Intermolecular bromoesterification of conjugated enynes: an efficient synthesis of bromoallenes†
Hao-Yuan
Wang
a,
Wei
Zhang
a,
Casi M.
Schienebeck
a,
Scott R.
Bennett
a and
Weiping
Tang
*ab
aSchool of Pharmacy, University of Wisconsin, Madison, WI 53705, USA
bDepartment of Chemistry, University of Wisconsin, Madison, WI 53706, USA. E-mail: wtang@pharmacy.wisc.edu
First published on 20th March 2014
Abstract
We have discovered that bromine electrophile and carboxylate nucleophile can be added to conjugated enynes intermolecularly in a 1,4-fashion with high diastereoselectivity. Highly functionalized bromoallenes with an adjacent stereogenic centre were prepared from readily available conjugated 1,3-enynes.
Halogen mediated addition of nucleophiles to alkenes is one of the fundamentally most important reactions.1,2 It provides useful building blocks with up to two adjacent new stereogenic centres. Halogen-mediated 1,4-addition to conjugated enynes can produce chiral allenes3–6 together with a stereogenic centre. This potentially very useful reaction has, however, received very little attention partly due to the complex regio- and diastereoselectivity issue as illustrated in Scheme 1.
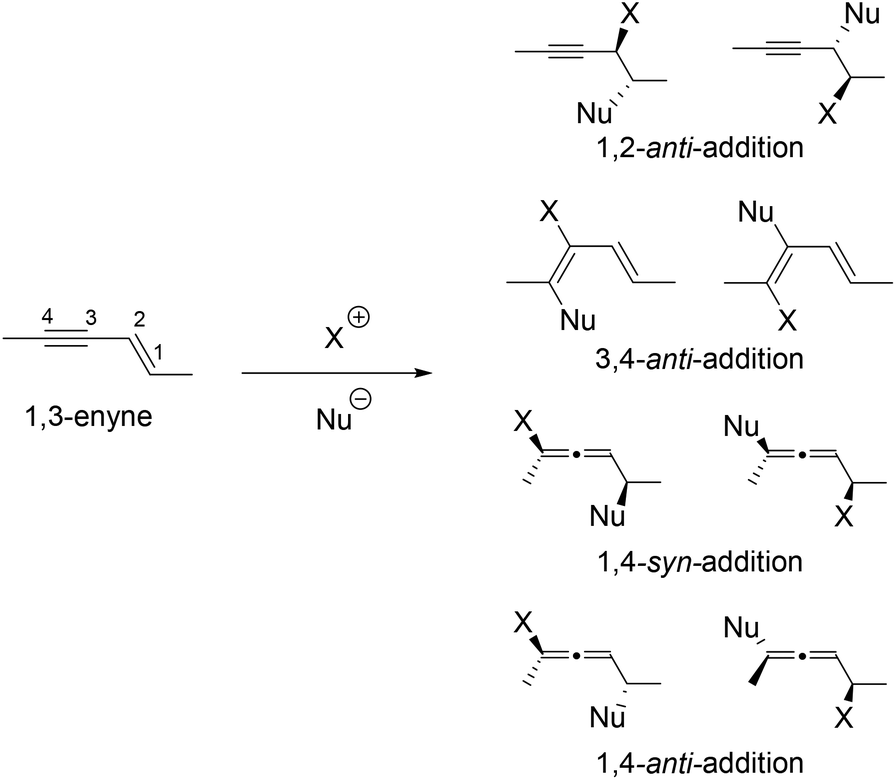 | ||
| Scheme 1 Potential isomeric products from halogen-mediated addition of nucleophiles to 1,3-enynes. | ||
The regioselectivity can be overcome partially by tethering the nucleophile with the 1,3-enyne. Indeed, examples of intramolecular halocyclizations in a 1,4-addition fashion have been documented in the literature. In 1982, the first intramolecular bromoetherification of 1,3-enynes was reported in a biomimetic synthesis of racemic panacene (Fig. 1).7,8 The diastereomeric ratio for this 1,4-addition was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. It was later found that the relative stereochemistry of panacene was assigned wrong.9 No or low diastereoselectivity was observed for similar intramolecular bromoetherification of 1,3-enynes in the synthesis of laurallene10 and kumausallene,11,12 with a few exceptions.13,14 The first stereoselective biomimetic synthesis of bromoallene-containing natural products was accomplished by us in 2011.15 Nearly perfect diastereoselectivity was observed in the biomimetic intramolecular 1,4-bromoetherification of 1,3-enynes in our enantioselective synthesis of kumausallene.
:1. It was later found that the relative stereochemistry of panacene was assigned wrong.9 No or low diastereoselectivity was observed for similar intramolecular bromoetherification of 1,3-enynes in the synthesis of laurallene10 and kumausallene,11,12 with a few exceptions.13,14 The first stereoselective biomimetic synthesis of bromoallene-containing natural products was accomplished by us in 2011.15 Nearly perfect diastereoselectivity was observed in the biomimetic intramolecular 1,4-bromoetherification of 1,3-enynes in our enantioselective synthesis of kumausallene.
