H functionalization has been well developed and used as a powerful method for carbon–carbon12 and carbon–heteroatom13 bond formation. Development toward the syntheses of phenanthridinones has also turned to palladium-catalyzed C–H bond activation. In particular, the palladium catalytic cyclization reactions of N-aryl-2-bromobenzamide14 and N-arylbenzamide15 are often reported. Domino processes for multiple C–H bond activations were also carried out by Wang16 and Cheng's17 research groups. However, these domino reactions were restricted to the synthesis of N-methoxyphenanthridinones and further photochemical reaction was required to provide the corresponding (NH)-phenanthridinones for other synthetic applications.
Very recently, novel procedures for the synthesis of phenanthridinones through an oxidative insertion of carbon monoxide to 2-aminobiaryls were disclosed independently by Orito, Zhu and Chuang.18 Their protocols selectively provide different sort of phenanthridinones. However, only one case among these reports is related to the synthesis of (NH)-phenanthridinones. Therefore, the development of novel methods to address this issue is still desirable. Our experience in catalytic coupling reactions involving nitriles19 encouraged us to explore the possibility of the synthesis of free (NH)-phenanthridinones by a coupling reaction involving a nitrile. Herein, we report the first example of an efficient and convenient synthetic pathway to form (NH)-phenanthridinones through a copper-catalyzed cyclization reaction involving C–O and C–N bond coupling of a nitrile.
Results and discussion
Our initial studies used 2′-bromo-[1,1′-biphenyl]-2-carbonitrile (1a) as a model substrate (Table 1, entry 1), which was treated with 5 mol% CuI and 3 equiv. of NaOH in 1.0 mL tBuOH at 120 °C for 24 h; and the corresponding phenanthridinone (2a) was obtained in 69% NMR yield. Product 2a was confirmed by 1H NMR, 13C NMR and HRMS analysis. We also observed a small amount of undefined side products and biphenylamide after working up the reaction.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Cu] | Base (n) | Solvent | Temp (°C) | Yieldb (%) | |
| 1a | 2a | |||||
| a Reactions were carried out using 0.1 mmol (1.0 equiv.) 2′-bromo-[1,1′-biphenyl]-2-carbonitrile (1a) with 5 mol% [Cu] and base (n equiv.) in 1.0 mL solvent at T °C for 24 h. b 1H NMR yield based on internal standard mesitylene. c Under N2. | ||||||
| 1 | CuI | NaOH (3) | tBuOH | 120 | 0 | 69 |
| 2 | CuBr | NaOH (3) | tBuOH | 120 | 0 | 54 |
| 3 | Cu2O | NaOH (3) | tBuOH | 120 | 0 | 62 |
| 4 | Cu(OAc)2 | NaOH (3) | tBuOH | 120 | 0 | 49 |
| 5 | CuI | KOH (3) | tBuOH | 120 | 0 | 66 |
| 6 | CuI | LiOH (3) | tBuOH | 120 | 6 | 52 |
| 7 | CuI | NaOtBu (4)–H2O (1.1) | tBuOH | 120 | 23 | 35 |
| 8 | CuI | NaOH (4) | tBuOH | 120 | 0 | 87 |
| 9 | CuI | NaOH (4) | tBuOH | 140 | 0 | 65 |
| 10 | CuI | NaOH (4) | tBuOH | 100 | 0 | 96 |
| 11 | CuI | NaOH (4) | tBuOH | 80 | 17 | 73 |
| 12 | CuI | NaOH (4) | DMF | 100 | 39 | 32 |
| 13 | CuI | NaOH (4) | DMSO | 100 | 21 | 25 |
| 14c | CuI | NaOH (4) | tBuOH | 100 | 0 | 93 |
| 15 | None | NaOH (4) | tBuOH | 100 | 47 | 0 |
To optimize the reaction conditions, the effect of solvent, base, reaction temperature and copper source were investigated (Table 1). We first examined the copper source for this reaction (entries 2–4). Among the various copper sources employed, CuI was found to be the most effective catalyst, providing the desired product 2a in 69% NMR yield (entry 1). Screening of the base revealed that NaOH and KOH gave similar results (entries 1 and 5). The cyclization reaction could not be completed within 24 h when using LiOH as base (entry 6). The use of a NaOtBu–H2O system (entry 7) also afforded the desired product 2a in lower yield. It was found that the amount of base significantly affected the yield of 2a, and 4.0 equiv. of NaOH provided the best yield for this cyclization reaction (entry 8). Some unidentified compounds and biphenylamide were detected when the temperature was increased to 140 °C (entry 9). However, only desired product 2a was obtained when the reaction temperature was decreased to 100 °C (entry 10).
