Palladacycle-catalyzed cascade reaction of bicyclic alkenes with alkynyl imines: synthesis of polycyclic 5H-benzo[b]azepines†
Guang-Cun
Ge
a,
Chang-Hua
Ding
a and
Xue-Long
Hou
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences (CAS), 345 Lingling Road, Shanghai 200032, China. E-mail: xlhou@sioc.ac.cn; Fax: +86 21 54925100; Tel: +86 21 54925144
bShanghai–Hong Kong Joint Laboratory in Chemical Synthesis, SIOC, CAS, Shanghai, China
First published on 20th March 2014
Abstract
A new methodology to prepare the unique six–seven membered [4,5,0] heterocyclic ring system, an important structure in natural products and medicinal chemistry, has been developed via a palladacycle-catalyzed cascade reaction of bicyclic alkenes and alkynyl imines. The unique catalytic activity of palladacycles as well as C–H activation under palladacycle catalysis, a new catalytic pattern of palladacycles, has been revealed.
Palladacycles as one type of palladium species feature easier availability, extra stability toward air and moisture, versatile frameworks as well as high catalytic activity. They have found many applications in a wide range of fields, such as biological chemistry, materials science, organic synthesis, and ligand resolution.1 More importantly, they have been used as catalysts in carbon–carbon and carbon–heteroatom bond formation2 as well as asymmetric reactions.3 Moreover, many studies have revealed that palladacycles have been recognized as reaction intermediates formed in many C–H functionalization reactions.4 In many cases, palladacycles are considered as pre-catalysts and Lewis acids in the reactions; nevertheless, recent achievements show the possibility for them to act as real catalysts.5 However, only a limited type of reaction could be catalyzed by palladacycles, and the understanding regarding the influence of palladacycles on the reaction is also limited. More knowledge of palladacycles in catalysis and exploration of more reactions using palladacycles are in high demand. During the studies on the applications of palladacycles in organic synthesis,6 we developed a reaction of in situ prepared organozinc halides with oxabicyclic alkenes in the presence of a palladacycle as the catalyst and revealed that in that reaction the palladacycle performed as the real catalyst.6a We also realized asymmetric synthesis using a chiral palladacycle as a catalyst6b,c as well as switched the chemoselectivity of the reaction under palladacycle-catalysis and provided a plausible explanation why different palladacycles led to different reaction selectivities.6e Further studies revealed that palladacycles could also serve as catalysts in the C–H activation reaction. In this paper, we would disclose our preliminary results of the palladacycle-catalyzed domino reaction of alkynyl imines with oxabicyclic alkenes to afford benzo[b]azepines via addition–cyclization-C–H activation sequences.
Recently, we reported a facile and regioselective synthesis of polysubstituted furans via the palladacycle-catalyzed reaction of bicyclic alkenes with terminal ynones.6d Based upon these results, alkynyl imine was adopted as a reactant to see whether pyrrole was afforded. The reaction of bicyclic alkene 1a and alkynyl imine 2a proceeded smoothly in the presence of palladacycle P1 (entry 1, Table 1). However, the expected product pyrrole was not obtained; instead, a polycyclic product 3a bearing a nitrogen-containing seven membered ring system, an important subunit in natural products and medicines,7 was afforded.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Palladacycle (mol%) | Additive (equiv.) | Temp (°C) | 3a (%) | 4a (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.1 mmol), ClCH2CH2Cl (2.0 mL). b Isolated yield. | |||||
