A versatile two-step method for the reductive alkylation and formal [4 + 2] annulation of secondary lactams: step economical syntheses of the ant venom alkaloids (2R,5S)-2-butyl-5-propylpyrrolidine and (+)-monomorine I†
Hui-Qiong
Deng
a,
Xiang-Yang
Qian
a,
Yu-Xiu
Li
a,
Jian-Feng
Zheng
a,
Linfeng
Xie
*a and
Pei-Qiang
Huang
*ab
aDepartment of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, China. E-mail: pqhuang@xmu.edu.cn
bState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, China
First published on 11th February 2014
Abstract
The reductive alkylation of secondary lactams is an important transformation in the total synthesis of alkaloids and pharmaceuticals. Methods for this transformation are scarce. We report in this paper a versatile two-step approach consisting of N-benzylation and one-pot reductive alkylation–debenzylation. With suitably functionalized Grignard reagents, a formal [4 + 2] annulation reaction has been achieved. Using this method, we have successfully synthesized several N-α-alkyl and N-α,α′-dialkyl heterocycles, including (±)-coniine (7) and the ant venom alkaloids (2R,5S)-cis-2-butyl-5-propylpyrrolidine (8) and (+)-monomorine I (10) each in two steps starting from readily available secondary lactams.
Introduction
Green and sustainable chemistry has attracted increasing attention in synthetic organic chemistry.1 One-pot procedures including tandem reactions,2 multi-component reactions,3 and domino reactions4 are valuable approaches towards that goal. A one-pot procedure combines at least two synthetic steps in one, thus increasing the efficiency of the synthesis and avoiding a tedious separation process and purification of the intermediates. In this context, the direct transformation of “inert” amides into amines with C–C bond formation is of particular significance and has attracted much attention lately.5,6 We have recently reported the first general one-pot transformation of tertiary lactams and amides into the corresponding amines by reductive bis-alkylation (Scheme 1, eqn (1))6a,b or mono-alkylation (Scheme 1, eqn (1)),6c–e and the direct transformations of secondary amides into ketones7a,b or amines7c (Scheme 1, eqn (2)). Although the latter method appears to be general for the reductive alkylation of secondary amides, it could not be extended to secondary lactams.7c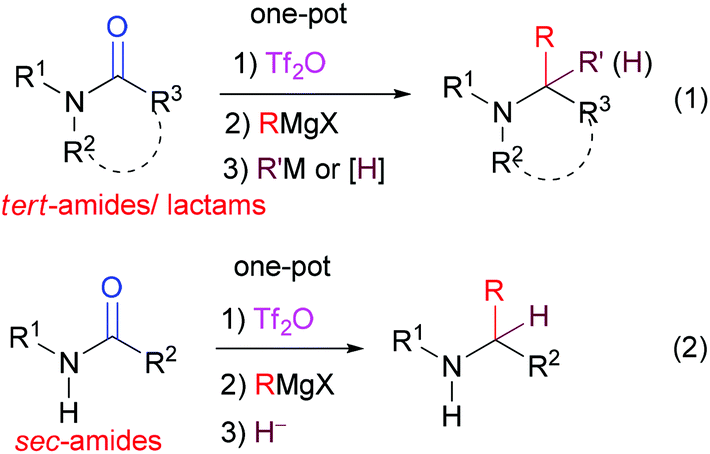 | ||
| Scheme 1 General methods for the direct transformation of tert-amides/lactams and sec-amides into amines by one-pot reductive bis-alkylation and reductive alkylation. | ||
Secondary lactams have been used quite frequently as the key intermediates in the total synthesis of alkaloids and pharmaceuticals.8–14 The desired transformation often involves a reductive alkylation of secondary lactams. In sharp contrast with tertiary lactams,6 few methods exist for the more challenging reductive alkylation of secondary lactams to give amines. To the best of our knowledge, the only example of such transformations has been the reductive cyanation reported by Ganem (Scheme 2, Method A).8,9 Stepwise transformations including reductive n-propylation (Scheme 2, Method B),10 1-pentynylation (Scheme 2, Method C1),11 and vinylation (Scheme 2, Approach C2)12 have been demonstrated. Since C1 and C2 approaches are restricted to sp-C and sp2-C nucleophiles, the need to transform a secondary lactam into a cyclic amine was fulfilled by adopting Oppolzer's procedure (Method B) in all the subsequent total and formal syntheses of pumiliotoxin C (cis-195A) and its diastereomers.10b–g Other transformations involving the use of amide-activating reagents (e.g. Cbz, Boc) and C–C bond formation methods13 require multiple steps.14c–h
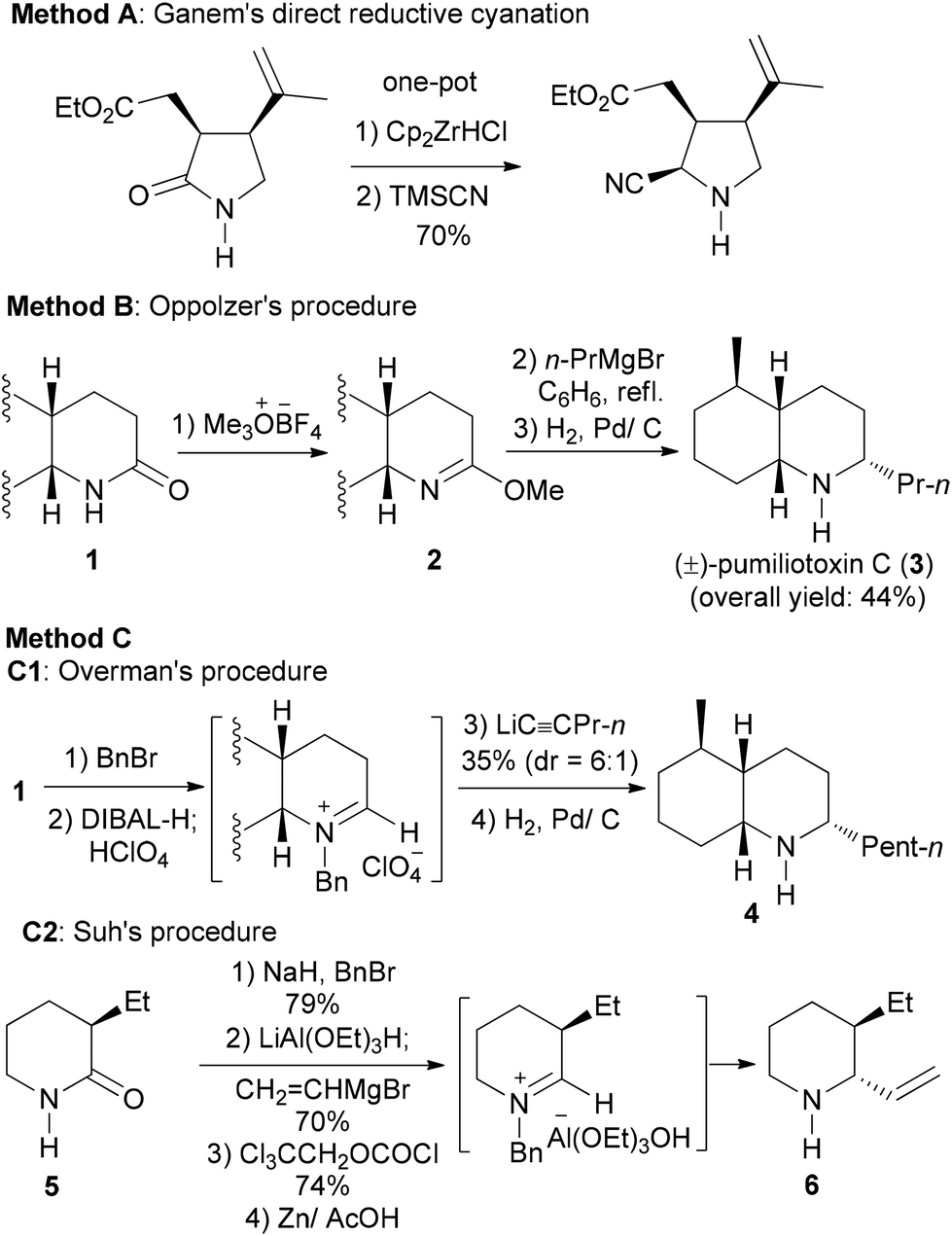 | ||
| Scheme 2 Reported examples of direct (A) and stepwise (B, C) reductive nucleophilic addition of secondary lactams. | ||
N-α-Alkyl and α,α′-dialkyl pyrrolidine, piperidine, pyrrolizidine, indolizidine and related ring systems are structural motifs of many bioactive alkaloids originating from plants, ants and animals.15 For example, coniine (7 in Fig. 1) is one of the major toxic alkaloids present in poison hemlock.16 Ant17 and poison frogs18 are rich sources of the abovementioned alkaloids with over eight hundred compounds being isolated so far.18b Several alkaloids have been isolated from the ant Myrmicaria melanogaster from Brunei in the Indonesian archipelago, including new ones such as cis-2-butyl-5-propylpyrrolidine (8) and (9Z)-3-propylindolizidine (9), and known members like 3-butyl-5-methylindolizidine (10) and (5Z,9Z)-3-butyl-5-propylindolizidine (11).19 The structures of 8 and 9 were elucidated by a combination of GC-MS and GC-IR analyses and confirmed by racemic synthesis. Their absolute configurations were not determined whereas the occurrence of only one enantiomer was assumed.19 Alkaloid (+)-10, isolated in 1973 and known as (+)-monomorine I, is a trail pheromone of the Pharaoh's ant Monomorium pharaonis L.20 Several 5-alkyl and 3,5-dialkylindolizidine alkaloids including (5Z,9Z)-223AB (11),21 alkaloid 167B (12),22 and alkaloid 209D (13)22 have subsequently been isolated. Due to the scarcity of the ant venom and amphibian alkaloids from the natural source, total synthesis becomes a dispensable tool for structure confirmation and determination. In addition, many of those alkaloids have been shown to possess important bioactivities.18c,d,22 The total synthesis provides the basis for the investigation of the biological activity of those alkaloids. Consequently, the enantioselective total synthesis of ant venom and amphibian alkaloids has continued to attract the attention of synthetic organic chemists.