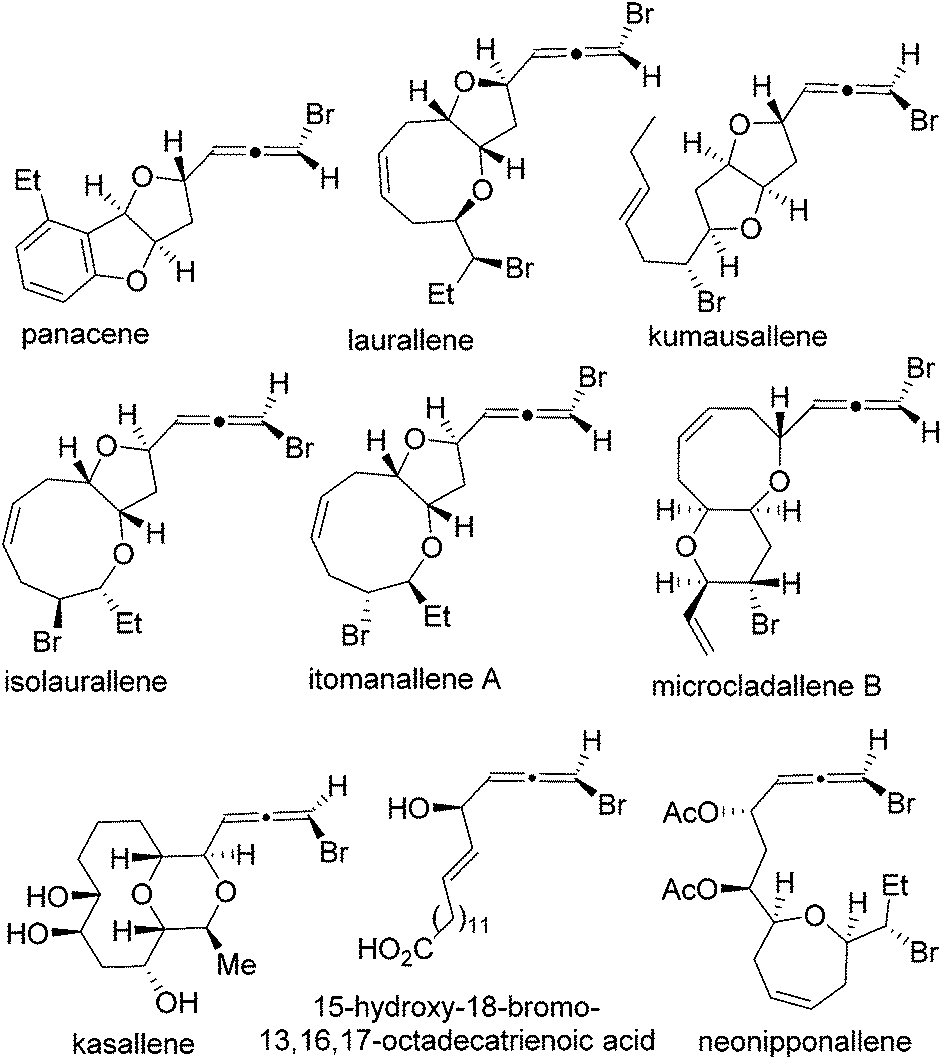 | ||
| Fig. 1 Selected bromoallene-containing natural products. | ||
In addition to panacene, laurallene, and kumausallene, the bromoallene moiety is also present in dozens of other natural products (Fig. 1).16,17 Only a small number of them have been synthesized to date.18–25 Interestingly, all haloallenes found in nature are disubstituted bromoallenes. Haloallene is also an important intermediate for the preparation of more complex allenes and other functional groups.26–44
In 2009, we reported the first 1,4-bromolactonization of 1,3-enynes (Scheme 2).45 Subsequently, the catalytic asymmetric version of this halocyclization was developed by us,46 which represents the first catalytic asymmetric halolactonization with more than 90% ee.47 A number of groups48–65 including us66 also developed different catalysts for asymmetric halolactonization of substituted alkenes and alkynes. In addition to carboxylate nucleophiles, we also demonstrated that high diastereoselectivity could be achieved for certain nitrogen nucleophiles in several halocyclizations.67 To the best of our knowledge, the much more challenging halogen-mediated intermolecular 1,4-addition to 1,3-enynes has never been reported for any nucleophiles. We herein describe the first example of intermolecular 1,4-addition of halogen and carboxylate to 1,3-enynes.