| 1 | P1 (10) | p-BrC6H4CO2H (0.5) | rt | 20 | 17 |
| 2 | P2 (10) | p-BrC6H4CO2H (0.5) | rt | 12 | 29 |
| 3 | P3 (10) | p-BrC6H4CO2H (0.5) | rt | 72 | — |
| 4 | P4 (10) | p-BrC6H4CO2H (0.5) | rt | 70 | — |
| 5 | P5 (10) | p-BrC6H4CO2H (0.5) | rt | Trace | 93 |
| 6 | P6 (10) | p-BrC6H4CO2H (0.5) | rt | Trace | — |
| 7 | P3 (5) | p-BrC6H4CO2H (0.5) | rt | 54 | — |
| 8 | P3 (10) | p-BrC6H4CO2H (0.5) | 0 | 37 | — |
| 9 | P3 (10) | p-BrC6H4CO2H (0.5) | 50 | 66 | — |
| 10 | P3 (10) | — | rt | 51 | — |
| 11 | P3 (10) | Et3N (0.5) | rt | — | — |
| 12 | P3 (10) | Cs2CO3 (0.5) | rt | — | — |
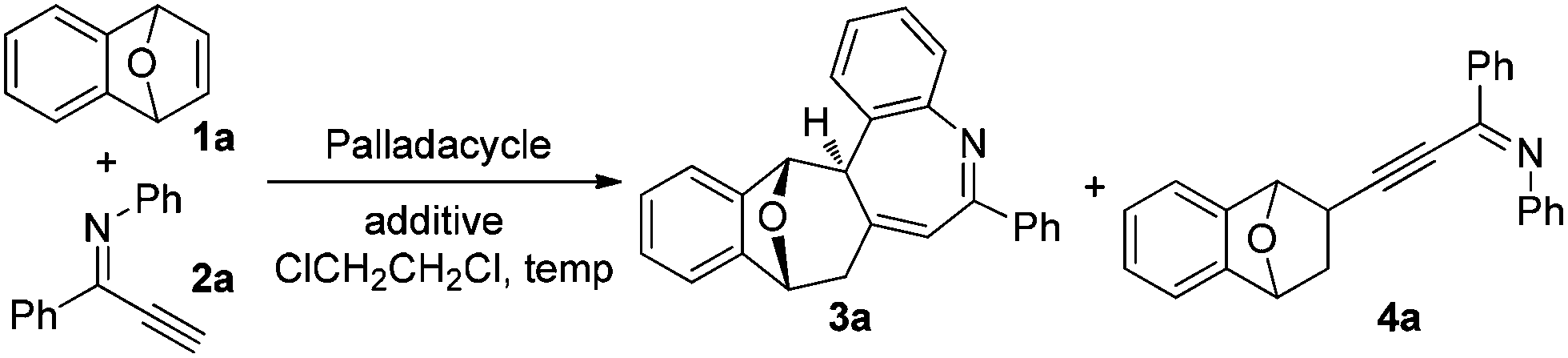
This unusual result intrigued us to clarify what affected the generation of compound 3a (Table 1). Firstly, the effect of palladium catalysts on the reaction was investigated. The common palladium catalysts, such as Pd(OAc)2, Pd(PPh3)Cl2, Pd(OAc)2/PPh3 and Pd2(dba)3, did not work in this reaction (not shown in the table), implying the unique catalytic activity of palladacycles. Further studies revealed that the different structure of palladacycles, including the coordination atoms, the framework as well as hybridization of carbon connected with Pd, has a great impact on the reaction (Fig. 1). Palladacycles P1–5 with a phosphine donor atom proved to be suitable catalysts for the transformation (entries 1–5), while that with nitrogen as a coordination atom P6 could not catalyze the reaction (entry 6). Importantly, palladacycles P3 and P4 with the sp2 C–Pd bond as catalysts led to the desired product 3a in 72% and 70% yield respectively (entries 3 and 4); in sharp contrast, palladacycle P5 containing the sp3 C–Pd bond afforded the addition product 4a in 93% yield (entry 5), which is in accordance with our findings before.6e It is rather surprising that much more different results were obtained using palladacycles P1 and P2 or P3 and P4 as catalysts (entries 1 and 2 vs. entries 3 and 4), though all of these four palladacycles have the sp2 C–Pd bond with phosphorus as the coordination atom. It can be seen that the differences between palladacycles P1, P2 and P3, P4 are that P3 and P4 have planar structures while P1 and P2 are non-planar; also the bond length of Pd–P in P1 and P2 and in P3 and P4 is not the same. These results reflect some worthwhile to be studied characteristics of palladacycles as catalysts. The effect of acid additives on the reaction was evaluated, which revealed the importance of the presence of acids as additives in the reaction (entry 3 vs. entry 10); meanwhile, the addition of a base such as Cs2CO3 or Et3N suppressed the reaction completely (entries 11 and 12). It was found that p-BrC6H4COOH is the best choice (for evaluation of other acid additives, see ESI†). The influence of temperature on the reaction was investigated with palladacycle P3 as the catalyst. Raising or lowering the reaction temperature did not improve the yield of product 3a (entries 8 and 9). The reaction is sensitive to the loading of the catalyst because the use of 5 mol% of P3 resulted in the decrease of the yield (entry 7 vs. entry 3). We also investigated the influence of solvents (not shown in the table). The reaction did not proceed in polar solvents such as N,N-dimethylformamide and dimethyl sulfoxide while in non-polar solvents like dichloromethane and toluene gave the target compound 3a in 53% yield and 63% yield respectively, which was lower than that when using 1,2-dichloroethane as a solvent. When the ratio of 1a to 2a was reduced from 2/1 to 1.3/1, the product 3a was still obtained in 72% yield.