The effect of solvent was investigated as well, and it was found that only highly polar solvents such as DMF and DMSO allowed the reaction to proceed successfully (entries 12 and 13). However, the substrate 1a could be fully converted only when using tBuOH as solvent. In addition, there was no significant difference when running the reaction under nitrogen atmosphere or under air (entry 14). Moreover, the present cyclization reaction could not afford any desired product 2a without a copper source (entry 15).
The copper-catalyzed cyclization reaction was successfully extended to various substrates (1), and the results are listed in Table 2. The reaction required at least 24 h to fully consume the substrates (1). As indicated, the reaction worked well for various substrates and both electron-donating and electron-withdrawing substituents on the aryl bromide moiety were well tolerated to give the corresponding products in moderate to good yields (2a–2j). Substituents para to the bromide (2h–2j) dramatically affected cyclization. Thus, a substrate with an electron-withdrawing group provided a higher yield of the corresponding product (2h) than those with an electron-donating group (2i, 2j). We frequently detected a small amount of the corresponding 2-bromobiarylamide for substrates with an electron-donating group on the aryl bromide moiety, which caused lower yields of the desired products (2c, 2d, 2i and 2j). Substituents on the benzonitrile moiety were also well tolerated (2k–2y); however, the yields of the desired products were not only decided by the substituents on the benzonitrile moiety but also by the substituents on the aryl bromide moiety. When hydrophilic substituents such as methoxy group, chloride or fluoride were introduced into the phenanthridinones, the yields of the desired products were generally lower (2n, 2o, 2p, 2q and 2r). This is probably due to the partial solubility of the products in water during the extraction. It is noteworthy that product 2k is a natural alkaloid known as phenaglydon, which has been isolated from the lipophilic leaf extract of Glycosmis cyanocarpa (Rutaceae).20
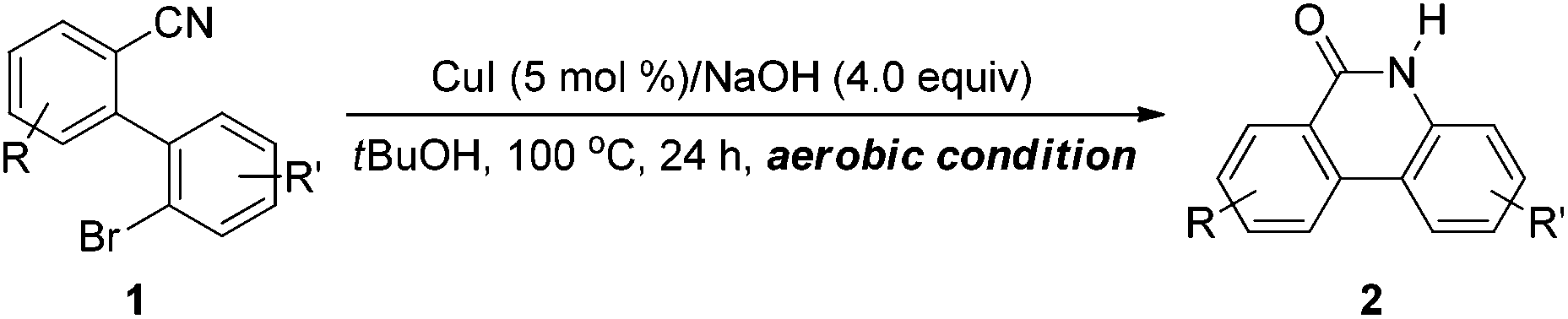
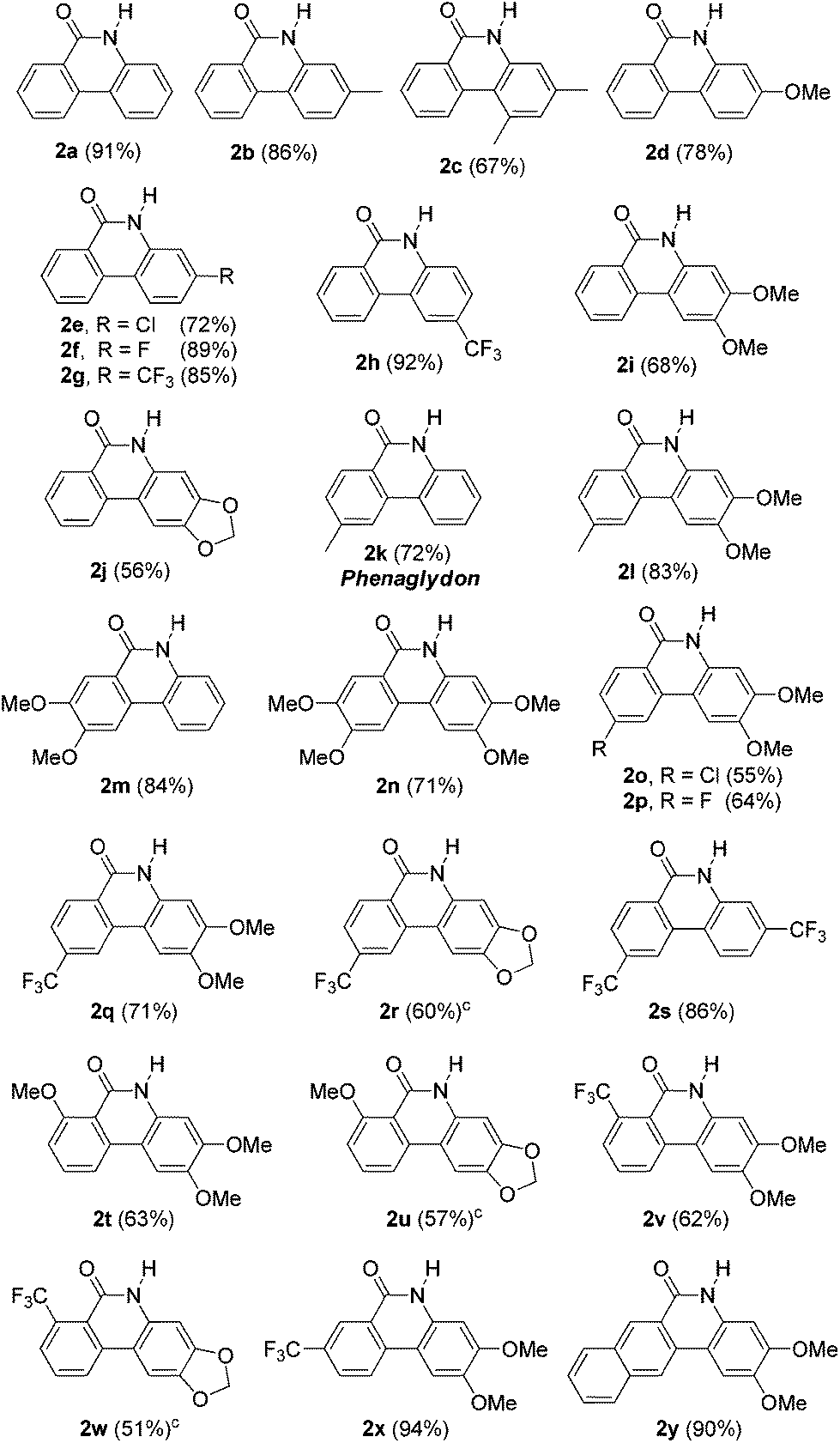
Reactions for substrates with a substituent ortho to the nitrile group (2t, 2u, 2v and 2w) did not go to completion even after a longer reaction time. The ortho substituents would retard the addition of hydroxide to nitrile and reduce the yields of the corresponding products. Substrates with a meta CF3 group on the benzonitrile moiety and with a naphthylnitrile moiety were also well tolerated to afford the corresponding products 2x and 2y in excellent yields. Substrates containing a picolinonitrile moiety or an electrophile such as a ketone group were not tolerated and the reactions provided messy crude spectra.