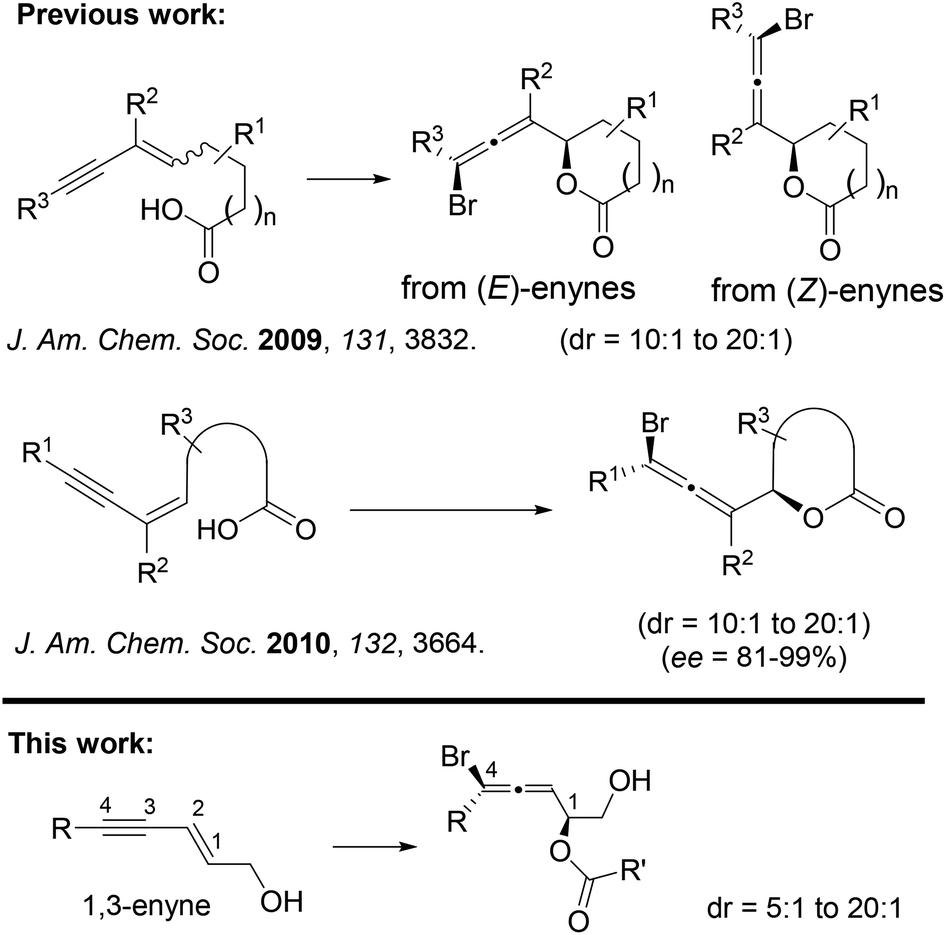 | ||
| Scheme 2 Intra- and intermolecular 1,4-bromoesterification of 1,3-enynes. | ||
Since enyne 1 is commercially available, we began our investigation on the intermolecular bromoesterification with this substrate. We first examined the source of halogen in the absence of any additive (Scheme 3). Around 30% yield of the desired 1,4-addition product 2a was observed with a 1:1 dr when DBDMH was employed, while no reaction occurred using NBS or TBCD.
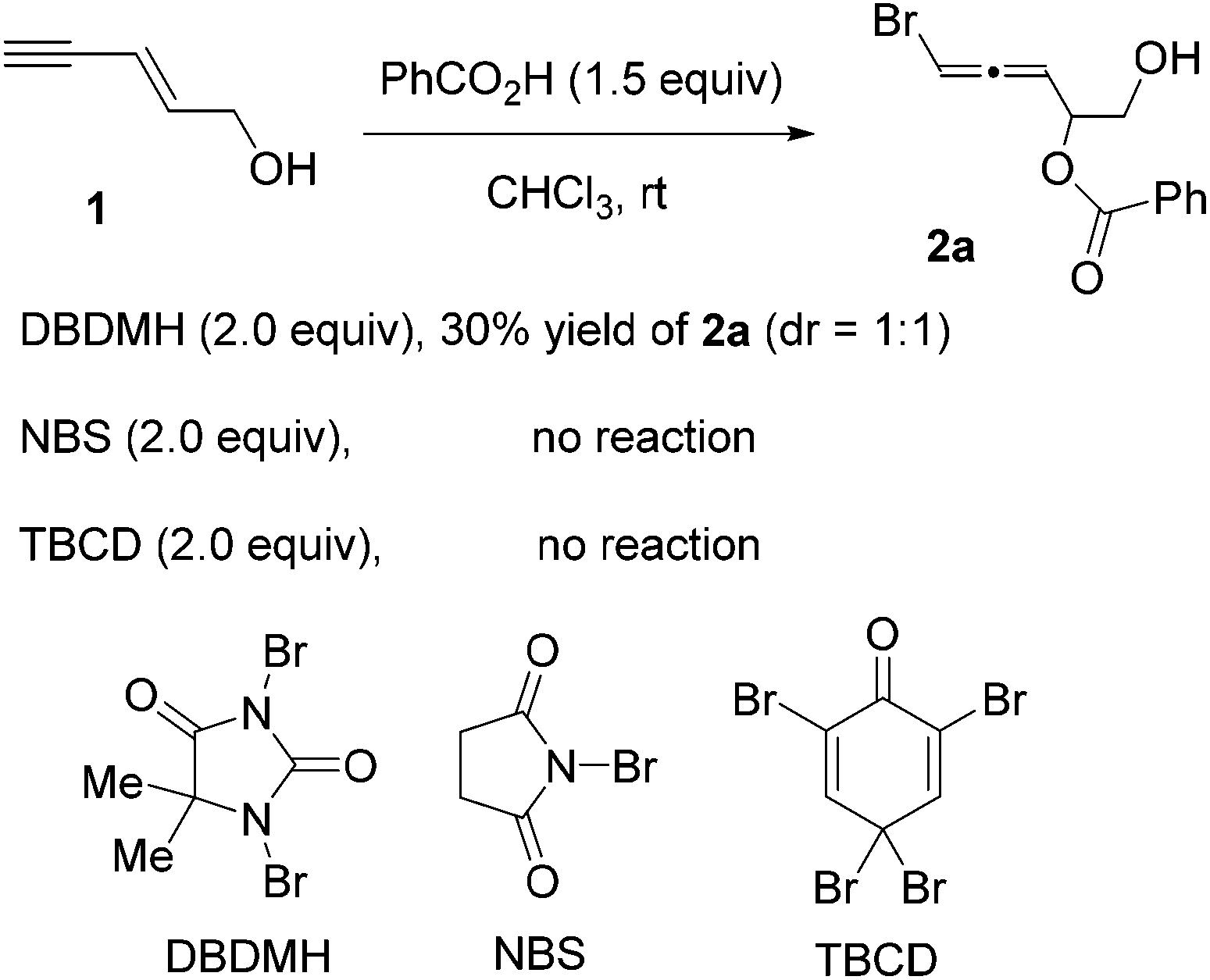 | ||
| Scheme 3 1,4-Bromoesterification of 1,3-enyne 1 with different halogenation reagents. | ||
To avoid the background reaction, which provides low diastereoselectivity, we then examined different catalysts that can activate NBS (entries 1–5, Table 1). Similar to the intramolecular reaction,45 DABCO afforded the highest diastereoselectivity (entry 1). The major diastereomer was assigned as the syn-addition product shown in Table 1 based on our previous studies on halocyclization of enynes.17,18,39 We next investigated the effect of the amount of NBS on the dr and yield in the presence of 1.1 equivalents of benzoic acid (entries 6–8). Both dr and yield were increased with less NBS reagent. Other solvents (entries 9 and 10) gave poor results. The best yield was obtained when the amount of benzoic acid was increased from 1.1 to 1.3 equivalents (entry 11). Although the yield of 2a could be improved further with an increased equivalent of benzoic acid (entry 12), the dr dropped from 10:1 to 7:1.