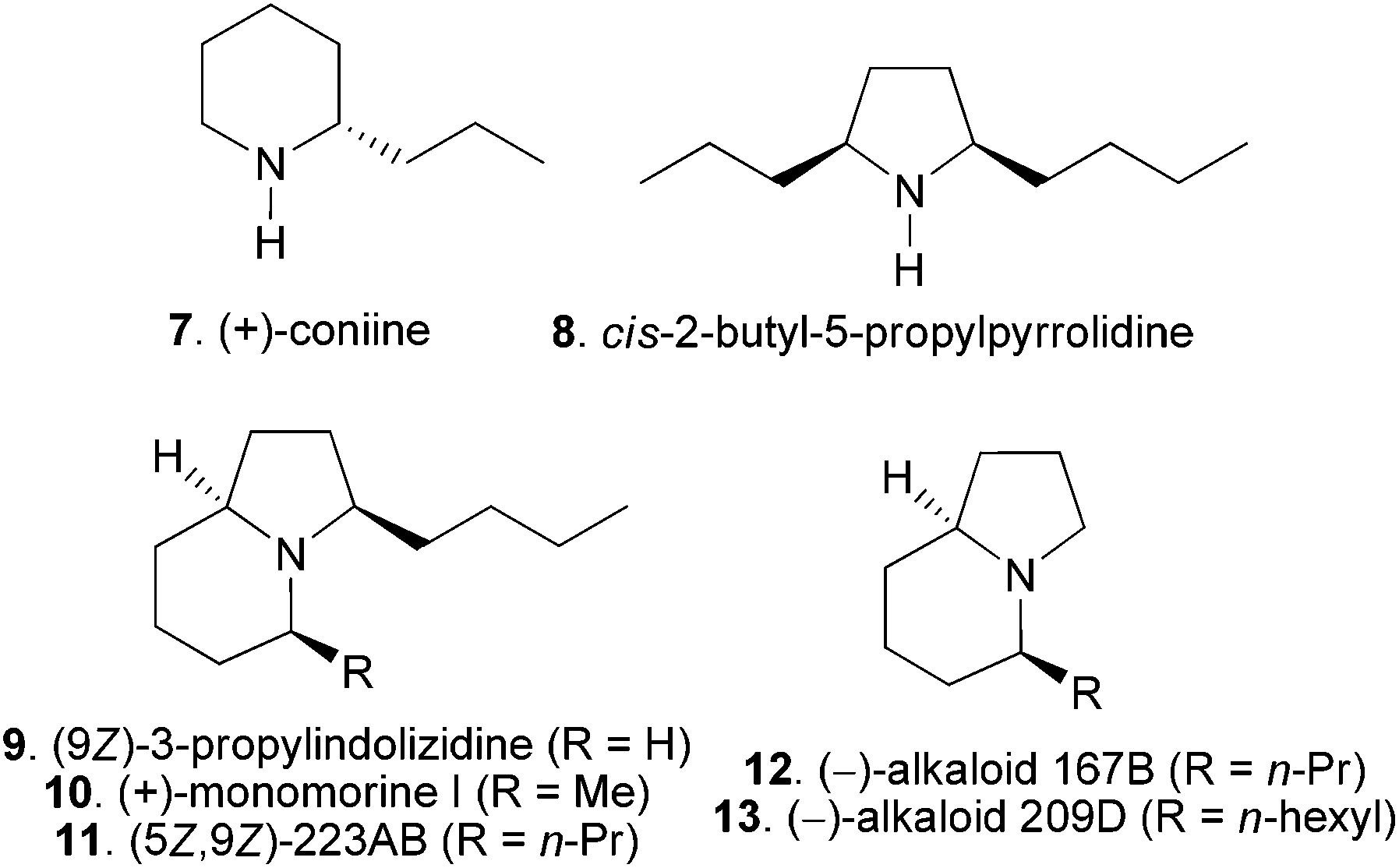 | ||
| Fig. 1 Selected examples of N-α-alkyl, N-α,α′-dialkylpyrrolidine, piperidine and indolizidine alkaloids. | ||
In conjunction with our own interest in the syntheses of ant venom and amphibian alkaloids,23 and in view of diverse alkyl groups in these alkaloids, we aimed to develop a concise and general method for the transformation of secondary lactams into N-α-alkyl, N-α,α′-dialkylpyrrolidine, piperidine, azepane and indolizidine alkaloids. Herein we report a two-step method, and its application to the concise racemic synthesis of plant alkaloid coniine, and the enantioselective syntheses of ant venom alkaloids cis-2-butyl-5-propylpyrrolidine (8) and (+)-monomorine I (10).
Results and discussion
Our synthetic plan was to convert a secondary lactam into a tertiary one by N-benzylation, and to subsequently undertake a direct reductive alkylation6c,d and debenzylation in one pot. It was further envisioned that with the use of suitably functionalized Grignard reagents, the reductive alkylation6c,d–debenzylation–deprotection–cyclization–reduction reactions could be combined into a one-pot procedure. This would allow for rapid access to bicyclic N-heterocycles in only two steps from the secondary lactams A (Scheme 3).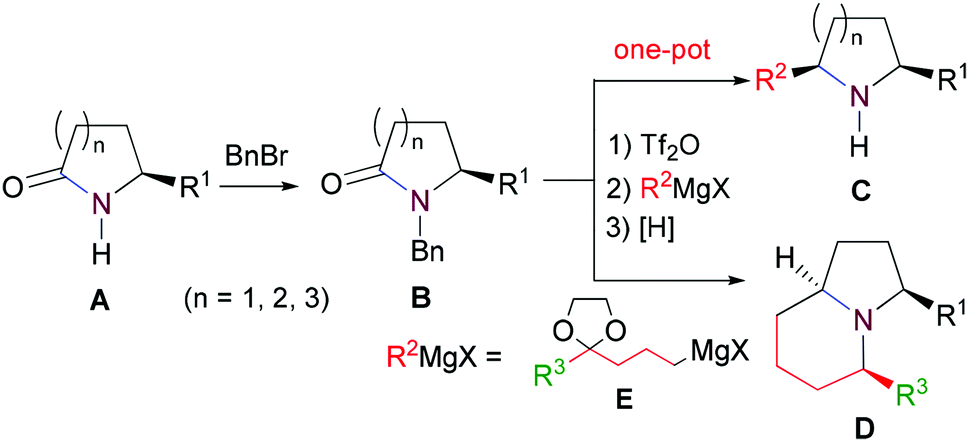 | ||
| Scheme 3 Synthetic plan. | ||
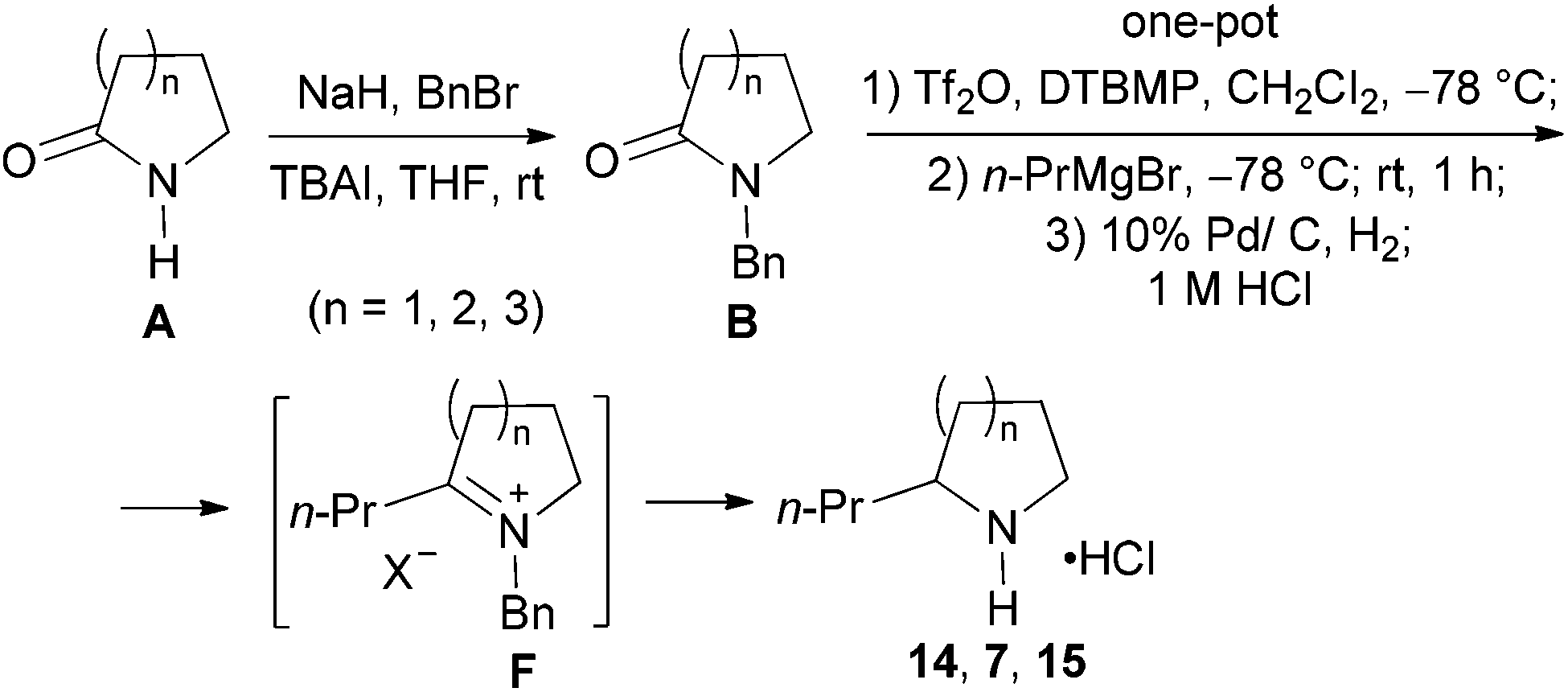
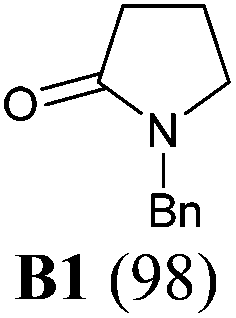
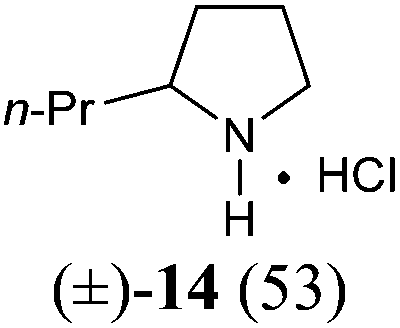
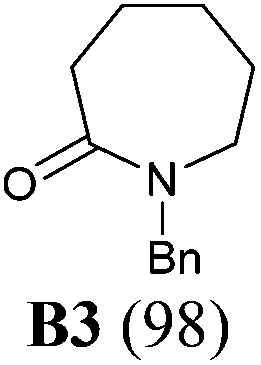
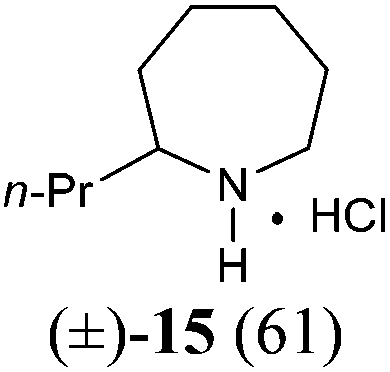
The investigation began with secondary lactams A1–A3, which were first benzylated (NaH, BnBr, n-Bu4NI)24 to give the corresponding N-benzyllactams B1–B3 in high yields (Table 1). For the subsequent reductive alkylation, the conditions that we established previously for tert-amides/lactams6c were adopted. Thus the triflic anhydride (Tf2O)/2,6-di-tert-butyl-4-methylpyridine (DTBMP) combination25 was selected as the amide-activating system, with Grignard reagents as alkylating agents, and catalytic hydrogenation6d employed for both reduction of an iminium intermediate and the debenzylation. Thus N-benzyl-γ-lactam B1 (1.0 equiv.) was successively treated with 1.2 equiv. of DTBMP, 1.2 equiv. of Tf2O (−78 °C, 45 min), and 1.0 equiv. of n-propylmagnesium bromide in tetrahydrofuran (−78 °C; rt, 1 h). The presumed iminium intermediate F1 (n = 1) was subjected to 10% Pd/C-catalyzed hydrogenation (H2, 1 atm, rt; 1 M HCl) to give the desired 2-n-propylpyrrolidine 14, isolated as a hydrochloride salt in 53% yield (Table 1, entry 1). Following the same procedure, except for the acidic conditions being used for the hydrogenation and debenzylation of B1, the one-pot reductive propylation–debenzylation of N-benzyl δ-lactam B2 gave (±)-coniine (7) in 63% yield (Table 1, entry 2). Similarly, one-pot reductive propylation–debenzylation of N-benzyl-ε-lactam B3 produced azepane 15 in 61% yield (Table 1, entry 3). We observed that with increasing ring size from five to seven, both hydrogenation and debenzylation reactions proceeded more readily in the absence of HCl.
Asymmetric synthesis of the alkaloid (2R,5S)-cis-2-butyl-5-propylpyrrolidine (8)
