Vanadium-catalyzed C(sp3)–H fluorination reactions†
Ji-Bao
Xia
,
Yuyong
Ma
and
Chuo
Chen
*
Division of Chemistry, Department of Biochemistry, The University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, Texas 75390-9038, USA. E-mail: Chuo.Chen@UTSouthwestern.edu
First published on 25th March 2014
Abstract
Vanadium(III) oxide catalyzes the direct fluorination of C(sp3)–H groups with Selectfluor. This reaction is operationally simple. The catalyst and the reaction by-product can be removed easily by filtration. Using this method, a fluorine atom can be introduced to the tertiary position of 1,4-cineole and L-menthone selectively.
Nature uses catalytic C–H oxidation reactions extensively to functionalize small molecules. Studying the structures and reactivities of hydroxylases has inspired the development of various iron, manganese, and copper catalysts for C–H oxidation reactions.1 Their reactivities often complement those of the palladium and rhodium catalysts commonly used for constructing C–C bonds.2 Vanadium-complexes are well-known for their ability to transfer an oxygen or a halogen atom to olefins.3 However, vanadium-catalyzed C–H oxidation has not been well-studied.4 Recently, we developed an efficient vanadium catalyst for selective benzylic C–H oxygenation with no competing aromatic oxidation.5 We report herein a vanadium catalyst system for C(sp3)–H fluorination (Fig. 1).
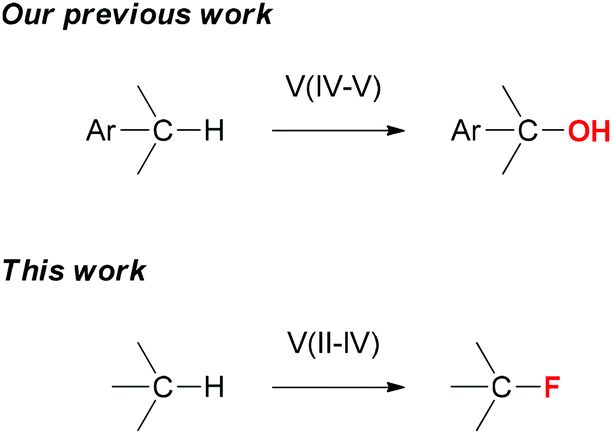 | ||
| Fig. 1 Development of vanadium-catalyzed oxidative C–H functionalization reactions. | ||
Mimoun reported in 1983 that vanadium complexes could catalyze C–H oxygenation through a radical mechanism.6 This area of research was later expanded by Shul'pin, Pombeiro, and others.7 While several efficient vanadium catalysts have been developed for C–H oxygenation, it has not been shown that vanadium complexes can catalyze C–H halogenation.
We seek to expand the scope of the vanadium-catalyzed C–H oxidation to C–H fluorination as incorporating fluorine atoms into small-molecules often improves their physical and biological properties.8 While there are many efficient methods for introducing fluorine atoms through functional group transformations,9 there are only a few studies of catalytic C–H fluorination reactions. The first example was reported by the Sanford group in 2006.10 They successfully developed a palladium(II/IV) catalyst system for catalyzing the quinoline/pyridine-directed benzylic C(sp3)–H fluorination and ortho C(sp2)–H fluorination. Subsequently, the Yu group developed a versatile palladium catalyst system for directed ortho C(sp2)–H fluorination in 2009.11 Methods for catalyzing C(sp3)–H fluorination without using a directing group have also emerged in literatures recently. The Groves group first developed a manganese catalyst system in 2012.12 The Lectka group then published a copper and an iron catalyst system.13 Subsequently, we disclosed a ketone-catalyzed photolytic method14 and the Inoue group an N-oxyl radical-catalyzed method.15 More recently, the Doyle group developed a palladium-catalyzed allylic C(sp3)–H fluorination reaction16 and the Hartwig group a silver-promoted oxidative method for C(sp2)–H fluorination of heterocycles.17 However, there is no report of vanadium-catalyzed C–H fluorination.