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | x | y | Solvent | dr | Yielda |
| a Yield was based on NMR using CH2Br2 as the internal standard. | ||||||
| 1 | DABCO | 2.0 | 1.5 | CHCl3 | 5:1 |
41% |
| 2 | DBU | 2.0 | 1.5 | CHCl3 | 1:1 |
<10% |
| 3 | DMAP | 2.0 | 1.5 | CHCl3 | 1:1 |
<10% |
| 4 | DMF | 2.0 | 1.5 | CHCl3 | 3:1 |
31% |
| 5 | PPh3 | 2.0 | 1.5 | CHCl3 | No reaction | |
| 6 | DABCO | 2.0 | 1.1 | CHCl3 | 3:1 |
46% |
| 7 | DABCO | 1.5 | 1.1 | CHCl3 | 5:1 |
63% |
| 8 | DABCO | 1.2 | 1.1 | CHCl3 | 10:1 |
65% |
| 9 | DABCO | 1.2 | 1.1 | DCE | 3:1 |
73% |
| 10 | DABCO | 1.2 | 1.1 | Toluene | No reaction | |
| 11 | DABCO | 1.2 | 1.3 | CHCl3 | 10:1 |
75% |
| 12 | DABCO | 1.2 | 1.5 | CHCl3 | 7:1 |
83% |
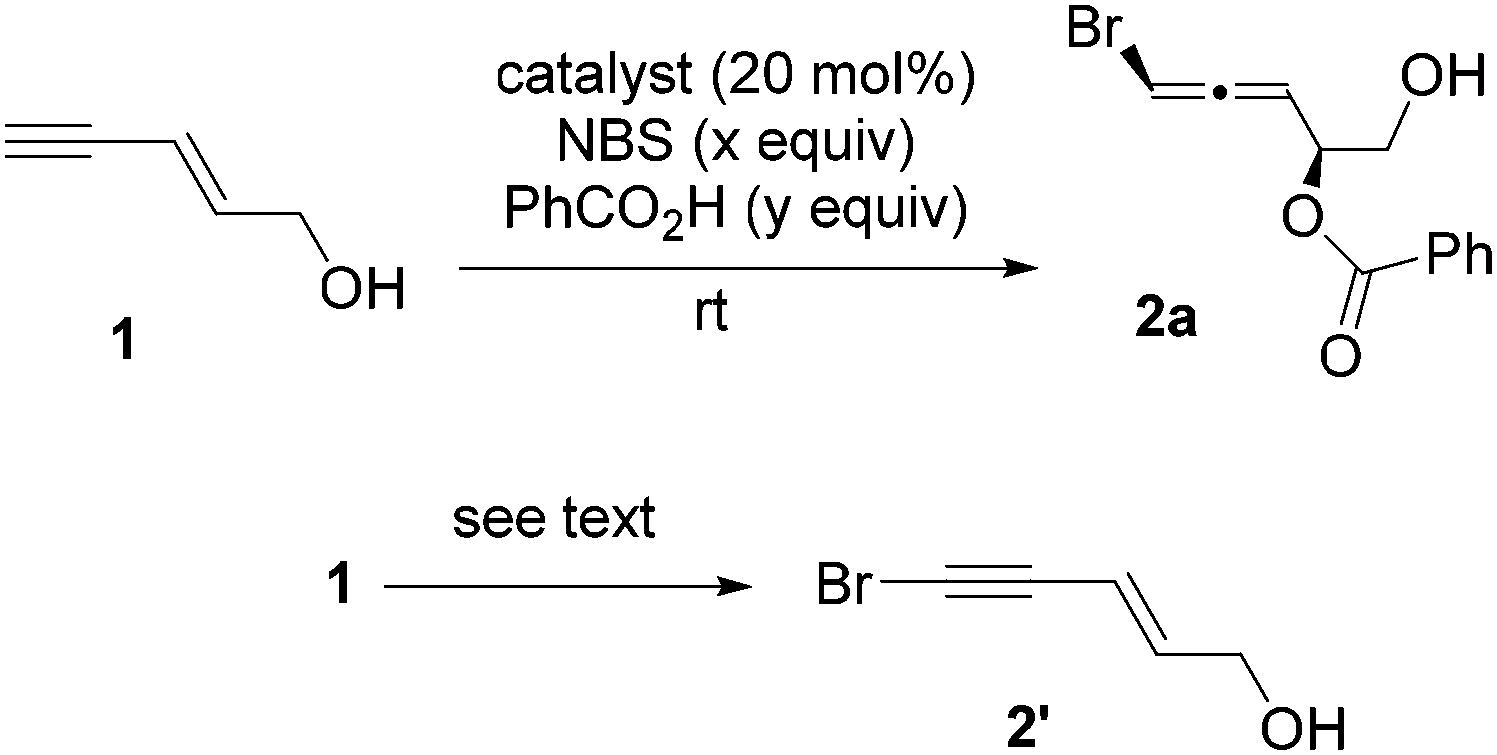
We also replaced NBS with TBCD under the conditions in entry 6 of Table 1. Interestingly, the only product we observed was bromoalkyne 2′, where the hydrogen atom on the terminal alkyne was replaced by a bromine atom.
With the optimized conditions (entry 11, Table 1) in hand, we then studied the scope of the carboxylic acids (Table 2). Similar results were obtained by using ortho- or para-methyl substituted benzoic acids (entries 2 and 3). A slower reaction was observed for benzoic acid with a strong electron-donating