We next addressed the enantioselective synthesis of the ant venom alkaloid cis-2-butyl-5-propylpyrrolidine (8). In addition to the racemic synthesis by Jones et al. for its structural confirmation,19 Coldham and Schwan have reported the asymmetric synthesis of (2R,5S)-26a and (2S,5R)-enantiomer,26b respectively. However, since the values of optical rotation of both enantiomers are zero, the absolute configuration of this natural product remains unknown. To this end, we plan to synthesize the 2R,5S-enantiomer of 8. The requisite precursor lactam (R)-17 was prepared from lactam (R)-16,27 readily available from L-pyroglutamic acid,14j by benzylation (NaH, BnBr, THF, rt, 12 h, yield: 97%) (Scheme 4). Lactam (R)-17 (1.0 equiv.) was successively treated with 0.6 equiv. of DTBMP, 1.2 equiv. of Tf2O (−78 °C, 1 h; 0 °C, 30 min) in CH2Cl2, and 1.0 equiv. of n-propylmagnesium bromide in THF (−78 °C, 1 h). The resulting mixture was subjected to 10% Pd/C (200% wt)-catalyzed hydrogenation (H2, 1 atm, 12 h; 1 M HCl, 24 h) to produce (2R,5S)-cis-2-butyl-5-propylpyrrolidine (8) in 51% yield as an 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11 cis/trans-diastereomeric mixture. The physical and spectral data of our synthetic sample are in agreement with those reported for the cis-diastereomer.26a,28 The observed cis-stereoselection is consistent with that reported by Rapoport et al.29 The overall yield of 8 from secondary lactam 16 was 44%.
:11 cis/trans-diastereomeric mixture. The physical and spectral data of our synthetic sample are in agreement with those reported for the cis-diastereomer.26a,28 The observed cis-stereoselection is consistent with that reported by Rapoport et al.29 The overall yield of 8 from secondary lactam 16 was 44%.
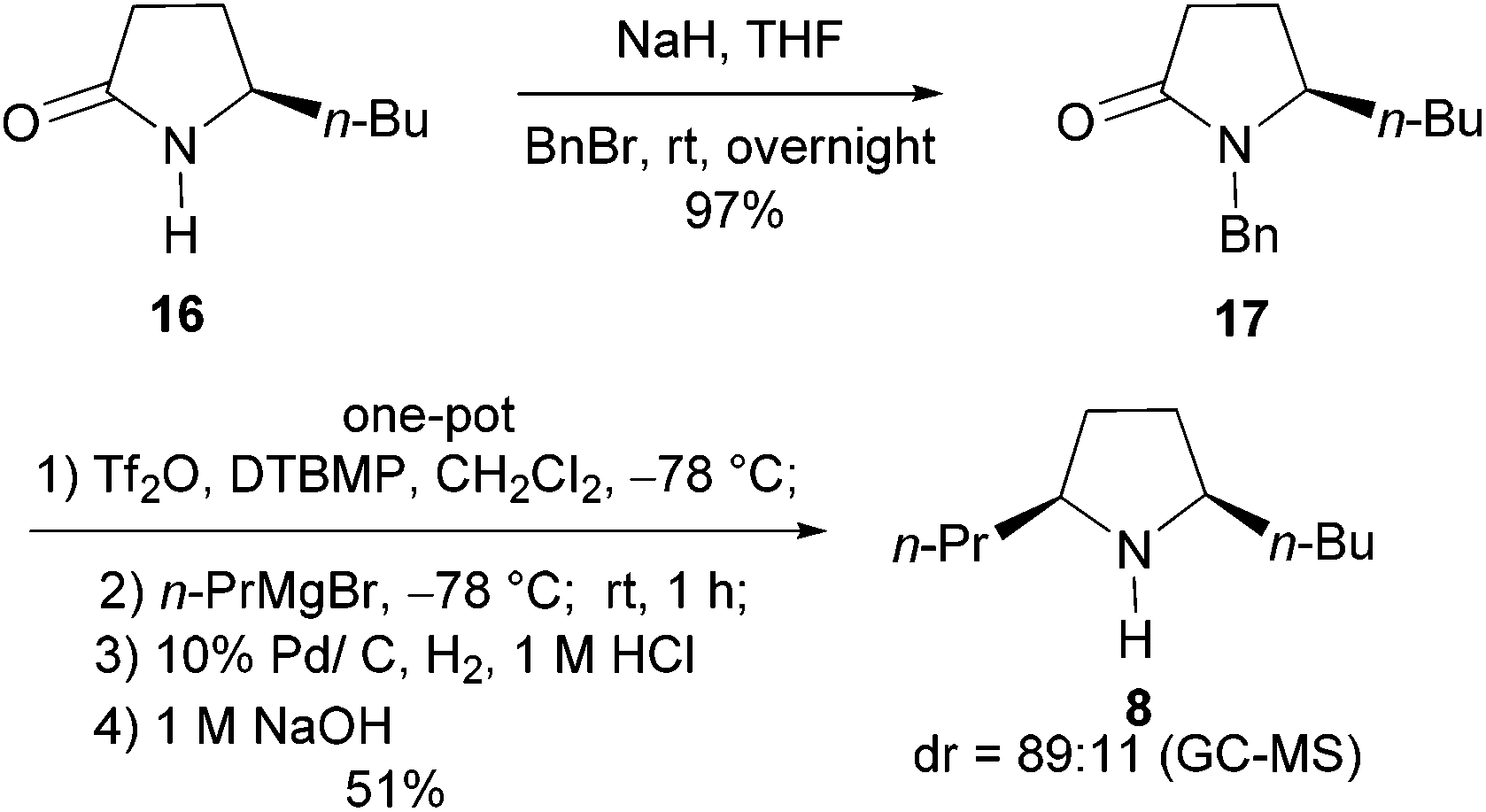 | ||
| Scheme 4 Two-step synthesis of the alkaloid (2R,5S)-cis-2-butyl-5-propylpyrrolidine (8) from lactam 16. | ||
Synthesis of (±)-5-methylindolizidine (21)
With the successful application of our newly developed reductive alkylation method in the aforementioned syntheses, the synthesis of indolizidine alkaloids was next undertaken. To test the feasibility of the strategy, 5-methylindolizidine 21, a lower homologue of alkaloids 167B (12) and 209D (13), was selected as our first target. The synthesis commenced with reductive alkylation of our existing N-benzylpyrrolidin-2-one (B1) (Scheme 5). Successive treatments of a CH2Cl2 solution of lactam B1 with DTBMP (0.6 equiv.), Tf2O (1.2 equiv.) (−78 °C, 1 h; 0 °C, 30 min), freshly prepared Grignard reagent 1830 (−78 °C, 1 h; rt, 1 h), and LiAlH4 (3.0 equiv.) produced the desired pyrrolidine 19. Deacetalization of compound 19 (4 M HCl, acetone, 50 °C, 2 h) gave keto-pyrrolidine 20 in 75% overall yield from lactam B1. Subjecting 20 to catalytic hydrogenolysis conditions (H2, 1 atm, 10% Pd/C; 1 M HCl, rt, 20 h) yielded the known 5-methylindolizidine (21),31 isolated as its hydrochloride salt in 70% yield (overall 52% from B1).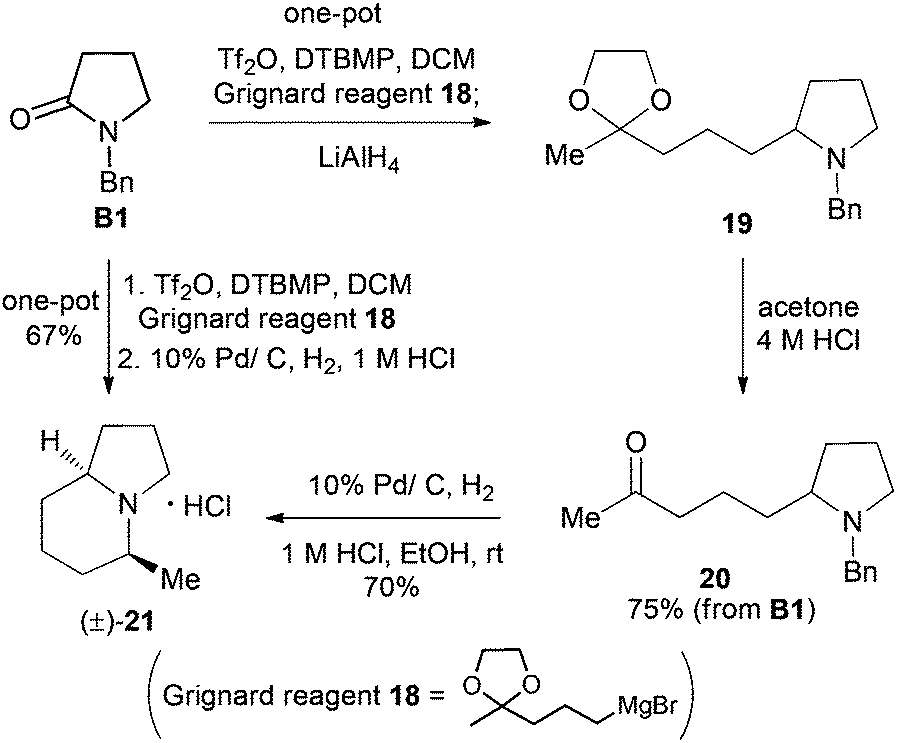 | ||
| Scheme 5 Three-pot and one-pot syntheses of indolizidine 21 from lactam B1. | ||
To further improve the efficiency of the current synthesis, a more ambitious one-pot transformation from lactam B1 to indolizidine 21 was attempted. Treatment of the reaction mixture of the Tf2O-activated lactam B1 and Grignard reagent 18 with 10% Pd/C under H2 (1 atm) for 12 h, followed by reaction in the presence of a 1 M HCl solution (2 equiv.) for 24 h gave, in one pot, the desired indolizidine 21 in 67% as a single diastereomer. The physical and spectral data of the synthetic product, as well as the observed cis-diastereoselection, are in agreement with those reported.22a,31 The overall yield from secondary lactam A1 was 65%.
Asymmetric synthesis of the alkaloid (+)-monomorine I (10)