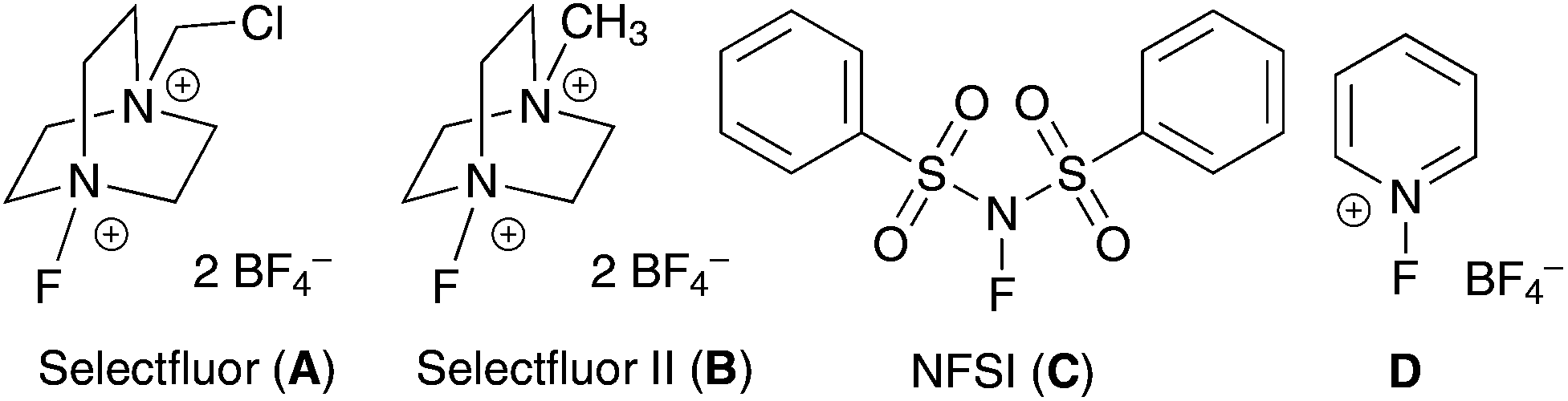
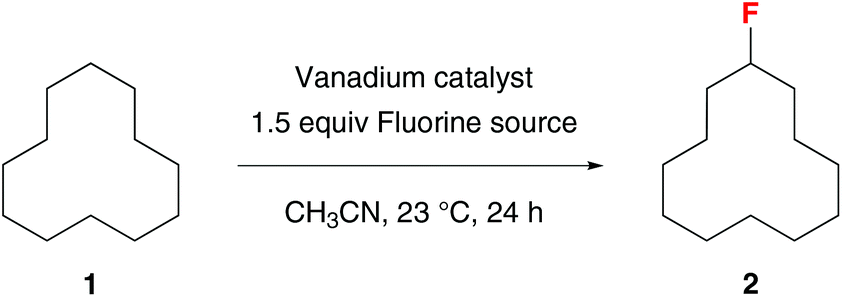
Because vanadium complexes are known to catalyze C–H oxygenation and olefin halogenation, we believe that they can also promote C–H fluorination. To search for an effective vanadium catalyst for C–H fluorination, we examined the reactivities of a series of vanadium complexes using cyclododecane (1) as the substrate and Selectfluor as the standard fluorinating reagent (Table 1). Unlike our previous benzylic C–H oxygenation reactions,5 vanadium(V) complexes did not catalyze the fluorination of 1 (entries 1–4), and vanadium(IV) complexes gave only a trace amount of fluorocyclodecane (2) (entries 5–7). In contrast, vanadium(III) and vanadium(II) complexes were more reactive (entries 8–12). For example, fluorination of 1 in the presence of 20 mol% V(acac)3 and 20 mol% Cp2V led to the formation of 2 in 21% and 13% yields, respectively (entries 10 and 12). Among all the vanadium complexes examined, vanadium(III) oxide (V2O3) was the most effective catalyst, giving 2 in 65% isolated yield at 10 mol% loading (20 mol% by vanadium) (entry 11). Although 2 could be further fluorinated under the reaction conditions, there were only ∼5% of the difluorination products based on 19F NMR analyses (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 monofluorination vs. difluorination). In terms of the fluorinating reagent, Selectfluor (F-TEDA, A) was the only effective fluorine atom donor. There was no reaction when using Selectfluor II (B), NFSI (C), or N-fluoropyridinium salts (D) as the source of fluorine (entries 13–15). We have also confirmed that a vanadium catalyst is required for this reaction (entry 16),18 and acetonitrile is the only suitable solvent.