group (entry 4), while benzoic acid with a strong electron-withdrawing group yielded a complex mixture (entry 5). Halogen substituted benzoic acids gave 44% to 70% yields of the desired products (entries 6–8). Lower yields for entries 7 and 8 are likely due to the poor solubility of the corresponding benzoic acids. Aliphatic carboxylic acids generally worked well with slightly lower drs (entries 9–11).
|
|
||||
|---|---|---|---|---|
| Entry | Carboxylic acid (R) | Product | dr | Yielda |
| a Isolated yield. b Based on recovered starting material. | ||||
| 1 | R = C6H5 | 2a | 10:1 |
73% |
| 2 | R = o-CH3C6H4 | 2b | 10:1 |
65% |
| 3 | R = p-CH3C6H4 | 2c | 10:1 |
65% |
| 4 | R = p-CH3OC6H4 | 2d | 10:1 |
45% |
| 5 | R = p-NO2C6H4 | Complex mixture | ||
| 6 | R = p-FC6H4 | 2e | 10:1 |
70% |
| 7 | R = p-ClC6H4 | 2f | 10:1 |
45% (60%)b |
| 8 | R = p-BrC6H4 | 2g | 10:1 |
44% (57%)b |
| 9 | R = CH3 | 2h | 8:1 |
67% |
| 10 | R = C6H5CH2 | 2i | 5:1 |
77% |
| 11 | R = CH3CH2 | 2j | 5:1 |
61% |
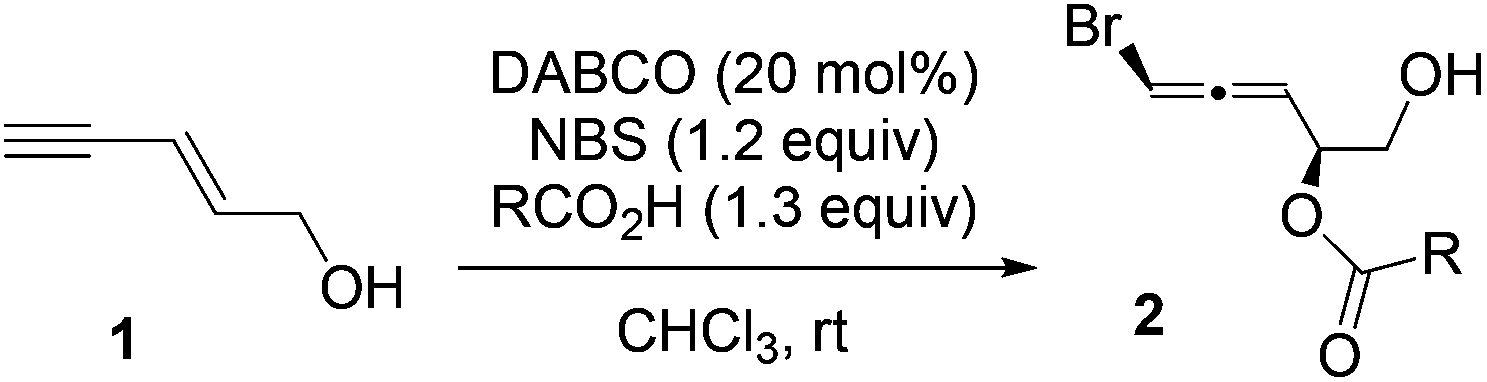
The scope of enynes was also examined (Table 3). Enynes with sterically bulky groups provided higher diastereoselectivity compared with 2 (entries 1 and 2). The dr and yield for enyne 3c with a long-chain aliphatic substituent (entry 3) were similar to those of the parent substrate 1. No reaction occurred for enynes with an aryl or cyclopropyl substituent (entries 4 and 5).