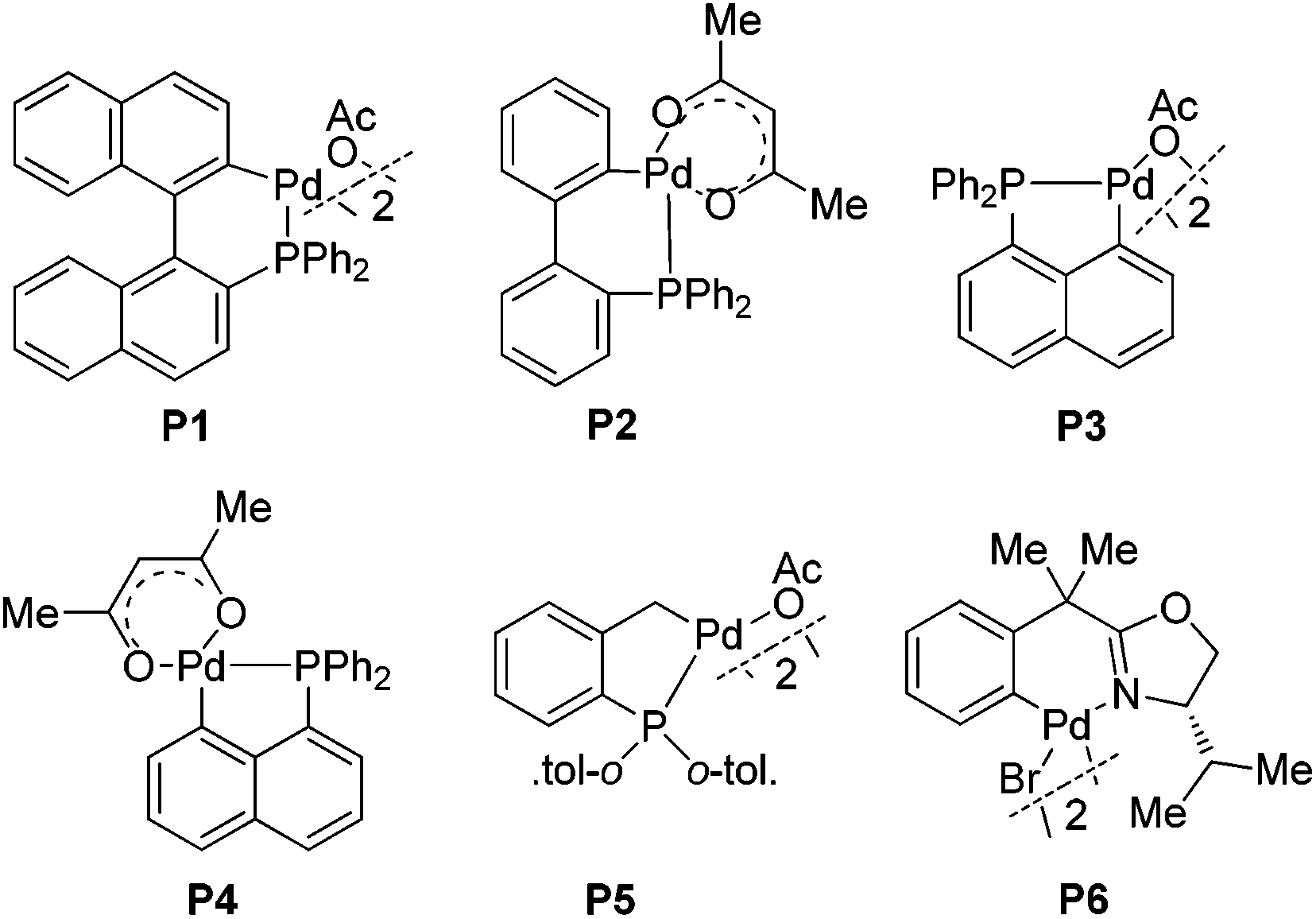 | ||
| Fig. 1 Palladacycles with different scaffolds and donor atoms. | ||
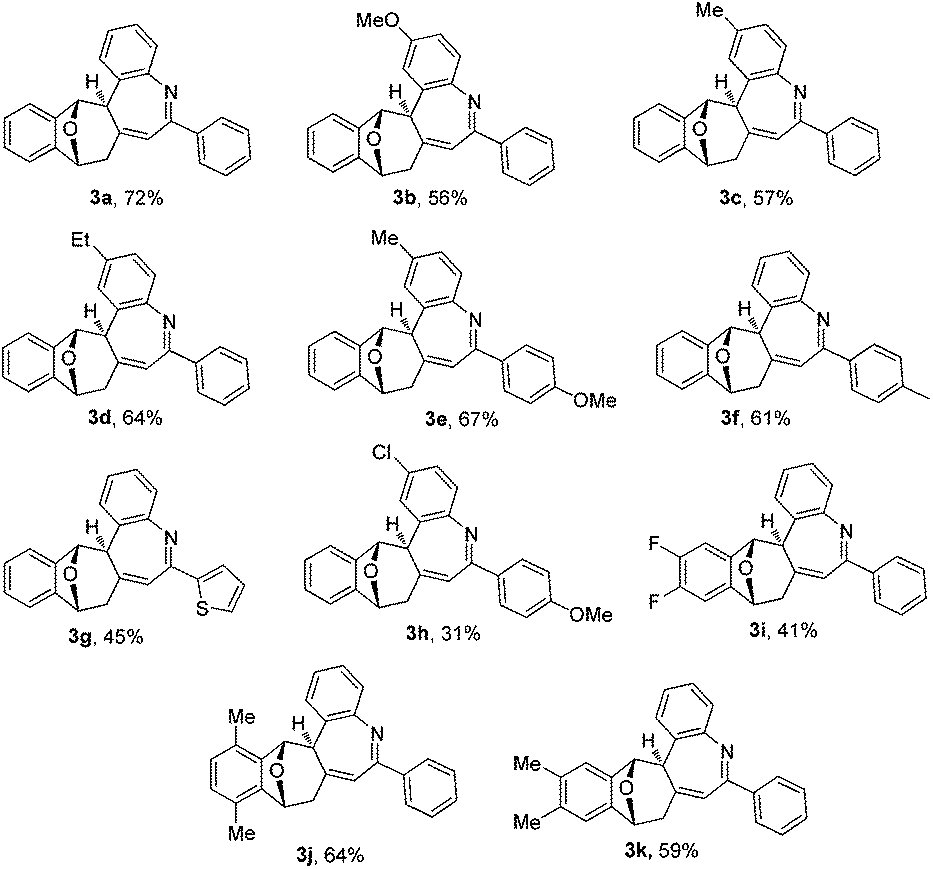
The substrate scope of bicyclic alkenes 1 and imine 2 was explored under the optimized reaction conditions, and the results are compiled in Table 2. It can be seen that many oxabicyclic alkenes 1 and alkynyl imines 2 are suitable substrates for the reaction, affording the corresponding 5H-benzo[b]azepines 3 in moderate to good yields. The alkynyl imines 2 with electron-donating substituents located at the 4-position of the aryl ring are tolerated, providing the corresponding products 3b–3f in moderate yield. When R3 was an electron-withdrawing group, the yield of the corresponding product 3h became low (31%). If R3 was a stronger electron-withdrawing group such as CF3, the reaction was fully retarded with the starting reactant being recovered (not shown in the table). A thienyl substituted imine is a suitable reactant, providing the corresponding 5H-benzo[b]azepine 3g in 45% yield. Substituents on the oxabicyclic alkenes 1 affected the reaction to some extent. Oxabicyclic alkenes 1 containing electron-donating groups worked well to give the products 3j and 3k in good yield. The incorporation of electron-withdrawing groups into oxabicyclic alkenes 1 led to a poor result (3i). Several other bicyclic alkenes, for example norbornene, 2,5-norbornadiene and dimethyl 7-oxabicyclo[2.2.1]hepta-2,5-diene-2,3-dicarboxylate, were tested, but the corresponding target compounds were not detected by GC-MS. The structure of this unique polycyclic ring system was further determined by X-ray analysis of single crystals of compound 3b (Fig. 2).