:1 monofluorination vs. difluorination). In terms of the fluorinating reagent, Selectfluor (F-TEDA, A) was the only effective fluorine atom donor. There was no reaction when using Selectfluor II (B), NFSI (C), or N-fluoropyridinium salts (D) as the source of fluorine (entries 13–15). We have also confirmed that a vanadium catalyst is required for this reaction (entry 16),18 and acetonitrile is the only suitable solvent.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst loading | Catalyst | Fluorine source | Yieldb |
| a Conditions: 1 (0.2 mmol), catalyst (0.04 mmol, or 0.02 mmol for V2O5 and V2O3), Selectfluor (0.3 mmol), CH3CN (2 mL), 23 °C. b Based on crude 19F NMR spectra using C6H5F as the external standard. c Isolated yield in parenthesis. | ||||
| 1 | 10 mol% | V2O5 | A | 0% |
| 2 | 20 mol% | VO(OiPr)3 | A | 0% |
| 3 | 20 mol% | VO(OSiPh3)3 | A | 0% |
| 4 | 20 mol% | VOF3 | A | 0% |
| 5 | 20 mol% | V(O)SO4 | A | 0% |
| 6 | 20 mol% | VO2 | A | <5% |
| 7 | 20 mol% | Cp2VCl2 | A | <5% |
| 8 | 20 mol% | VF3 | A | <5% |
| 9 | 20 mol% | VBr3 | A | <5% |
| 10 | 20 mol% | V(acac)3 | A | 21% |
| 11 | 10 mol% | V2O3 | A | 73% (65%)c |
| 12 | 20 mol% | Cp2V | A | 13% |
| 13 | 10 mol% | V2O3 | B | 0% |
| 14 | 10 mol% | V2O3 | C | 0% |
| 15 | 10 mol% | V2O3 | D | 0% |
| 16 | 10 mol% | — | A | 0% |
|
||||
The substrate scope of this vanadium(III)-oxide-catalyzed C–H fluorination reaction is shown in Table 2. Fluorination of cyclohexane and cyclodecane gave 3 and 4 in good yields. 1-Adamantanol and 1-adamantanecarboxylic acid reacted selectively at the tertiary positions to give 5 and 6 in 74% and 70% isolated yields, respectively. We observed only less than 5% of the difluorination products. The second fluorination also occurred at the tertiary positions of adamantane, suggesting that the tertiary C–H groups are significantly more reactive than the secondary C–H groups. Consistently, fluorination of 2-pentanone gave 7 in 47% NMR yield while fluorination of 4-methyl-2-pentanone and isovaleric acid gave 8 and 9 in 78% NMR yield and 63% isolated yield, respectively. In contrast to electrophilic fluorination reactions, this vanadium-catalyzed reaction did not functionalize the α-position of carbonyl compounds. The presence of an aromatic ester group significantly affected the reaction efficiency. While fluorination of methyl isovalerate gave 10 in 85% NMR yield, reaction of phenyl isovalerate gave 11 in only 12% NMR yield. This method could also be used to prepare β-fluoro-α-amino ester 12 directly from N-phthaloyl valine methyl ester in 46% isolated yield.