Now we are in a position to attempt the synthesis of the ant venom alkaloid (+)-monomorine I (10)14d,g,h,32 from lactam (R)-17, available in one step from (R)-16 (Schemes 4 and 6). Following the conditions established for the one-pot conversion of lactam B1 to indolizidine 21, the reductive alkylation, deacetalization, and N-debenzylation of 17 proceeded smoothly. However, the final cyclization did not take place. This might be attributed to the acidic conditions which prevented the desired intramolecular substitution reaction from occurring. To address this problem, the reaction mixture was basified with NaHCO3 (8 equiv.), and hydrogenated under 1 atm of H2 for one day. To our delight, (+)-monomorine I (10) and its C-9 diastereomer were obtained in a remarkable yield of 60% from lactam 17. The diastereoselectivity was 86:14 in favour of (+)-monomorine I (10). Thus, (+)-monomorine I (10) was synthesized from lactam 16 in two steps with a 50% overall yield. The physical and spectral data of our synthetic product are in agreement with those reported {[α]20D +32.6 (c 0.4, hexane); lit.32m [α]24D +33.3 (c 0.39, hexane)}. The stereoselectivity in establishing the C-9 stereogenic center (9Z, dr = 86:14) is in agreement with that observed in the synthesis of pyrrolidine 8 (dr = 89:11), while formation of the 5Z stereochemistry in excellent diastereoselectivity is consistent with that observed for 5-methylindolizidine (21).
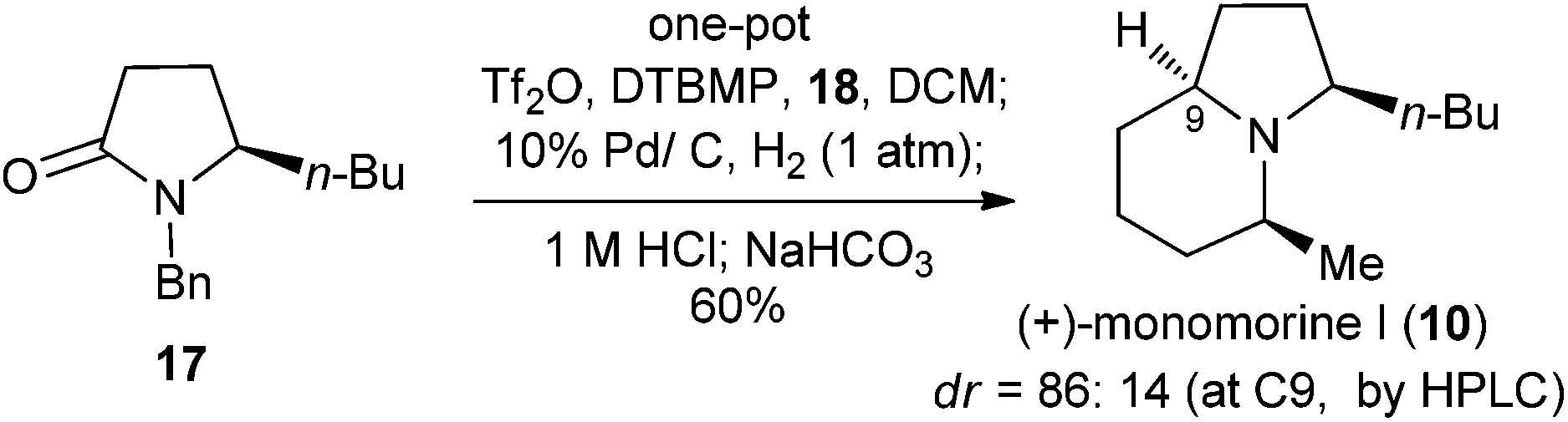 | ||
| Scheme 6 One-pot synthesis of (+)-monomorine I (10) from lactam 17. | ||
Conclusion
In summary, by combining the direct reductive alkylation of tertiary lactams, a method that we developed recently, with in situ N-debenzylation, a versatile two-step method for the efficient conversion of secondary lactams into N-α-substituted and N-α,α′-disubstituted heterocycles has been achieved. Using suitably functionalized Grignard reagents, fused piperidino bicyclic ring systems can be built, which constitutes a formal [4 + 2] annulation reaction. On the basis of this method, we synthesized several N-α-alkyl-heterocycles including (±)-coniine (7), the ant venom alkaloids (2R,5S)-cis-2-butyl-5-propylpyrrolidine (8), and (+)-monomorine I (10) in two steps starting from secondary lactams. The two-pot stereoselective synthesis of (+)-monomorine I (10) from the lactam 16 was achieved in an overall yield of 50%. Compared with the previous 5- and 10-step approaches14e,g,h for the same transformation, our two-step method is remarkably concise. Moreover, the method is versatile and flexible in terms of ring size and substituents, which opens an avenue for the concise synthesis of other mono- and bi-cyclic alkaloids and N-heterocyclic pharmaceuticals.Experimental section
General methods
1H NMR and 13C NMR spectra were recorded on a Bruker 400 (1H/400 MHz, 13C/100 MHz) spectrometer. Chemical shifts are expressed in parts per million (δ) relative to an internal standard of residual chloroform (7.26 ppm for 1H NMR and 77.0 ppm for 13C NMR). Data for 1H NMR are reported as chemical shift (multiplicity, coupling constant, number of protons). ESI-mass spectra were recorded on a Bruker Dalton ESquire 3000 plus LC-MS apparatus. GC-MS were recorded on a Shimadzu GCMS-QP2010 Plus. Optical rotations were measured with a Perkin-Elmer 341 automatic polarimeter or an Anton Paar MCP 500 polarimeter. Melting points were determined on a Büchi M560 Automatic Melting Point apparatus. Infrared spectra were recorded with a Nicolet Avatar 330 FT-IR spectrometer using the film or KBr pellet technique.Silica gel (300–400 mesh) was used for flash column chromatography. Trifluoromethanesulfonic anhydride (Tf2O) was distilled over phosphorous pentoxide and used within a week. THF was distilled over sodium benzophenone ketyl under N2. Dichloromethane was distilled over calcium hydride under N2. Grignard reagents were titrated33 before use.