In order to extend the application of the present method, we selectively synthesized two natural alkaloids crinasiadine and trisphaeridine by using our developed protocol as the key step (Scheme 1). These two natural alkaloids represent the basic skeletons of the Amaryllidaceae alkaloids, which appear in a wide range of natural alkaloids and bioactive compounds. Our synthetic route began from a commercially available compound 6k. Transformation of the aldehyde to nitrile and the subsequent Suzuki coupling reaction provided the substrate 1z. Under the standard conditions of the present copper catalysis, 1z was then converted to crinasiadine 2z in 4 steps with 36% overall yield. Further transformation of 2z to its corresponding phenanthridine afforded another natural alkaloid trisphaeridine 3b with two more steps and 25% overall yield. Conversion of 2z or 3b to other natural alkaloids can be easily achieved by N-alkylation. For example, N-methylation of 2z and 3b can lead to other two natural alkaloids N-methylcrinasiadine21 and bicolorine.22 In addition, more complicated crinasiadine analogues (C1, C2, C3 and C4) could be synthesized by this pathway in moderate to good yields.
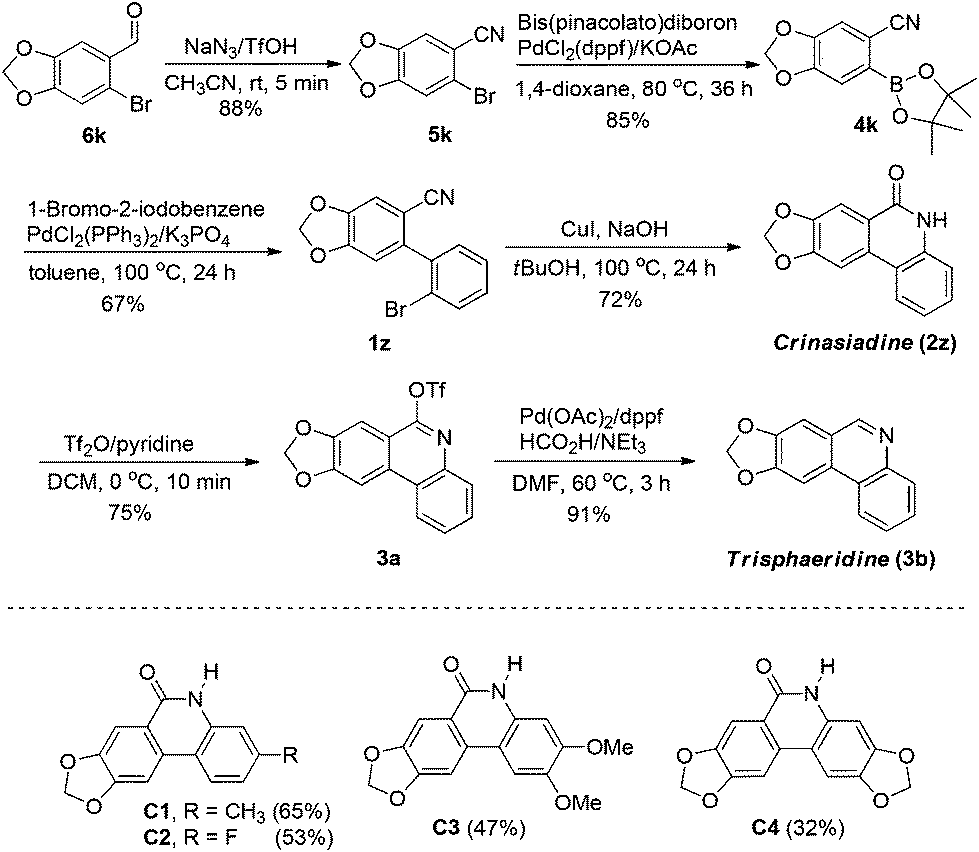 | ||
| Scheme 1 Syntheses of crinasiadine, trisphaeridine and analogues of crinasiadine. | ||
The present methodology could be conducted for gram-scale synthesis as well. As shown in Scheme 2, conversion of 1x to 2x under the standard conditions was carried out on a 5 mmol scale with 76% isolated yield, which implied the potential applications in industry. We also synthesized variously poly-substituted phenanthridine derivatives via formation of the corresponding triflate compound 3c as an important building block in excellent yield, and various 6-substituents could be introduced into the phenanthridine through palladium or nickel catalytic coupling reactions. 6-Aryl and 6-alkyl phenanthridines could be respectively afforded by Suzuki and Kumada type coupling reactions, and 6-H-phenanthridine could be provided by palladium catalyzed reduction with formic acid. Thus, the 6-phenylphenanthridine (3d), 6-ethylphenanthridine (3e) and 6-H-phenanthridine (3f) were successfully generated in 74%, 28% and 81% yields, respectively.