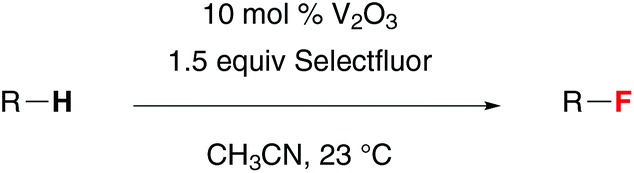
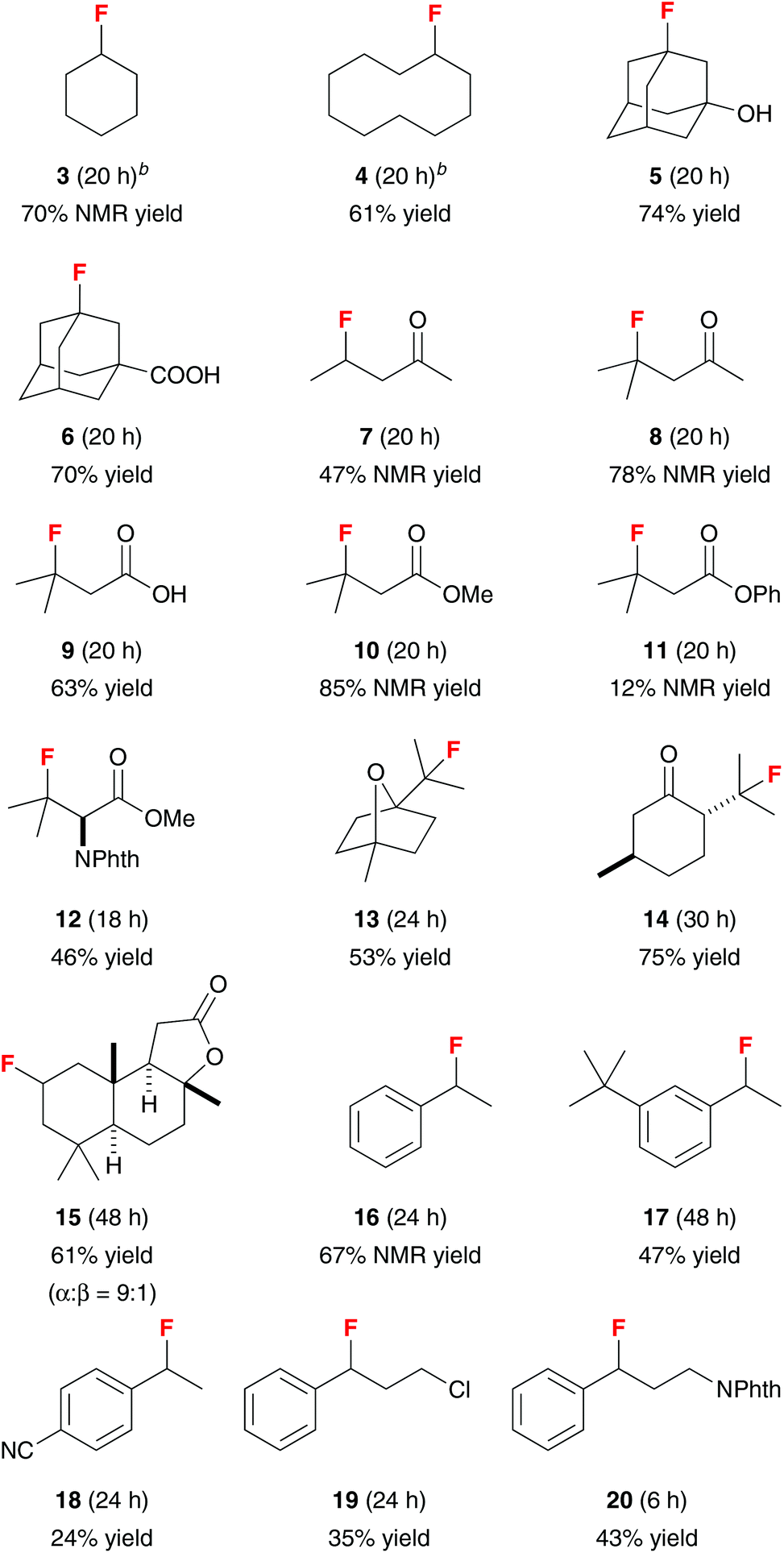
The utility of this reaction was further demonstrated by the selective fluorination of monoterpenes 1,4-cineole and L-menthone. The C–H fluorination preferentially occurred at the tertiary positions to give 13 and 14 in 53% and 75% isolated yields, respectively. Vanadium(III) oxide also catalyzed the fluorination of sesquiterpenoid sclareolide with improved efficiency and selectivity compared to the manganese-porphyrin catalyst system.12a The C-2 fluoride 15 (α:β = 9:1) was obtained in 61% yield along with the C-3 α-fluoride in 15% yield (C-2:C-3 = 4:1) as an inseparable mixture of isomers.