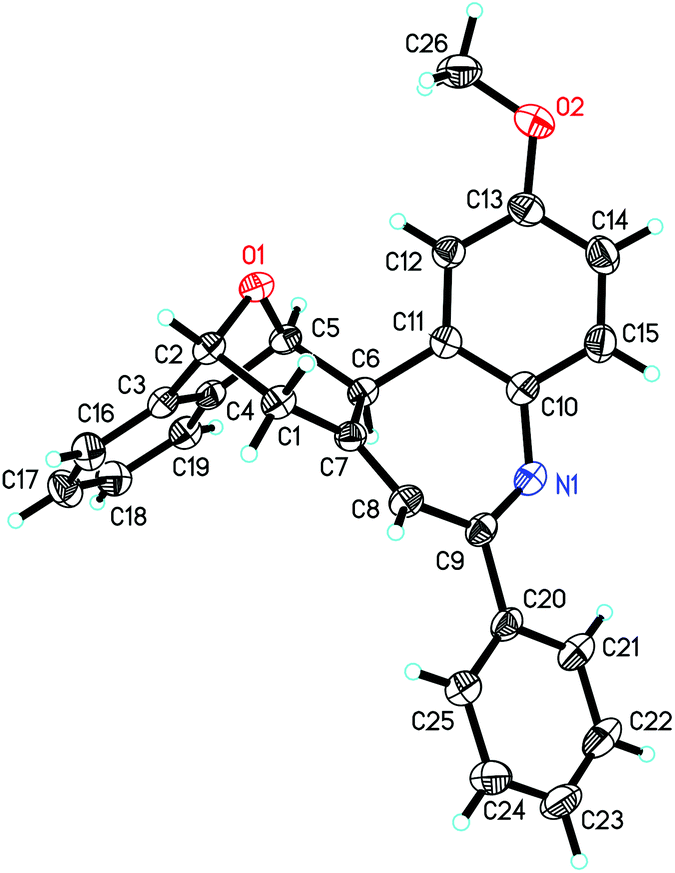 | ||
| Fig. 2 ORTEP drawing of compound 3b. | ||
|
|
|---|
| a Reaction conditions: 1 (0.26 mmol), 2 (0.2 mmol), ClCH2CH2Cl (2.0 mL); isolated yields. |
|
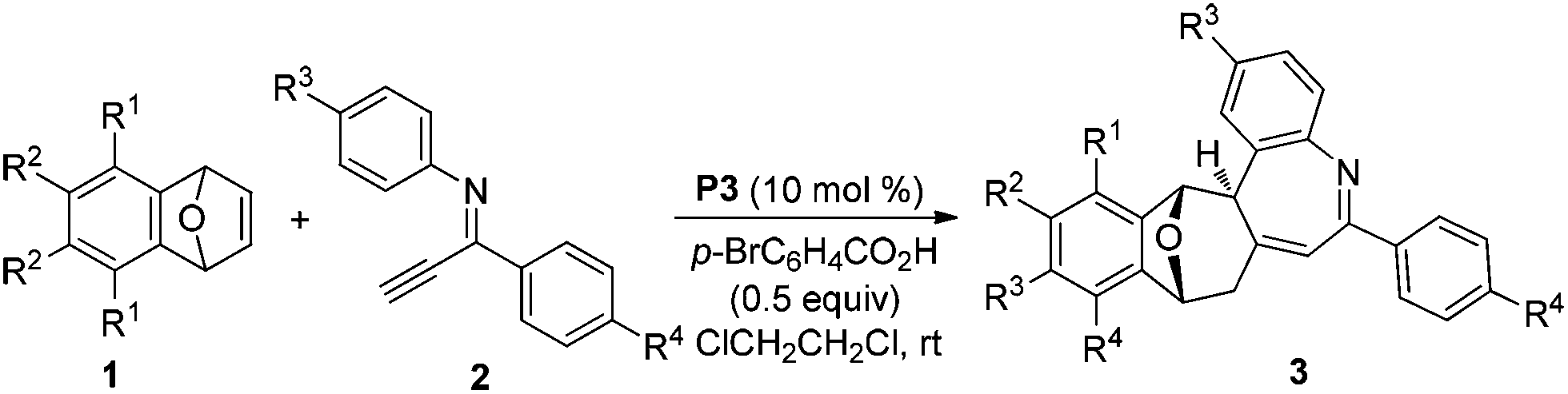
To understand the reaction mechanism and the formation of a six–seven membered ring [4,5,0] heterobicyclic system, some experiments were carried out. Initially, the addition product 4a was subjected to the reaction conditions of Table 2, however no reaction occurred, which suggested that it is not the reaction intermediate (eqn (1)). We have found that an alkylidenecyclopropane serves as a possible intermediate in the furan formation reaction.6d Thus, we monitored the progress of the reaction of oxabicyclic alkene 1a and imine 2avia1H- and 13C NMR spectroscopy (eqn (2)). Two new peaks at δ 1.52 (dd, J = 0.4, 7.2 Hz) and 1.81 (d, J = 7.2 Hz) appeared in 1H NMR when the reaction was run for 6 minutes. These two peaks could be assigned to two protons on the cyclopropane ring of the intermediate 5 by comparison with that of compound 7 (δ 2.00, d, J = 7.2 Hz and 2.24, dd, J = 1.2, 7.2 Hz), which was produced in the control experiment, the reaction of bicyclic alkene 1a with alkynyl pyridine 6 (eqn (3)). These two peaks weakened, and the protons of compound 3a appeared when the reaction was allowed to proceed for a longer time. 13C NMR experiments gave similar information on the formation of an alkylidenecyclopropane. Two peaks appeared at δ 26.7 and 28.9 in 6 minutes, which have similar chemical shifts with the two carbons of the cyclopropane ring of compound 7. These NMR experiments indicated that the alkylidenecyclopropane 5 might be a reaction intermediate in the formation of compound 3a (Scheme 1).