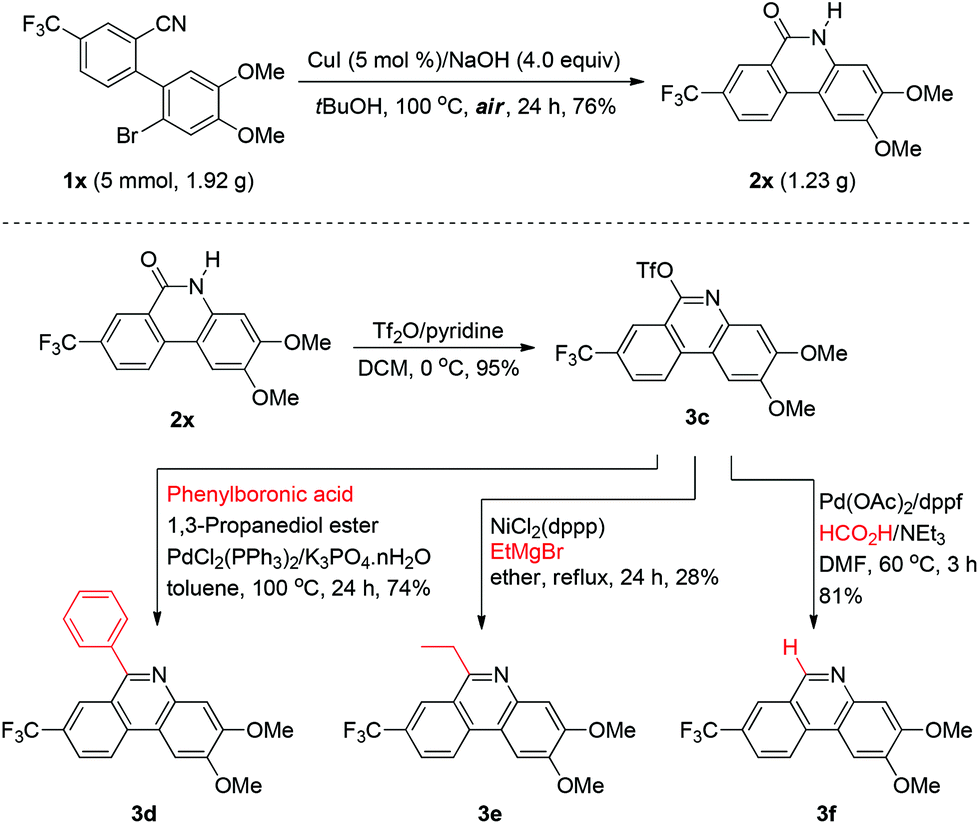 | ||
| Scheme 2 Application to large-scale synthesis and syntheses of various phenanthridines. | ||
Although a more detailed study might be required to fully understand the mechanism of this copper-catalyzed annulation, a tentative pathway can be proposed according to the above results and previous report (Scheme 3).19b Thus, the catalytic reaction is likely to be initiated by the coordination of the nitrile on compound 1 to a Cu(I) complex, which accelerates the following nucleophilic addition by hydroxide to form complex A. The oxidative addition of complex A in an intramolecular manner then occurred to generate Cu(III) species (complex B). The subsequent reductive elimination provides compound 3 and regenerates the Cu(I) species. Tautomerization of 3 affords the desired product 2.