General procedure for the N-benzylation of lactams A1–A3
To a cooled (0 °C) suspension of NaH (1.5 mmol, 60% in mineral oil) and n-Bu4NI (0.01 mmol) in THF (7 mL) was added dropwise a solution of lactam A1, A2, or A3 (1 mmol) in THF (3 mL). The mixture was stirred at 0 °C for 1 h. Then benzyl bromide (2 mmol) was added dropwise and the resulting mixture stirred at rt for 12 h. The reaction was quenched with water (1 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: EtOAc–hexane) to afford the known compounds B1, B2, or B3, respectively.1-Benzylpyrrolidin-2-one (B1)34
Yield: 98%. Colourless oil. IR (film) νmax: 2918, 2851, 1679, 1493, 1426, 1019, 735, 699 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.94–2.02 (m, 2H), 2.43 (t, J = 8.1 Hz 2H), 3.25 (t, J = 7.3 Hz, 2H), 4.44 (s, 2H), 7.17–7.40 (m, 5H) ppm; 13C NMR (100 MHz, CDCl3) δ 17.7, 30.9, 46.5 (2C), 127.5, 128.0, 128.6, 136.5, 174.9 ppm.1-Benzyl-piperidin-2-one (B2)35
Yield: 96%. Colourless oil. IR (film) νmax: 2941, 2865, 1638, 1493, 1449, 1349, 1257, 1171, 1070, 706 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.72–1.84 (m, 4H), 2.47 (t, J = 6.3 Hz, 2H), 3.20 (t, J = 5.8 Hz, 2H), 4.60 (s, 2H), 7.22–7.35 (m, 5H) ppm; 13C NMR (100 MHz, CDCl3) δ 21.3, 23.1, 32.4, 47.1, 50.0, 127.2, 128.0, 128.5, 137.3, 169.8 ppm.1-Benzyl-azepan-2-one (B3)36
Yield: 98%. White solid. Mp: 58–59 °C; IR (film) νmax: 2928, 2855, 1643, 1485, 1445, 1358, 1263, 1193, 1143, 1080, 968, 703, 507 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.44–1.52 (m, 2H), 1.67–1.76 (m, 4H), 2.57–2.64 (m, 2H), 3.26–3.31 (m, 2H), 4.59 (s, 2H), 7.23–7.34 (m, 5H) ppm; 13C NMR (100 MHz, CDCl3) δ 23.4, 28.0, 30.0, 37.1, 48.9, 51.0, 127.2, 128.1, 128.5, 137.9, 175.9 ppm.(±)-2-Propylpyrrolidine hydrochloride salt (14)37
To a cooled (−78 °C) solution of compound B1 (88 mg, 0.50 mmol) and 2,6-di-tert-butyl-4-methylpyridine (123 mg, 0.60 mmol) in CH2Cl2 (5 mL) was added dropwise Tf2O (101 μL, 0.60 mmol). The resulting mixture was stirred at −78 °C for 45 min and then at rt for 5 min. The mixture was re-cooled to −78 °C, and a freshly prepared n-propylmagnesium bromide (1.18 mL, 0.50 mmol, 0.42 M in THF) was added dropwise over a period of 1 h. The resulting mixture was warmed to rt and stirred for 1 h. The mixture was transferred to a Schlenk flask containing 10% Pd/C (176 mg, 200 wt%) and stirred at rt under 1 atm of H2 for 12 h [completion of the reduction of the iminium intermediate was monitored by MS (ESI) analysis]. Afterwards a solution of 1 M HCl (1.0 mL, 1.0 mmol) was added, and the resulting mixture was stirred for 24 h. The mixture was filtered through a pad of Celite, and the filtrate washed with a solution of 1 M NaOH (2.0 mL, 2.0 mmol), and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel [eluent: EtOAc–hexane = 4:1 containing 1.5% (v/v) of 25% ammonia aqueous solution, dried over anhydrous MgSO4]. After being concentrated under reduced pressure to about 1/3 of the original volume, 2 mL of 10% HCl in MeOH was added, and the resulting mixture concentrated under reduced pressure to afford 2-propylpyrrolidine hydrochloride salt 14 (39 mg, yield: 52%) as a wax. IR (film) νmax: 3433, 2960, 2753, 2536, 1616, 1459, 1255, 1208, 1010, 756 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.98 (t, J = 7.3 Hz, 3H), 1.41–1.55 (m, 2H), 1.65–1.76 (m, 2H), 1.91–2.21 (m, 4H), 3.25–3.44 (m, 2H), 3.44–3.56 (m, 1H), 9.28 (br s, 1H), 9.76 (br s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.7, 20.2, 23.5, 30.3, 34.1, 44.4, 60.2 ppm; HRMS (ESI) calcd for [C7H16ClN] (M − Cl−): 114.1283; found: 114.1285.
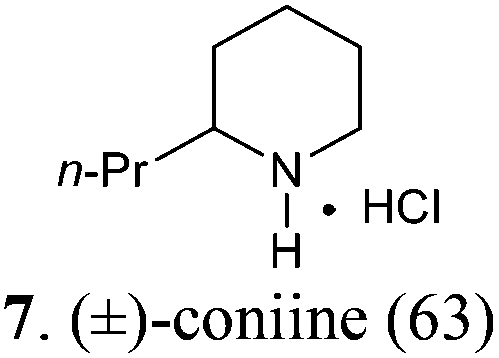
(±)-2-Propylpiperidine hydrochloride salt (7)
To a cooled (−78 °C) solution of compound B2 (95 mg, 0.50 mmol) and 2,6-di-tert-butyl-4-methylpyridine (123 mg, 0.60 mmol) in CH2Cl2 (10 mL) was added dropwise Tf2O (101 μL, 0.60 mmol). The resulting mixture was stirred at −78 °C for 45 min and then at rt for 5 min. After the reaction mixture was re-cooled to −78 °C, a freshly prepared n-propylmagnesium bromide (1.18 mL, 0.50 mmol, 0.42 M in THF) was added dropwise over a period of 1 h. Then the mixture was slowly warmed to rt and stirred for 1 h before being transferred to a Schlenk flask containing 10% Pd/C (190 mg, 200 wt%). The reaction mixture was stirred at rt under 1 atm of H2 for 48 h [completion of the reaction was monitored by MS (ESI) analysis]. Afterwards the mixture was filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel [eluent: EtOAc–hexane = 3:1, containing 1.5% (v/v) of 25% ammonia aqueous solution, dried over anhydrous MgSO4]. After concentration, acidification with HCl, and re-concentration, (±)-coniine hydrochloride salt (7·HCl)6c (52 mg, yield: 63%) was obtained as a white solid. Mp: 213–214 °C. IR (film) νmax: 3411, 2947, 2814, 2735, 1586, 1451, 1312, 1092, 1030, 801 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.90 (t, J = 7.3 Hz, 3H), 1.30–1.52 (m, 3H), 1.54–2.02 (m, 7H), 2.65–2.84 (m, 1H), 2.84–3.02 (m, 1H), 3.31–3.49 (m, 1H), 9.20 (br s, 1H), 9.43 (br s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.7, 18.5, 22.2, 22.4, 28.1, 35.3, 44.7, 57.1 ppm; MS (ESI): m/z 128.4 (M − Cl−, 100%).
(±)-2-Propylazepane hydrochloride salt (15)
Following the procedure described for the synthesis of compound 7, after the reductive alkylation of lactam B3 (102 mg, 0.5 mmol) with n-propylmagnesium bromide, 10% Pd/C (204 mg, 200 wt%) was added. The mixture was stirred under H2 (1 atm) for 30 h (the completion of the reaction was monitored by MS (ESI) analysis). The mixture was filtered and the filtrate concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel [eluent: EtOAc–hexane = 1:1, containing 1.5% (v/v) of 25% ammonia aqueous solution, dried over anhydrous MgSO4]. After concentration, acidification with HCl, and re-concentration, the azepane hydrochloride salt 1538 (54 mg, yield: 61%) was obtained as a white solid. Mp: 167–168 °C; IR (film) νmax: 3428, 2955, 2855, 2819, 2760, 1580, 1455, 1093 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.93 (t, J = 7.3 Hz, 3H), 1.38–1.52 (m, 2H), 1.54–1.81 (m, 5H), 1.81–2.98 (m, 5H), 3.09–3.23 (m, 2H), 3.23–3.32 (m, 1H), 9.20 (br s, 1H), 9.47 (br s, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 13.7, 19.1, 24.8, 24.9, 27.0, 30.5, 35.8, 45.0, 58.7 ppm; HRMS (ESI) calcd for [C9H20ClN] (M − Cl−) 142.1596; found: 142.1592.
(R)-1-Benzyl-5-butylpyrrolidin-2-one (17)
To a suspension of NaH (145 mg, 3.6 mmol, 60% in mineral oil) in THF (25 mL) at 0 °C was added dropwise a solution of compound 1627,14j (340 mg, 2.4 mmol) in THF (5 mL). The mixture was stirred at 0 °C for 1 h. Then benzyl bromide (825 mg, 4.8 mmol) was added dropwise to the mixture, and the reaction was kept at rt for 12 h. Afterwards the reaction was quenched with water (5 mL) and extracted with EtOAc (3 × 15 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: EtOAc–hexane = 1:1) to afford compound 17 (538 mg, yield: 97%) as a colourless oil. IR (film) νmax: 3028, 2858, 1702, 1678, 1432, 1425, 1225 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.87 (t, J = 7.1 Hz, 3H), 1.38–1.08 (m, 5H), 1.62–1.74 (m, 2H), 2.03–2.14 (m, 1H), 2.33–2.53 (m, 2H), 3.37–3.46 (m, 1H), 3.96 (d, J = 15.0 Hz, 1H), 4.98 (d, J = 15.0 Hz, 1H), 7.35–7.20 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 14.0, 22.6, 23.9, 26.5, 30.3, 32.5, 44.0, 55.9, 127.4, 128.0, 128.6, 136.9, 175.1; HRMS (ESI) calcd for [C15H21NO + Na+]: 254.1515; found: 254.1517.