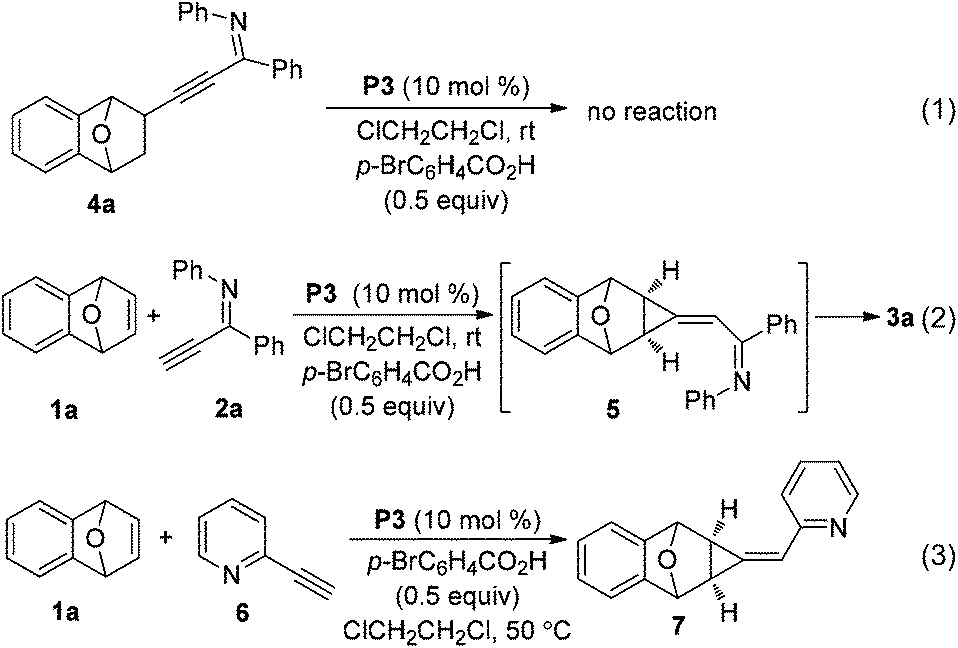 | ||
| Scheme 1 Experiments for identifying the reaction intermediate 5. | ||
Based upon the clues we have and literature reports,4,5a,6d a plausible reaction mechanism for the formation of compound 3 could be proposed (Scheme 2). An alkynylpalladium(II) Int-A was generated via the reaction of the imine 2 with palladacycles, which underwent a carbopalladation with bicyclic alkene 1 to give Int-B, which undergoes an intramolecular addition and a subsequent protonation to afford an alkylidenecyclopropane Int-C. An intramolecular C–H bond activation of Int-C furnishes Int-D, which is converted to compound 3 accompanying the release of the palladacycle catalyst. Further experiments are needed to have the real reaction mechanism, especially the transformation of intermediate C to the final product 3.