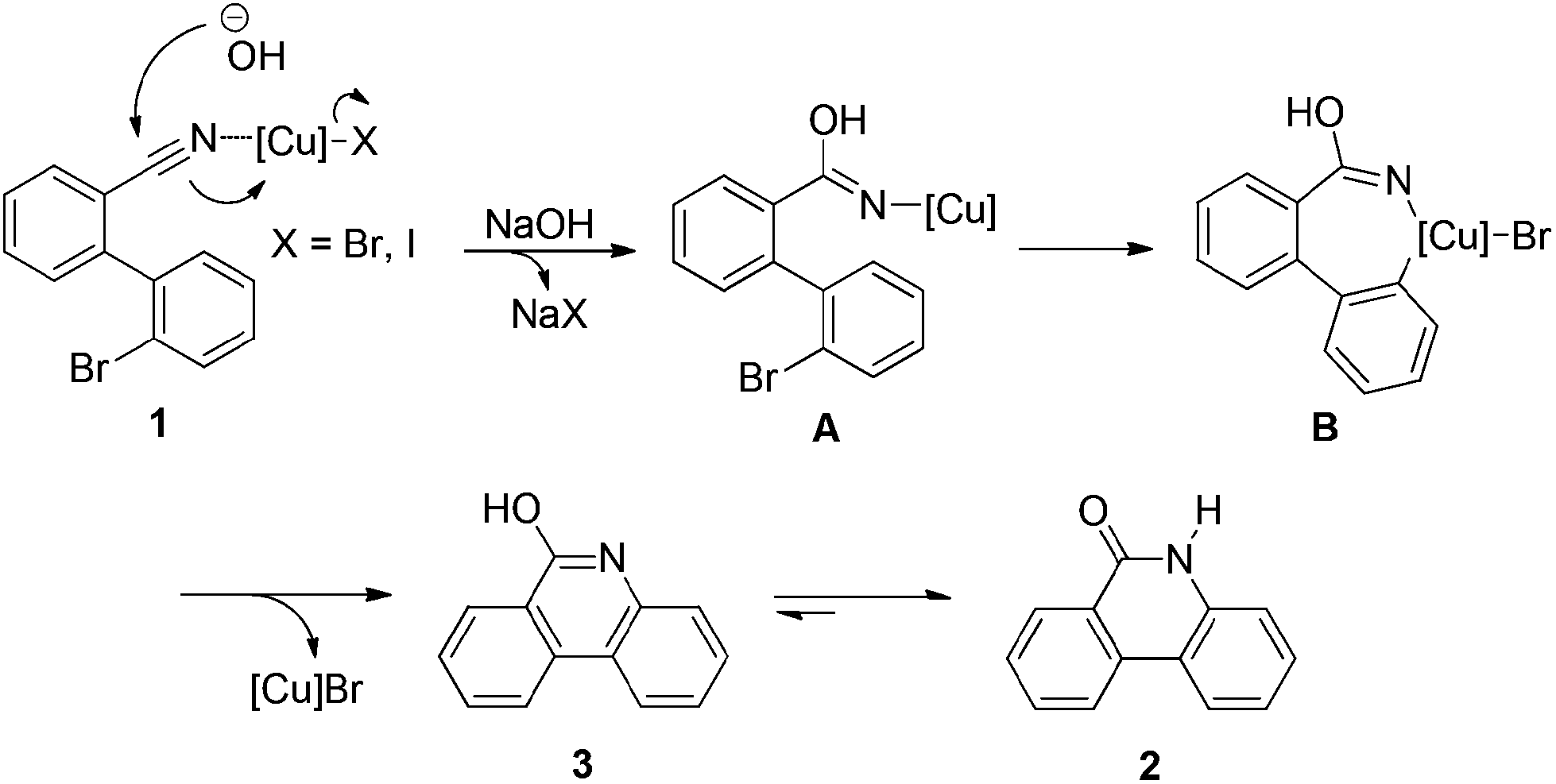 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a novel method for the copper-catalyzed cascade reaction of C–O and C–N bond coupling. This method efficiently provides poly-substituted (NH)-phenanthridinones in moderate to good yields with tolerance of a wide variety of substrates. In addition, this method could be also applied to synthesize three natural alkaloids in a short number of steps with good overall yields. Moreover, conversion of the (NH)-phenanthridinone to the corresponding phenanthridines with various 6-substituents was carried out as well. Further studies to explore the possibility to extend the applications of this catalytic system are currently underway.Experimental
General procedure for the copper-catalyzed cyclization
To a screw-capped vial (10 mL) were added CuI (0.025 mmol, 4.8 mg, 5 mol%), NaOH (2.0 mmol, 80 mg, 4.0 equiv.), and substrate 1 (0.5 mmol, 1.0 equiv.) in tBuOH (5 mL). The vial was then sealed with a cap and allowed to stir at 100 °C for 24 h. The crude reaction mixture was diluted with ethyl acetate (20 mL) and H2O (10 mL). The mixture was then kept stirring at 70 °C for 30 min then the aqueous layer was removed and the organic layer was concentrated in vacuo. The residue was allowed to quickly flow through a short flash column chromatography by using ethyl acetate as eluent and then concentrated in vacuo, following washed by CH2Cl2 to provide the pure product. Products 2 were obtained according to this procedure.Acknowledgements
We thank the National Science Council of the Republic of China (NSC-101-2113-M-032-004-MY2) for the financial support of this research.Notes and references
- For selected papers: (a) Z. Jin, Nat. Prod. Rep., 2011, 28, 1126 RSC; (b) S. Ghosal, P. H. Rao, D. K. Jaiswal, Y. Kumar and A. W. Frahm, Phytochemistry, 1981, 20, 2003 CrossRef CAS; (c) J. M. Llabrés, F. Viladomat, J. Bastida, C. Codina and M. Rubiralta, Phytochemistry, 1986, 25, 2637 CrossRef; (d) S. Ghosal, K. S. Saini and A. W. Frahm, Phytochemistry, 1983, 22, 2305 CrossRef CAS; (e) J. Hu, W.-D. Zhang, Y.-H. Shen, C. Zhang, L. Xu, R.-H. Liu, B. Wang and X.-K. Xu, Biochem. Syst. Ecol., 2007, 35, 114 CrossRef CAS PubMed.