(2R,5S)-2-Butyl-5-propylpyrrolidine (8)
To a cooled (−78 °C) solution of compound 17 (105 mg, 0.45 mmol) and 2,6-di-tert-butyl-4-methylpyridine (55 mg, 0.27 mmol) in CH2Cl2 (5 mL) was added dropwise Tf2O (90 μL, 0.55 mmol). The mixture was stirred at −78 °C for 1 h and then at 0 °C for 30 min. After the reaction mixture was re-cooled to −78 °C, a freshly prepared propylmagnesium bromide (1.26 mL, 0.45 mmol, 0.36 M in THF) was added dropwise over a 1 h period. Then the mixture was slowly warmed to rt and stirred for 1 h before being transferred to a suspension of 10% Pd/C (210 mg) in CH2Cl2. The reaction was kept at rt under 1 atm of H2 for 12 h. A solution of 1 M HCl (0.9 mL, 0.9 mmol) was then added and the mixture stirred for 24 h. The mixture was filtered through a pad of Celite, and the filtrate was washed with 1 M NaOH (1.8 mL, 1.8 mmol) and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel [eluent: EtOAc–hexane = 3:1, containing 1.5% (v/v) of 25% ammonia aqueous solution, dried over anhydrous MgSO4] to give (2R,5S)-2-butyl-5-propylpyrrolidine (8) (39 mg, yield: 51%) as a cis/trans-diastereomeric mixture in a 89:11 ratio. A sample of pure cis-diastereomer was obtained by column flash chromatographic separation. (2R,5S)-2-Butyl-5-propylpyrrolidine (8): colourless oil; [α]20D +0.6 (c 0.19, CHCl3); lit.26a [α]21D 0.0 (c 0.6, CHCl3). IR (film) νmax: 2955, 2926, 2852, 1459, 1260, 1095, 1024 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.86–0.94 (m, 6H), 1.25–1.41 (m, 10H), 1.47–1.58 (m, 2H), 1.79–1.90 (m, 2H), 2.05 (br s, 1H), 2.88–3.09 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 14.0 (CH3), 14.2 (CH3), 20.6 (CH2), 22.9 (CH2), 29.7 (CH2), 31.2 (CH2), 36.3 (CH2), 38.8 (CH2), 59.2 (CH), 59.4 (CH); HRMS (ESI) calcd for [C11H23N + H+]: 170.1909; found: 170.1909.
1-Benzyl-2-(3-(2-methyl-1,3-dioxolan-2-yl)propyl)pyrrolidine (19)
To a cooled (−78 °C) solution of compound B1 (100 mg, 0.57 mmol) and 2,6-di-tert-butyl-4-methylpyridine (70 mg, 0.34 mmol) in CH2Cl2 (6 mL) was added dropwise Tf2O (100 μL, 0.68 mmol). The mixture was stirred at −78 °C for 1 h and then at 0 °C for 30 min. After the reaction mixture was re-cooled to −78 °C, a freshly prepared Grignard reagent 18 (1.5 mL, 0.57 mmol, 0.38 M in THF) was added dropwise over a period of 1 h. The mixture was then allowed to warm slowly to rt and stirred for another 1 h. After the reaction mixture was re-cooled to −78 °C, LiAlH4 (65 mg, 1.71 mmol) was added and the suspension was stirred for 1 h. The reaction was quenched with water and an aqueous solution of 15% NaOH, and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (basic Al2O3, eluent: EtOAc–hexane = 50:1) to give crude acetal 19 as a colourless oil. The crude product was directly used in the next step without further purification. IR (film) vmax: 3061, 3025, 2959, 2874, 2785, 1452, 1375, 1260, 1067, 947 cm−1. 1H NMR (500 MHz, CDCl3) δ 1.31 (s, 3H), 1.34–1.42 (m, 2H), 1.45–1.55 (m, 2H), 1.61–1.75 (m, 5H), 1.90–1.99 (m, 1H), 2.11 (dd, J = 9.3, 17.5 Hz, 1H), 2.31–2.39 (m, 1H), 2.87–2.96 (m, 1H), 3.17 (d, J = 12.9 Hz, 1H), 3.88–3.98 (m, 4H), 4.03 (d, J = 12.9 Hz, 1H), 7.17–7.37 (m, 5H). 13C NMR (125 MHz, CDCl3) δ 21.0 (CH2), 21.9 (CH2), 23.8 (CH3), 30.4 (CH2), 34.3 (CH2), 39.5 (CH2), 54.1 (CH2), 58.6 (CH2), 64.5 (CH), 64.61 (CH2), 64.62 (CH2), 110.1 (C), 126.8 (CH), 128.2 (CH), 129.0 (CH), 139.4 (C); HRMS (ESI) calcd for [C18H27NO2]+ (M + H+): 290.2115; found: 290.2117.
5-(1-Benzylpyrrolidin-2-yl)pentan-2-one (20)
To a solution of crude acetal 19 in acetone (5 mL) at 0 °C was added 4 M HCl (2.9 mL), and the mixture was stirred at 50 °C for 2 h. The solvent was removed under reduced pressure. The residue was diluted with CH2Cl2, then quenched with a saturated aqueous Na2CO3 (5 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (eluent: CH2Cl2–MeOH = 15:1) to give ketone 20 (105 mg, yield: 75% from lactam B1) as a colourless oil. IR (film) vmax 3026, 2922, 2852, 2786, 1715, 1452, 1356, 1158, 1028 cm−1. 1H NMR (500 MHz, CDCl3) δ 1.31–1.38 (m, 1H), 1.50–1.62 (m, 2H), 1.64–1.78 (m, 4H), 1.93–2.02 (m, 1H), 2.16 (s, 3H), 2.15 (dd, J = 9.2, 17.5 Hz, 1H), 2.35–2.42 (m, 1H), 2.47 (t, J = 7.2 Hz, 1H), 2.97–2.91 (m, 1H), 3.22 (d, J = 12.9 Hz, 1H), 4.03 (d, J = 12.9 Hz, 1H), 7.21–7.43 (m, 5H). 13C NMR (125 MHz, CDCl3) δ 20.6 (CH2), 22.0 (CH2), 29.9 (CH3), 30.3 (CH2), 33.5 (CH2), 44.0 (CH2), 54.1 (CH2), 58.6 (CH2), 64.1 (CH), 126.8 (CH), 128.2 (CH), 129.0 (CH), 139.3 (C), 208.9 (C); HRMS (ESI) calcd for [C16H23NO]+ (M + H+): 246.1852; found: 246.1856.
5-Methyloctahydroindolizine hydrochloride salt (21)
Method A: To a suspension of 10% Pd/C (10 mg) in EtOH (1 mL) was added a solution of compound 20 (13 mg, 0.05 mmol) in EtOH (1 mL) and 1 M HCl (0.08 mL, 0.08 mmol) successively. The reaction mixture was stirred at rt under 1 atm of H2 for 20 h. Afterwards the mixture was filtered through a pad of Celite, the residue washed with EtOH, and the solvent removed under reduced pressure. The residue was purified by column chromatography on silica gel (eluent: CH2Cl2–MeOH = 15:1) to give compound 21 (5 mg, yield: 70%, colourless wax) as a single diastereomer.
Method B: To a cooled (−78 °C) solution of compound B1 (170 mg, 0.97 mmol) and 2,6-di-tert-butyl-4-methylpyridine (120 mg, 0.58 mmol) in CH2Cl2 (10 mL) was added dropwise Tf2O (190 μL, 1.17 mmol). The mixture was stirred at −78 °C for 1 h and then at 0 °C for 30 min. The reaction mixture was re-cooled to −78 °C, and a freshly prepared Grignard reagent 18 (3.0 mL, 1.26 mmol, 0.42 M in THF) was added dropwise over a 1 h period. The resulting mixture was warmed to rt and stirred for 1 h before being transferred to a suspension of 10% Pd/C (170 mg) in CH2Cl2. The mixture was stirred at rt under 1 atm of H2 for 12 h. A solution of 1 M HCl (1.95 mL, 1.95 mmol) was then added and the mixture stirred for 24 h. The mixture was filtered through a pad of Celite and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel (eluent: CH2Cl2–MeOH = 15:1) to give compound 2122a,31 (90 mg, yield: 67%) as a single diastereomer. Colourless wax; IR (film) νmax: 2919, 2848, 2613, 2580, 2536, 1450, 1389, 1032 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.41–1.50 (m, 1H), 1.53 (d, J = 6.5 Hz, 3H), 1.84 (dm, J = 14.7 Hz, 1H), 1.92–2.07 (m, 3H), 2.10–2.31 (m, 5H), 2.62–2.74 (m, 1H), 2.75–2.91 (m, 2H), 3.86–3.92 (m, 1H), 11.73 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 18.8 (CH3), 20.0 (CH2), 23.7 (CH2), 27.2 (CH2), 28.9 (CH2), 31.0 (CH2), 51.9 (CH2), 61.9 (CH), 68.6 (CH); HRMS (ESI) calcd for [C9H18ClN] (M − Cl−): 140.1434; found: 140.1436.