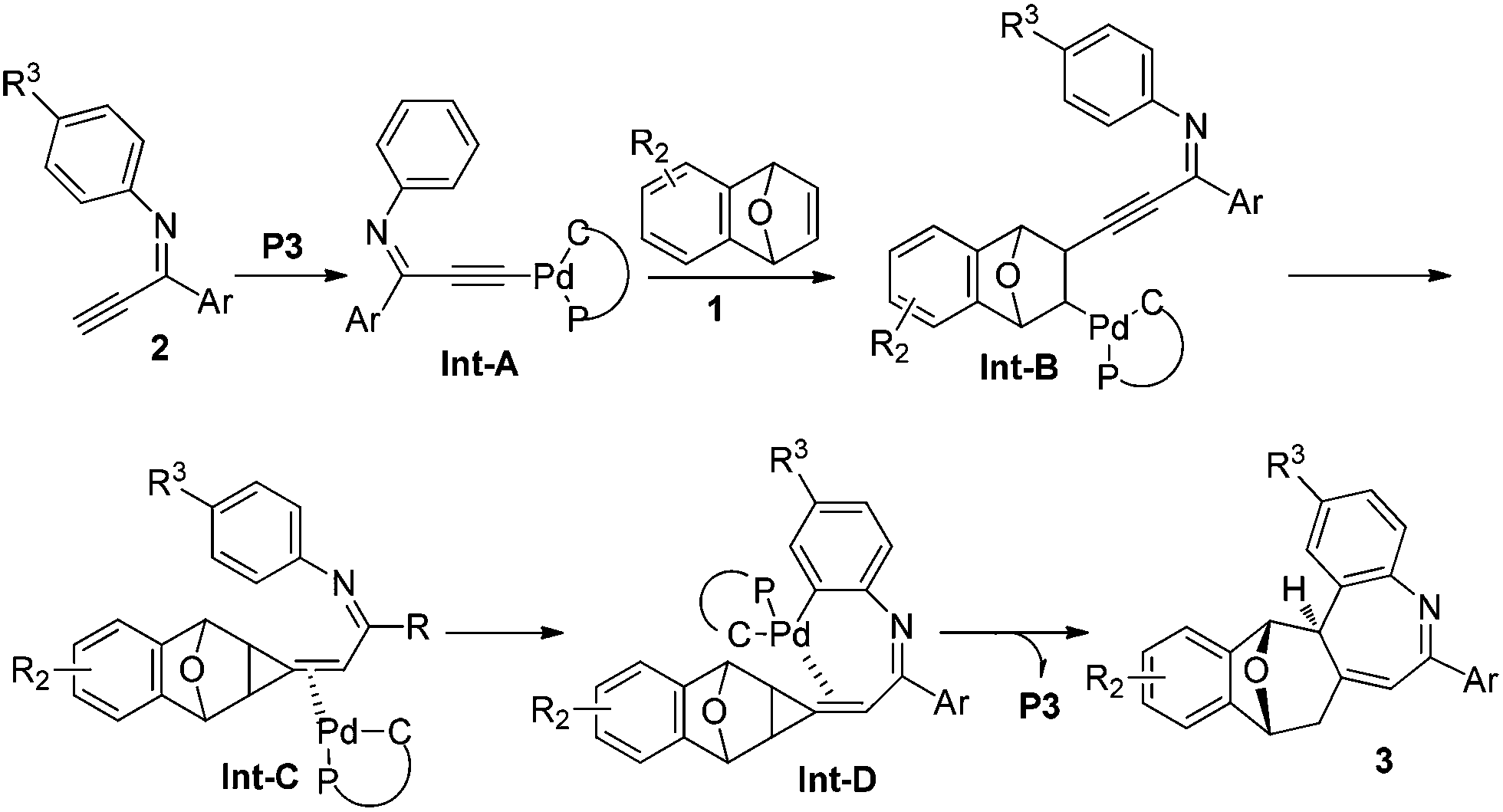 | ||
| Scheme 2 Proposed reaction mechanism. | ||
In conclusion, we have developed a new methodology to prepare the six–seven membered [4,5,0] heterocyclic ring system, an important structure in natural products and medicinal chemistry, via the cascade reaction of bicyclic alkenes and alkynyl imines. A possible reaction mechanism was proposed. Also, the unique catalytic activity of palladacycles as well as C–H activation under palladacycle-catalysis, a new catalytic pattern of palladacycles, has been revealed. Further studies for understanding the characteristics of palladacycles as the real catalysts as well as their applications in organic synthesis are ongoing in our lab.
This work was financially supported by the Major Basic Research Development Program (2011CB808706), the National Natural Science Foundation of China (21121062, 21272251, and 21032007), the External Cooperation Program of the Chinese Academy of Sciences, the Technology Commission of Shanghai Municipality and the Croucher Foundation of Hong Kong. This paper is dedicated to Professor Cheng Ye Yuan on the occasion of his 90th birthday.
Notes and references
- For some reviews: (a) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527 CrossRef CAS PubMed; (b) V. Farina, Adv. Synth. Catal., 2004, 346, 1553 CrossRef CAS; (c) R. B. Bedford, Chem. Commun, 2003, 1787 RSC; (d) M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS PubMed; (e) I. P. Beletskaya and A. V. Cheprakov, J. Organomet. Chem., 2004, 689, 4055 CrossRef CAS.