- (a) S. Patil, S. Kamath, T. Sanchez, N. Neamati, R. F. Schinazi and J. K. Buolamwini, Bioorg. Med. Chem., 2007, 15, 1212 CrossRef CAS PubMed; (b) R. K.-Y. Zee-Cheng, S.-J. Yan and C. C. Cheng, J. Med. Chem., 1978, 21, 199 CrossRef CAS; (c) M. Banasik, H. Komura, M. Shimoyama and K. Ueda, J. Biol. Chem., 1992, 267, 1569 CAS; (d) D. Bellocchi, A. Macchiarulo, G. Costantino and R. Pellicciari, Bioorg. Med. Chem., 2005, 13, 1151 CrossRef CAS PubMed.
- (a) J. J. S. Lamba and J. M. Tour, J. Am. Chem. Soc., 1994, 116, 11723 CrossRef CAS; (b) T. Watanabe, Y. Ohashi, R. Yoshino, N. Komano, M. Eguchi, S. Maruyama and T. Ishikawa, Org. Biomol. Chem., 2003, 1, 3024 RSC; (c) P. Lv, K. Huang, L. Xie and X. Xu, Org. Biomol. Chem., 2011, 9, 3133 RSC.
- (a) M. A. Yawer, I. Hussain, I. Iqbal, A. Spannenberg and P. Langer, Tetrahedron Lett., 2008, 49, 4467 CrossRef CAS PubMed; (b) A. Riahi, M. Shkoor, O. Fatunsin, M. A. Yawer, I. Hussain, C. Fischer and P. Langer, Tetrahedron, 2009, 65, 9300 CrossRef CAS PubMed; (c) M. G. Banwell, D. W. Lupton, X. Ma, J. Renner and M. O. Sydnes, Org. Lett., 2004, 6, 2741 CrossRef CAS PubMed; (d) C. Genès, G. Lenglet, S. Depauw, R. Nhili, S. Prado, M.-H. David-Cordonnier, S. Michel, F. Tillequin and F.-H. Porée, Eur. J. Med. Chem., 2011, 46, 2117 CrossRef PubMed.
- B. S. Bhakuni, A. Kumar, S. J. Balkrishna, J. A. Sheikh, S. Konar and S. Kumar, Org. Lett., 2012, 14, 2838 CrossRef CAS PubMed.
- C. C. Woodroofe, B. Zhong, X. Lu and R. B. Silverman, J. Chem. Soc., Perkin Trans. 2, 2000, 55 RSC.
- E. C. Horning, V. L. Stromberg and H. A. Lloyd, J. Am. Chem. Soc., 1952, 74, 5153 CrossRef CAS.
- (a) T. Cailly, F. Fabis and S. Rault, Tetrahedron, 2006, 62, 5862 CrossRef CAS PubMed; (b) E. Dubost, R. Magnelli, T. Cailly, R. Legay, F. Fabis and S. Rault, Tetrahedron, 2010, 66, 5008 CrossRef CAS PubMed.