(3R,5S,8aS)-(+)-3-Butyl-5-methyloctahydroindolizine [(+)-monomorine I, 10]
To a cooled (−78 °C) solution of compound 17 (60 mg, 0.26 mmol) and 2,6-di-tert-butyl-4-methylpyridine (32 mg, 0.16 mmol) in CH2Cl2 (10 mL) was added dropwise Tf2O (50 μL, 0.31 mmol). The mixture was stirred at −78 °C for 1 h and then at 0 °C for 30 min. After the reaction mixture was re-cooled to −78 °C, a freshly prepared Grignard reagent 18 (0.97 mL, 0.34 mmol, 0.35 M in THF) was added dropwise over a period of 1 h. Then the mixture was slowly warmed to rt and stirred for 1 h before being transferred to a suspension of 10% Pd/C (60 mg) in CH2Cl2. The reaction mixture was stirred at rt under 1 atm of H2 for 12 h. A solution of 1 M HCl (0.52 mL, 0.52 mmol) was then added and the mixture stirred for 24 h. Afterwards, NaHCO3 (218 mg, 2.6 mmol) was added and the mixture stirred for 24 h. Upon filtration through a pad of Celite and evaporation of the solvent under reduced pressure, the residue was purified by column chromatography (Al2O3, basic, eluent: Et2O–hexane = 1:9) to give compound 10 (30.5 mg, yield: 60%) as a C-9 diastereomeric mixture in a ratio of 86:14 (determined by HPLC). The major diastereomer was isolated (27 mg, yield: 52%) after further separation by column chromatography (basic Al2O3, eluent: Et2O–hexane = 1:9) to give a colourless oil. [α]20D +32.6 (c 0.4, hexane) {lit.32m [α]24D +33.3 (c 0.39, hexane)}; IR (film) νmax: 2955, 2926, 2855, 1693, 1461, 1378 cm−1; 1H NMR (400 MHz, CDCl3) δ 0.88 (t, J = 7.1 Hz, 3H), 1.12 (d, J = 6.4 Hz, 3H), 1.17–1.35 (m, 8H), 1.36–1.46 (m, 2H), 1.49–1.55 (m, 1H), 1.62–1.75 (m, 4H), 1.77–1.85 (m, 1H), 2.01–2.10 (m, 1H), 2.15–2.25 (m, 1H), 2.42–2.50 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 14.2 (CH3), 22.88 (CH2), 22.91 (CH3), 24.9 (CH2), 29.4 (CH2), 29.8 (CH2), 30.4 (CH2), 30.9 (CH2), 35.9 (CH2), 39.8 (CH2), 60.2 (CH), 62.9 (CH), 67.2 (CH); HRMS (ESI) calcd for [C13H25N]+ (M + H+): 196.2060; found: 196.2068.
Acknowledgements
The authors are grateful to the National Basic Research Program (973 Program) of the MOST of China (grant no. 2010CB833200), the Natural Science Foundation of China (NSFC, grant no. 21332007; NFFTBS, grant no. J1210014), and the Program for Changjiang Scholars and Innovative Research Team in University of the MOE of China for financial support.Notes and references
- For a recent leading review, see: C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197 CrossRef CAS PubMed.
- For selected recent reviews on tandem reactions, see: (a) K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551 RSC; (b) J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001 CrossRef CAS PubMed; (c) H.-C. Guo and J.-A. Ma, Angew. Chem., Int. Ed., 2006, 45, 354 CrossRef CAS PubMed.
- For selected recent reviews on multicomponent reactions, see: (a) J.-P. Zhu and H. Bienaymé, Multicomponent Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) G. Guillena, D. J. Ramón and M. Yus, Tetrahedron: Asymmetry, 2007, 18, 693 CrossRef CAS; (c) B. Ganem, Acc. Chem. Res., 2009, 42, 463 CrossRef CAS PubMed.
- For selected recent reviews on domino reactions, see: (a) L. F. Tietze, G. Brasche and K. Gerike, Domino Reactions in Organic synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed; (b) H. Pellissier, Tetrahedron, 2006, 62, 1619 and 2143 Search PubMed; (c) A. Grossman and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314 CrossRef PubMed.
- For a recent review on the developments in amide synthesis, see: R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453 CrossRef CAS.
- For reductive bis- or mono-alkylation of amides/lactams, see: (a) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037 CrossRef CAS PubMed; (b) K.-J. Xiao, J.-M. Luo, X.-E. Xia, Y. Wang and P.-Q. Huang, Chem.–Eur. J., 2013, 19, 13075 CrossRef CAS PubMed; (c) K.-J. Xiao, Y. Wang, K.-Y. Ye and P.-Q. Huang, Chem.–Eur. J., 2010, 16, 12792 CrossRef CAS PubMed; (d) K.-J. Xiao, Y. Wang, Y.-H. Huang, X.-G. Wang, J.-C. Liao and P.-Q. Huang, J. Org. Chem., 2013, 78, 8305 CrossRef CAS PubMed; (e) S.-Y. Huang, Z. Chang, S.-C. Tuo, L.-H. Gao, A.-E. Wang and P.-Q. Huang, Chem. Commun., 2013, 49, 7088 RSC; (f) Y. Oda, T. Sato and N. Chida, Org. Lett., 2012, 14, 950 CrossRef CAS PubMed ; For reductive phosphination of amides, see: ; (g) Y.-Z. Gao, Z.-B. Huang, R.-Q. Zhuang, J. Xu, P.-B. Zhang, G. Tang and Y.-F. Zhao, Org. Lett., 2013, 15, 4214 CrossRef CAS PubMed ; For reductive nucleophilic addition of N-alkoxyamides, see: ; (h) K. Shirokane, Y. Kurosaki, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2010, 49, 6369 CrossRef CAS PubMed; (i) G. Vincent, R. Guillot and C. Kouklovsky, Angew. Chem., Int. Ed., 2011, 50, 1350 CrossRef CAS PubMed; (j) Y. Oda, T. Sato and N. Chida, Org. Lett., 2012, 14, 950 CrossRef CAS PubMed; (k) Y. Yanagita, H. Nakamura, K. Shirokane, Y. Kurozaki, T. Sato and N. Chida, Chem.–Eur. J., 2013, 19, 678 CrossRef CAS PubMed; (l) M. Jäkel, J. Qu, T. Schnitzer and G. Helmchen, Chem.–Eur. J., 2013, 19, 16746 CrossRef PubMed; (m) K. Shirokane, T. Wada, M. Yoritate, R. Minamikawa, N. Takayama, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2014, 53, 512 CrossRef CAS PubMed ; for a highlight, see: ; (n) V. Pace and W. Holzer, Aust. J. Chem., 2013, 66, 507 CAS.
- (a) K.-J. Xiao, A.-E. Wang, Y.-H. Huang and P.-Q. Huang, Asian J. Org. Chem., 2012, 1, 130 CrossRef CAS; (b) K.-J. Xiao, A.-E. Wang, Y.-H. Huang and P.-Q. Huang, Acta Chim. Sin., 2012, 70, 1917 CrossRef CAS; (c) K.-J. Xiao, A.-E. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2012, 51, 8314 CrossRef CAS PubMed ; For Tf2O-activated reduction of tertiary and secondary amides, see: ; (d) S.-H. Xiang, J. Xu, H.-Q. Yuan and P.-Q. Huang, Synlett, 2010, 1829 CAS.
- For direct reductive cyanation of secondary γ-lactams, see: (a) Q. Xia and B. Ganem, Org. Lett., 2001, 3, 485 CrossRef CAS PubMed; (b) Q. Xia and B. Ganem, Tetrahedron Lett., 2002, 43, 1597 CrossRef CAS.
- For direct reductive diallylation of secondary lactams, see: (a) Y. N. Bubnov, F. V. Pastukhov, I. V. Yampolsky and A. V. Ignatenko, Eur. J. Org. Chem., 2000, 1503 CrossRef CAS; (b) Z.-F. Li and Y.-M. Zhang, Tetrahedron, 2002, 58, 5301 CrossRef CAS.
- (a) W. Oppolzer, C. Fehr and J. Wameke, Helv. Chim. Acta, 1977, 60, 48 CrossRef CAS PubMed ; for the application of Oppolzer's method to the total synthesis of pumiliotoxin C and its diastereomers, see: ; (b) K. Paulvannan and J. R. Stille, Tetrahedron Lett., 1993, 34, 6673 CrossRef CAS; (c) A. Padwa, T. M. Heidelbaugh and J. T. Kuethe, J. Org. Chem., 2000, 65, 2368 CrossRef CAS PubMed; (d) M. Saeki and M. Toyota, Tetrahedron Lett., 2010, 51, 4620 CrossRef CAS; (e) G. Mehta and M. Praveen, J. Org. Chem., 1995, 60, 279 CrossRef CAS; (f) S.-I. Murahashi, S. Sasao, E. Saito and T. Naota, Tetrahedron, 1993, 49, 8805 CrossRef CAS; (g) M. Toyota, T. Asoh, M. Matsuura and K. Fukumoto, J. Org. Chem., 1996, 61, 8687 CrossRef CAS.
- L. E. Overman, D. Lesuisse and M. Hashimoto, J. Am. Chem. Soc., 1983, 105, 5373 CrossRef CAS.
- Y.-G. Suh, S.-A. Kim, J.-K. Jung, D.-Y. Shin, K.-H. Min, B.-A. Koo and H.-S. Kim, Angew. Chem., Int. Ed., 1999, 38, 3545 CrossRef CAS.
- F. A. Davis, P. K. Mohanty, D. M. Burns and Y. W. Andemichael, Org. Lett., 2000, 2, 3901 CrossRef CAS PubMed.
- For reviews on the synthetic application of pyroglutamic acid, see: (a) S. K. Panday, J. Prasad and D. K. Dikshit, Tetrahedron: Asymmetry, 2009, 20, 1581 CrossRef CAS; (b) C. Nájera and M. Yus, Tetrahedron: Asymmetry, 1999, 10, 2245 CrossRef ; for selective examples, see: ; (c) N. Toyooka, D. Zhou, H. Nemoto, Y. Tezuka, S. Kadota, N. R. Andriamaharavo, H. M. Garraffo, T. F. Spande and J. W. Daly, J. Org. Chem., 2009, 74, 6784 CrossRef CAS PubMed; (d) G. Lesma, A. Colombo, A. Sacchetti and A. Silvani, J. Org. Chem., 2009, 74, 590 CrossRef CAS PubMed; (e) N. Toyooka, D. Zhou and H. Nemoto, J. Org. Chem., 2008, 73, 4575 CrossRef CAS PubMed; (f) N. Toyooka, D. Zhou, H. Nemoto, Y. Tezuka, S. Kadota, T. H. Jones, H. M. Garraffo, T. F. Spande and J. W. Daly, Synlett, 2008, 1894 CrossRef CAS; (g) E. Conchon, Y. Gelas-Mialhe and R. Remuson, Tetrahedron: Asymmetry, 2006, 17, 1253 CrossRef CAS; (h) C. Saliou, A. Fleurant, J. P. Célérier and G. Lhommet, Tetrahedron Lett., 1991, 32, 3365 CrossRef CAS ; see also: ; (i) G. Kim and E.-j. Lee, Tetrahedron: Asymmetry, 2001, 12, 2073 CrossRef CAS; (j) R. A. Pilli, C. L. Dias and A. O. Maldaner, J. Org. Chem., 1995, 60, 717 CrossRef CAS.