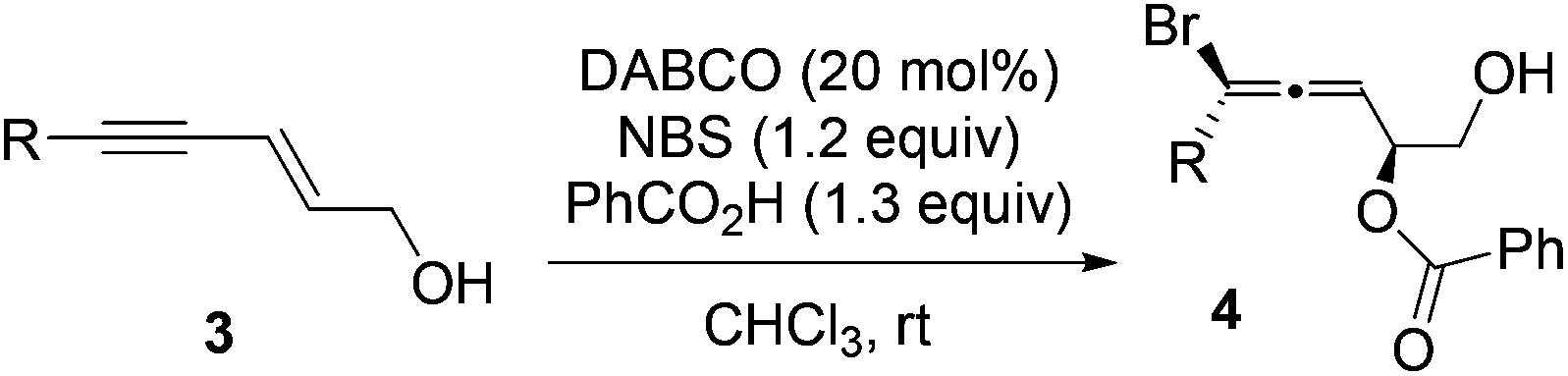
We also found that the free hydroxyl group in 1 was required since no reaction occurred for substrate 3f, where the OH group was masked as benzyl ether (Scheme 4). Surprisingly, enynes 3g and 3h with a cis-alkene also did not afford any desired products. Only a trace amount of the product was observed for secondary alcohol 3i under standard conditions.
 | ||
| Scheme 4 Failed substrates. | ||
Similar to the previously reported intramolecular 1,4-addition of halogen and nucleophile to 1,3-enynes,17,18,39 the overall syn-addition is likely due to the interaction between the negatively charged carboxylate and the partially positively charged electrophile. The free OH group may facilitate the addition by forming a hydrogen-bond with the carboxylate.
In summary, we have developed the first intermolecular 1,4-bromoesterification of conjugated 1,3-enynes. Functionalized bromoallenes were prepared efficiently from relatively simple starting materials diastereoselectively. A broad range of carboxylic acids and enynes with either a terminal or internal alkyne can participate in the 1,4-addition reaction.
Experimental section
General procedure for the intermolecular 1,4-bromoesterification of conjugated enynes
To a 6 mL vial was added enyne 1 (0.1 mmol, 8.2 mg), DABCO (0.02 mmol, 2.2 mg), and benzoic acid (0.13 mmol, 15.9 mg). To the above mixture was added 1 mL of CHCl3. Subsequently, NBS (0.12 mmol, 21.4 mg) was added to the above solution. The reaction mixture was stirred at room temperature until enyne 1 was consumed as indicated by TLC. The reaction was filtered through a short silica gel column. 1H NMR analysis of the crude mixture was performed to obtain the dr. The mixture was then purified by flash column chromatograph using hexane and ethyl acetate (4:1) as the eluent. Product 2a was obtained as a colorless oil in 73% yield (21.0 mg, dr = 10:1). The reaction was scaled up to 1 mmol for the preparation of 2a (dr = 10:1, 68% yield).