- (a) M.-J. Wu, C.-F. Lin and S.-H. Chen, Org. Lett., 1999, 1, 767 CrossRef CAS; (b) M.-J. Wu, C.-F. Lin and W.-D. Lu, J. Org. Chem., 2002, 67, 5907 CrossRef CAS PubMed.
- (a) I. Moreno, I. Tellitu, J. Etayo, R. SanMartín and E. Domínguez, Tetrahedron, 2001, 57, 5403 CrossRef CAS; (b) M. D. Ganton and M. A. Kerr, Org. Lett., 2005, 7, 4777 CrossRef CAS PubMed.
- C. Lu, A. V. Dubrovskiy and R. C. Larock, J. Org. Chem., 2012, 77, 8648 CrossRef CAS PubMed.
- (a) J. J. Mousseau and A. B. Charette, Acc. Chem. Res., 2013, 46, 412 CrossRef CAS PubMed; (b) B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885 CrossRef CAS PubMed; (c) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (d) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (e) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed.
- (a) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (b) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed; (c) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981 CrossRef CAS PubMed; (d) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- (a) T. Harayama, T. Akiyama, Y. Nakano, H. Nishioka, H. Abe and Y. Takeuchi, Chem. Pharm. Bull., 2002, 50, 519 CrossRef CAS; (b) T. Harayama, Y. Kawata, C. Nagura, T. Sato, T. Miyagoe, H. Abe and Y. Takeuchi, Tetrahedron Lett., 2005, 46, 6091 CrossRef CAS PubMed; (c) Z. Ma, Z. Xiang, T. Luo, K. Lu, Z. Xu, J. Chen and Z. Yang, J. Comb. Chem., 2006, 8, 696 CrossRef CAS PubMed; (d) R. Bernini, S. Cacchi, G. Fabrizi and A. Sferrazza, Synthesis, 2008, 729 CAS; (e) G. Zhang, X. Zhao, Y. Yan and C. Ding, Eur. J. Org. Chem., 2012, 669 Search PubMed.
- (a) N. Borduas, A. J. Lough and V. M. Dong, Inorg. Chim. Acta, 2011, 369, 247 CrossRef CAS PubMed; (b) C. S. Yeung, X. Zhao, N. Borduas and V. M. Dong, Chem. Sci., 2010, 1, 331 RSC; (c) N. Ishida, Y. Nakanishi, T. Moriya and M. Murakami, Chem. Lett., 2011, 40, 1047 CrossRef CAS.
- G.-W. Wang, T.-T. Yuan and D.-D. Li, Angew. Chem., Int. Ed., 2011, 50, 1380 CrossRef CAS PubMed.
- J. Karthikeyan and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 9880 CrossRef CAS PubMed.
- (a) E. Kumazawa, T. Tokuhashi, A. Horibata, N. Kurono, H. Senboku, M. Tokuda, T. Ohkuma and K. Orito, Eur. J. Org. Chem., 2012, 4622 CrossRef CAS; (b) D. Liang, Z. Hu, J. Peng, J. Huang and Q. Zhu, Chem. Commun., 2013, 49, 173 RSC; (c) V. Rajeshkumar, T.-H. Lee and S.-C. Chuang, Org. Lett., 2013, 15, 1468 CrossRef CAS PubMed.
- (a) J.-C. Hsieh, Y.-C. Chen, A.-Y. Cheng and H.-C. Tseng, Org. Lett., 2012, 14, 1282 CrossRef CAS PubMed; (b) J.-C. Hsieh, A.-Y. Cheng, J.-H. Fu and T.-W. Kang, Org. Biomol. Chem., 2012, 10, 6404 RSC; (c) J.-C. Wan, J.-M. Huang, Y.-H. Jhan and J.-C. Hsieh, Org. Lett., 2013, 15, 2742 CrossRef CAS PubMed.
- G. Wurz, O. Hofer and H. Greger, Nat. Prod. Lett., 1993, 3, 177 CrossRef CAS.
- R. Suau, A. I. Gómez and R. Rico, Phytochemistry, 1990, 29, 1710 CrossRef CAS.
- F. Viladomat, J. Bastida, G. Tribo, C. Codina and M. Rubiralta, Phytochemistry, 1990, 29, 1307 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data, spectral data and copies of all compounds. See DOI: 10.1039/c3qo00082f |
| This journal is © the Partner Organisations 2014 |