- For reviews on the chemistry of pyrrolidine, piperidine and indolizidine alkaloids, see: (a) J. P. Michael, Nat. Prod. Rep., 1999, 16, 675 RSC; (b) J. P. Michael, Nat. Prod. Rep., 2007, 24, 191 RSC; (c) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC.
- S. T. Lee, B. T. Green, K. D. Welch, J. A. Pfister and K. E. Panter, Chem. Res. Toxicol., 2008, 21, 2061 CrossRef CAS PubMed.
- For two reviews, see: (a) D. J. Robins, Five-membered monoheterocyclic compounds alkaloids (continued): Pyrrolidine alkaloids, in Second Supplements to the 2nd Edition of Rodd's Chemistry of Carbon Compounds, 1991, ch. 7, vol. IV, pp. 1–19 Search PubMed; (b) A. Numata and T. Ibuka, Alkaloids from Ants and Other Insects, in The Alkaloids: Chemistry and Pharmacology, 1987, ch. 6, vol. 31, pp. 193–315 Search PubMed.
- For recent reviews, see: (a) R. A. Saporito, T. F. Spande, H. M. Garraffo and M. A. Donnelly, Heterocycles, 2009, 79, 277 CrossRef CAS; (b) J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556 CrossRef CAS PubMed; (c) J. W. Daly, J. Med. Chem., 2003, 46, 445 CrossRef CAS PubMed; (d) M. Hesse, Alkaloids. Nature's Curse or Blessing, Verlag Helvetica Chimica Acta, Zürich, Switzerland, and Wiley-VCHWeinheim, Germany, 2002 Search PubMed.
- T. H. Jones, H. L. Voegtle, H. M. Miras, R. G. Weatherford, T. F. Spande, H. M. Garraffo, J. W. Daly, D. W. Davidson and R. R. Snelling, J. Nat. Prod., 2007, 70, 160 CrossRef CAS PubMed.
- F. J. Ritter, I. E. M. Rotgans, E. Tulman, P. E. Verwiel and F. Stein, Experientia, 1973, 29, 530 CrossRef CAS.
- For the isolation of (5Z,9Z)-223AB (9), see: H. M. Garraffo, T. F. Spande, J. W. Daly, A. Baldessari and E. G. Gros, J. Nat. Prod., 1993, 56, 357 CrossRef CAS.
- For the first enantioselective total syntheses of alkaloids 167B and 209D, see: (a) R. P. Polniaszek and S. E. Belmont, J. Org. Chem., 1990, 55, 4688 CrossRef CAS ; For the synthesis and bioactivity of 209D and its diastereomers, see: ; (b) H. Takahata, M. Kubota, K. Ihara, N. Okamoto, T. Momose, N. Azer, A. T. Eldefrawi and M. E. Eldefrawi, Tetrahedron: Asymmetry, 1998, 9, 3289 CrossRef CAS.
- (a) H.-K. Zhang, X. Li, H. Huang and P.-Q. Huang, Scientia Sinica Chim., 2011, 41, 732 ( Science China-Chem. , 2011 , 54 , 737 ) Search PubMed; (b) G.-J. Lin and P.-Q. Huang, Org. Biomol. Chem., 2009, 7, 4491 RSC; (c) X. Zheng, P.-Q. Huang, Y.-P. Ruan, A. W. M. Lee and W. H. Chan, Heterocycles, 2001, 55, 1505 CrossRef CAS; (d) X. Zheng, P.-Q. Huang, Y.-P. Ruan, A. W. M. Lee and W. H. Chan, Nat. Prod. Lett., 2002, 16, 53 CrossRef CAS PubMed.
- S. Czernecki, C. Georgoulis, C. Provelenghiou and G. Fusey, Tetrahedron Lett., 1976, 17, 3535 CrossRef.
- (a) P. J. Stang and T. Warren, Synthesis, 1980, 283 CrossRef CAS ; for a recent example, see: ; (b) M. Claire, V. Viviana and M. Nuno, Angew. Chem., Int. Ed., 2010, 49, 1583 CrossRef PubMed; (c) V. Valerio, D. Petkova, C. Madelaine and N. Maulide, Chem.–Eur. J., 2013, 19, 2606 CrossRef CAS PubMed.
- (a) I. Coldham and D. Leonori, J. Org. Chem., 2010, 75, 4069 CrossRef CAS PubMed; (b) S. C. Scöderman and A. L. Schwan, Org. Lett., 2013, 15, 4434 CrossRef PubMed.
- This lactam is commercially available from AKos Consulting and Solutions Deutschland GmbH, Germany, and also readily available from L-pyroglutamic acid, cf.: ref. 14j.
- The 1H NMR data of our product match those recorded by Coldham et al. for cis-diastereomer,26a while the 13C NMR data matched those of Coldham's trans-diastereomer. Rapoport et al. noted that for 2,5-dialkylpyrrolidines, in the 1H NMR, the proton at C-5 of the cis-isomer appears at higher field (δH-5 2.89–3.07 for our cis-isomer; 2.88–3.09 for Coldham's cis-isomer) than that of the trans-isomer (δH-5 3.47–3.65 for Coldham's trans-isomer).29 These led us to assume that Coldham et al. took trans-isomer's 13C NMR data for the cis-isomer.
- K. Shiosaki and H. Rapoport, J. Org. Chem., 1985, 50, 1229 CrossRef CAS.
- B. Tullio and F. Alberto, Gazz. Chim. Ital., 1953, 83, 1037 Search PubMed.
- (a) S. Gracia, R. Jerpan, S. Pellet-Rostaing, F. Popowycz and M. Lemaire, Tetrahedron Lett., 2010, 51, 6290 CrossRef CAS; (b) K. A. Tehrani, M. D'hooghe and N. De Kimpe, Tetrahedron, 2003, 59, 3099 CrossRef CAS.
- For the seminal first stereoselective synthesis, see: (a) R. V. Stevens and A. W. M. Lee, J. Chem. Soc., Chem. Commun., 1982, 102 RSC ; For selected enantioselective syntheses of the natural (+)-enantiomer, see: ; (b) C. R. Reddy, B. Latha and N. N. Rao, Tetrahedron, 2012, 68, 145 CrossRef CAS; (c) F. A. Davis, J. Zhang and Y. Wu, Tetrahedron Lett., 2011, 52, 2054 CrossRef CAS; (d) C. R. Reddy and B. Latha, Tetrahedron: Asymmetry, 2011, 22, 1849 CrossRef CAS; (e) M. C. Paderes and S. R. Chemler, Org. Lett., 2009, 11, 1915 CrossRef CAS PubMed; (f) Y.-G. Wang, T. Kumano, T. Kano and K. Maruoka, Org. Lett., 2009, 11, 2027 CrossRef CAS PubMed; (g) Ref. 14d; ; (h) Ref. 14e; ; (i) M. B. Berry, D. Craig, P. S. Jones and G. J. Rowlands, Beilstein J. Org. Chem., 2007, 3, 39 CrossRef PubMed; (j) S. Randl and S. Blechert, J. Org. Chem., 2003, 68, 8879 CrossRef CAS PubMed; (k) G. Kim, S.-D. Jung, E.-J. Lee and N. Kim, J. Org. Chem., 2003, 68, 5395 CrossRef CAS PubMed; (l) M. Amat, N. H. Llor, E. C. Jose and J. Bosch, J. Org. Chem., 2003, 68, 1919 CrossRef CAS PubMed; (m) M. Ito and C. Kibayashi, Tetrahedron, 1991, 47, 9329 CrossRef CAS and references cited therein. For unnatural (−)-enantiomer, see: ; (n) N. Ortega, D.-T. D. Tang, S. Urban, D. Zhao and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 9500 CrossRef CAS PubMed; (o) R. W. Bates and P. Song, Synthesis, 2009, 655 CrossRef CAS; (p) L. S. Pattenden, H. Adams, S. A. Smith and J. P. A. Harrity, Tetrahedron, 2008, 64, 2951 CrossRef CAS; (q) S. H. Zhang, L. Xu, L. Miao, H. Shu and M. L. Trudell, J. Org. Chem., 2007, 72, 3133 CrossRef CAS PubMed; (r) J. Royer and H. P. Husson, J. Org. Chem., 1985, 50, 670 CrossRef CAS (the first enantioselective synthesis).
- B. E. Love and E. G. Jones, J. Org. Chem., 1999, 64, 3775 CrossRef.
- (a) J. Mun and M. B. Smith, Synth. Commun., 2007, 37, 813 CrossRef CAS; (b) A. M. R. Smith, D. Billen and K. K. Hii, Chem. Commun., 2009, 3925 RSC.
- (a) B. Li, J. R. Harjani, N. S. Cormier, H. Madarati, J. Atkinson, G. Cosa and D. A. Pratt, J. Am. Chem. Soc., 2013, 135, 1394 CrossRef CAS PubMed; (b) K. Thai, W. Li, T. Dudding, F. Bilodeau and M. Gravel, Org. Lett., 2010, 12, 5708 CrossRef CAS PubMed.
- S. Hanada, E. Tsutsumi, Y. Motoyama and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 15032 CrossRef CAS PubMed.
- A. R. Katritzky, X.-L. Cui, B.-Z. Yang and P. J. Steel, J. Org. Chem., 1999, 64, 1979 CrossRef CAS PubMed [(2R)-2-propylpyrrolidine hydrochloride salt].
- G. Ehrhart and G. Seidl, Chem. Ber., 1964, 97, 1994 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of compounds 7, 8, 10, 14–16, and 19–21. See DOI: 10.1039/c3qo00065f |
| This journal is © the Partner Organisations 2014 |