Acknowledgements
We thank the University of Wisconsin for financial support and a Young Investigator Award from Amgen (to W. T.).Notes and references
- K. E. Harding and T. H. Tiner, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 4, p. 363 Search PubMed.
- J. Rodriguez and J. P. Dulcere, Synthesis, 1993, 1177 CrossRef CAS PubMed.
- N. Krause and A. S. K. Hashmi, Modern Allene Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- S. Ma, Chem. Rev., 2005, 105, 2829 CrossRef PubMed.
- K. M. Brummond and J. E. DeForrest, Synthesis, 2007, 795 CrossRef CAS PubMed.
- S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384 RSC.
- K. S. Feldman, C. C. Mechem and L. Nader, J. Am. Chem. Soc., 1982, 104, 4011 CrossRef CAS.
- K. S. Feldman, Tetrahedron Lett., 1982, 23, 3031 CrossRef CAS.
- J. Boukouvalas, M. Pouliot, J. Robichaud, S. MacNeil and V. Snieckus, Org. Lett., 2006, 8, 3597 CrossRef CAS PubMed.
- J. Ishihara, Y. Shimada, N. Kanoh, Y. Takasugi, A. Fukuzawa and A. Murai, Tetrahedron, 1997, 53, 8371 CrossRef CAS.
- M. T. Crimmins and E. A. Tabet, J. Am. Chem. Soc., 2000, 122, 5473 CrossRef CAS.
- P. A. Evans, V. S. Murthy, J. D. Roseman and A. L. Rheingold, Angew. Chem., Int. Ed., 1999, 38, 3175 CrossRef CAS.
- D. C. Braddock, R. Bhuva, Y. Perez-Fuertes, R. Pouwer, C. A. Roberts, A. Ruggiero, E. S. E. Stokes and A. J. P. White, Chem. Commun., 2008, 1419 RSC.
- C. Sabot, D. Berard and S. Canesi, Org. Lett., 2008, 10, 4629 CrossRef CAS PubMed.
- J. B. Werness and W. Tang, Org. Lett., 2011, 13, 3664 CrossRef CAS PubMed.
- A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196 CrossRef PubMed.
- V. M. Dembitsky and T. Maoka, Prog. Lipid Res., 2007, 46, 328 CrossRef CAS PubMed.
- T. A. Grese, K. D. Hutchinson and L. E. Overman, J. Org. Chem., 1993, 58, 2468 CrossRef CAS.
- J. Wang and B. L. Pagenkopf, Org. Lett., 2007, 9, 3703 CrossRef CAS PubMed.
- T. Saitoh, T. Suzuki, M. Sugimoto, H. Hagiwara and T. Hoshi, Tetrahedron Lett., 2003, 44, 3175 CrossRef CAS.
- M. T. Crimmins and K. A. Emmitte, J. Am. Chem. Soc., 2001, 123, 1533 CrossRef CAS.
- M. T. Crimmins, K. A. Emmitte and A. L. Choy, Tetrahedron, 2002, 58, 1817 CrossRef CAS.
- J. Park, B. Kim, H. Kim, S. Kim and D. Kim, Angew. Chem., Int. Ed., 2007, 46, 4726 CrossRef PubMed.
- W. Jeong, M. J. Kim, H. Kim, S. Kim, D. Kim and K. J. Shin, Angew. Chem., Int. Ed., 2010, 49, 752 CrossRef CAS PubMed.
- M. J. Kim, T.-i. Sohn, D. Kim and R. S. Paton, J. Am. Chem. Soc., 2012, 134, 20178 CrossRef CAS PubMed.
- J. A. Marshall and N. D. Adams, J. Org. Chem., 1997, 62, 8976 CrossRef CAS.
- H. Ohno, H. Hamaguchi and T. Tanaka, Org. Lett., 2001, 3, 2269 CrossRef CAS PubMed.
- H. Ohno, K. Ando, H. Hamaguchi, Y. Takeoka and T. Tanaka, J. Am. Chem. Soc., 2002, 124, 15255 CrossRef CAS PubMed.
- H. Ohno, H. Hamaguchi, M. Ohata, S. Kosaka and T. Tanaka, J. Am. Chem. Soc., 2004, 126, 8744 CrossRef CAS PubMed.
- H. Hamaguchi, S. Kosaka, H. Ohno and T. Tanaka, Angew. Chem., Int. Ed., 2005, 44, 1513 CrossRef CAS PubMed.
- B. Xu and G. B. Hammond, Angew. Chem., Int. Ed., 2005, 44, 7404 CrossRef CAS PubMed.
- B. M. Trost and D. T. Stiles, Org. Lett., 2005, 7, 2117 CrossRef CAS PubMed.
- S. Ma and H. Xie, Tetrahedron, 2005, 61, 251 CrossRef CAS PubMed.
- L. C. Shen, R. P. Hsung, Y. S. Zhang, J. E. Antoline and X. J. Zhang, Org. Lett., 2005, 7, 3081 CrossRef CAS PubMed.