- (a) W. A. Herrmann, C. Brobmer, K. Öfele, C.-P. Reisinger, T. Priermeier, M. Beller and H. Fischer, Angew. Chem., Int. Ed. Engl., 1995, 34, 1844 CrossRef CAS; (b) D. Zim and S. L. Buchwald, Org. Lett., 2003, 5, 2413 CrossRef CAS PubMed; (c) W. A. Herrmann, V. P. W. Böhm and C. P. Reisinger, J. Organomet. Chem., 1999, 576, 23 CrossRef CAS; (d) M. S. Viciu, R. A. Kelly III, E. D. Stevens, F. Nuad, M. Studer and S. P. Nolan, Org. Lett., 2003, 5, 1479 CrossRef CAS PubMed; (e) L. H. Wang, J. Y. Li, X. L. Cui, Y. S. Wu, Z. W. Zhu and Y. J. Wu, Adv. Synth. Catal., 2010, 352, 2002 CrossRef CAS; (f) Q. L. Luo, J. P. Tan, Z. F. Li, W. H. Nan and D. R. Xiao, J. Org. Chem., 2012, 77, 8332 CrossRef CAS PubMed; (g) J. F. Cívicos, D. A. Alonso and C. Nájera, Eur. J. Org. Chem., 2012, 3670 CrossRef; (h) K. Karami, M. Ghasemi and N. H. Naeini, Tetradedron Lett., 2013, 54, 1352 CrossRef CAS; (i) Y. Yang, N. J. Oldenhuis and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 615 CrossRef CAS PubMed.
- (a) S. F. Kirsch, L. E. Overman and M. P. Watson, J. Org. Chem., 2004, 69, 8101 CrossRef CAS PubMed; (b) M. P. Watson, L. E. Overman and R. G. Bergman, J. Am. Chem. Soc., 2007, 129, 5031 CrossRef CAS PubMed; (c) H. Nomura and C. J. Richards, Chem. – Eur. J., 2007, 13, 10216 CrossRef CAS PubMed; (d) T. K. Zhang, D. L. Mo, L. X. Dai and X. L. Hou, Org. Lett., 2008, 10, 5337 CrossRef CAS PubMed; (e) M. Weber, S. Jautze, W. Frey and R. Peters, J. Am. Chem. Soc., 2010, 132, 12222 CrossRef CAS PubMed; (f) H. Nomura and C. J. Richards, Chem. – Asian J., 2010, 5, 1726 CrossRef CAS PubMed; (g) C. Pi, Y. Li, X. Cui, H. Zhang, Y. Han and Y. Wu, Chem. Sci., 2013, 4, 2675 RSC.
- For some reviews: (a) J. Q. Yu, R. Giri and X. Chen, Org. Biomol. Chem., 2006, 4, 4041 RSC; (b) O. Daugulis, V. G. Zaitsev, D. Shabashov, Q. N. Pham and A. Lazareva, Synlett, 2006, 3382 CrossRef CAS; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- (a) A. Tenaglia, L. Giordano and G. Buono, Org. Lett., 2006, 8, 4315 CrossRef CAS PubMed; (b) P. He, Y. Lu, C. G. Dong and Q. S. Hu, Org. Lett., 2007, 9, 343 CrossRef CAS PubMed; (c) Y. Suzuma, T. Yamamoto, T. Ohta and Y. Ito, Chem. Lett., 2007, 36, 470 CrossRef CAS; (d) D. Gatineau, L. Giordano and G. Buono, J. Am. Chem. Soc., 2011, 133, 10728 CrossRef CAS PubMed; (e) A. Tenaglia, K. L. Jeune, L. Giordano and G. Buono, Org. Lett., 2011, 13, 636 CrossRef CAS PubMed.
- (a) T. K. Zhang, K. Yuan and X.-L. Hou, J. Organomet. Chem., 2007, 692, 1912 CrossRef CAS; (b) T. K. Zhang, D.-L. Mo, X.-L. Hou and L.-X. Dai, Org. Lett., 2008, 10, 3689 CrossRef CAS PubMed; (c) X.-J. Huang, D.-L. Mo, C.-H. Ding and X.-L. Hou, Synlett, 2011, 943 CAS; (d) G.-C. Ge, D.-L. Mo, C.-H. Ding, X.-L. Hou and L.-X. Dai, Org. Lett., 2012, 14, 5756 CrossRef CAS PubMed; (e) D.-L. Mo, B. Chen, C.-H. Ding, L.-X. Dai, G.-C. Ge and X.-L. Hou, Organometallics, 2013, 32, 4465 CrossRef CAS.
- (a) A. R. Pinder, Nat. Prod. Rep., 1989, 6, 67 RSC; (b) S. M. Grant and D. Faulds, Drugs, 1992, 43, 873 CrossRef CAS PubMed; (c) D. O'Hagan, Nat. Prod. Rep., 1997, 14, 637 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 983615. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00030g |
| This journal is © the Partner Organisations 2014 |