- C. J. Tang and Y. K. Wu, Tetrahedron, 2007, 63, 4887 CrossRef CAS PubMed.
- B. Vaz, M. Dominguez, R. Alvarez and A. R. de Lera, Chem.–Eur. J., 2007, 13, 1273 CrossRef CAS PubMed.
- H. Hamaguchi, S. Kosaka, H. Ohno, N. Fujii and T. Tanaka, Chem.–Eur. J., 2007, 13, 1692 CrossRef CAS PubMed.
- Y. Z. Xia, A. S. Dudnik, V. Gevorgyan and Y. H. Li, J. Am. Chem. Soc., 2008, 130, 6940 CrossRef CAS PubMed.
- Y. Tang, L. Shen, B. J. Dellaria and R. P. Hsung, Tetrahedron Lett., 2008, 49, 6404 CrossRef CAS PubMed.
- A. K. A. Persson and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2010, 49, 4624 CrossRef CAS PubMed.
- T. Jiang, A. K. A. Persson and J.-E. Bäckvall, Org. Lett., 2011, 13, 5838 CrossRef CAS PubMed.
- A. K. A. Persson, T. Jiang, M. T. Johnson and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2011, 50, 6155 CrossRef CAS PubMed.
- Y. Deng, T. Bartholomeyzik, A. K. A. Persson, J. Sun and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2012, 51, 2703 CrossRef CAS PubMed.
- H. Chiba, Y. Sakai, A. Ohara, S. Oishi, N. Fujii and H. Ohno, Chem.–Eur. J., 2013, 19, 8875 CrossRef CAS PubMed.
- W. Zhang, H. D. Xu, H. Xu and W. Tang, J. Am. Chem. Soc., 2009, 131, 3832 CrossRef CAS PubMed.
- W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664 CrossRef CAS PubMed.
- G. Chen and S. Ma, Angew. Chem., Int. Ed., 2010, 49, 8306 CrossRef CAS PubMed.
- D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298 CrossRef CAS PubMed.
- R. Yousefi, D. C. Whitehead, J. M. Mueller, R. J. Staples and B. Borhan, Org. Lett., 2011, 13, 608 CrossRef CAS PubMed.
- R. Yousefi, K. D. Ashtekar, D. C. Whitehead, J. E. Jackson and B. Borhan, J. Am. Chem. Soc., 2013, 135, 14524 CrossRef CAS PubMed.
- L. Zhou, C. K. Tan, X. Jiang, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474 CrossRef CAS PubMed.
- C. K. Tan, L. Zhou and Y.-Y. Yeung, Org. Lett., 2011, 13, 2738 CrossRef CAS PubMed.
- J. Chen, L. Zhou, C. K. Tan and Y.-Y. Yeung, J. Org. Chem., 2012, 77, 999 CrossRef CAS PubMed.
- C. K. Tan, C. Le and Y.-Y. Yeung, Chem. Commun., 2012, 48, 5793 RSC.
- X. Jiang, C. K. Tan, L. Zhou and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2012, 51, 7771 CrossRef CAS PubMed.
- G. E. Veitch and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 7332 CrossRef CAS PubMed.
- K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura and H. Fujioka, Angew. Chem., Int. Ed., 2010, 49, 9174 CrossRef CAS PubMed.
- K. Murai, A. Nakamura, T. Matsushita, M. Shimura and H. Fujioka, Chem.–Eur. J., 2012, 18, 8448 CrossRef CAS PubMed.
- K. Murai, T. Matsushita, A. Nakamura, N. Hyogo, J. Nakajima and H. Fujioka, Org. Lett., 2013, 15, 2526 CrossRef CAS PubMed.
- M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068 CrossRef CAS PubMed.
- J. E. Tungen, J. M. J. Nolsoe and T. V. Hansen, Org. Lett., 2012, 14, 5884 CrossRef CAS PubMed.
- D. H. Paull, C. Fang, J. R. Donald, A. D. Pansick and S. F. Martin, J. Am. Chem. Soc., 2012, 134, 11128 CrossRef CAS PubMed.
- C. Fang, D. H. Paull, J. C. Hethcox, C. R. Shugrue and S. F. Martin, Org. Lett., 2012, 14, 6290 CrossRef CAS PubMed.
- K. Ikeuchi, S. Ido, S. Yoshimura, T. Asakawa, M. Inai, Y. Hamashima and T. Kan, Org. Lett., 2012, 14, 6016 CrossRef CAS PubMed.
- M. Wilking, C. Mueck-Lichtenfeld, C. G. Daniliuc and U. Hennecke, J. Am. Chem. Soc., 2013, 135, 8133 CrossRef CAS PubMed.
- W. Zhang, N. Liu, C. M. Schienebeck, K. Decloux, S. Zheng, J. B. Werness and W. Tang, Chem.–Eur. J., 2012, 18, 7296 CrossRef CAS PubMed.
- N. Liu, J. B. Werness, I. A. Guzei and W. Tang, Tetrahedron, 2011, 67, 4385 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, HRMS, and IR data and copies of NMR spectra for all starting materials and products. See DOI: 10.1039/c3qo00088e |
| This journal is © the Partner Organisations 2014 |