Selective benzylic fluorination could also be achieved. Although vanadium(III) bromide and vanadium(III) acetoacetate were effective catalysts for benzylic fluorination at 5 mol% loading, bromine atom-transfer occurred with vanadium(III) bromide, and the yields obtained with vanadium(III) acetoacetate were irreproducible due to facile ligand fluorination. In contrast, vanadium(III) oxide provided reliable results despite higher catalyst loading. Under the standard reaction conditions (10 mol% V2O3), benzylic fluorides 16 and 17 could be obtained in 67% NMR yield and 47% isolated yield, respectively. However, the fluorine atom of benzylic fluorides could eliminate under the reaction conditions via a SN1-type mechanism, leading to low yields of benzylic fluorides for the more electron-rich substrates. In contrast, the electron-deficient 4-ethylbenzonitrile was rather unreactive, giving 18 in only 24% isolated yield. Finally, fluorination of propylbenzene with a chlorine or protected nitrogen atom at the terminal position of the side-chain gave 19 and 20 in 35% and 43% isolated yields, respectively.
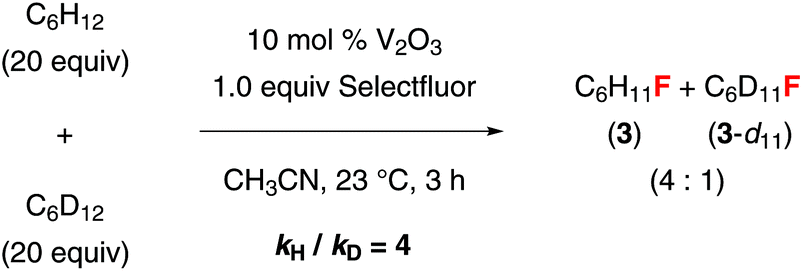
We have also conducted some preliminary mechanistic studies. A competition experiment using a 1:1 ratio of cyclohexane and cyclohexane-d12 gave a 4:1 mixture of 3 and 3-d11 (Fig. 2). The primary kinetic isotope effect (KIE) (kH/kD = 4) indicates that C–H abstraction is the rate-limiting step. This fluorination reaction is highly oxygen-sensitive, suggesting that it may proceed through a radical mechanism. While the nature of the active catalyst is not clear, we believe that a vanadium(II/III) or (III/IV) cycle rather than a vanadium(III/V) cycle is involved because vanadium(V) complexes could not promote this reaction. It is also likely that vanadium(III) oxide was first oxidized to a vanadium(IV) species, which served as the active catalyst. The fluorination reaction proceeded equally well in wet acetonitrile. Since we did not observe any Ritter reaction products or ketones, we believed that the alkyl radical initially generated was not oxidized to a carbocation before being trapped by a fluoride. However, there was no reaction when using water as a cosolvent. It is likely that the addition of a large excess amount of water deactivated the catalyst by altering its structure.
 | ||
| Fig. 2 KIE study of the vanadium-catalyzed C–H fluorination. | ||
In summary, we have found that commercially available vanadium(III) oxide can catalyze the direct conversion of a C–H group to C–F. This heterogeneous catalyst can be easily removed by filtration along with the Selectfluor by-product H-TEDA. This operationally simple method provides improved efficiency for C–H fluorination at non-benzylic positions compared to existing methods. We are now exploring further utilities of vanadium complexes in catalytic oxidation reactions.
Experimental section
General procedure for the vanadium(III) oxide-catalyzed C(sp3)–H fluorination reaction
To a 4 mL clear vial charged with vanadium(III) oxide (V2O3, 3.0 mg, 0.02 mmol, 10 mol%) and Selectfluor (106.3 mg, 0.3 mmol, 1.5 equiv.) was added anhydrous acetonitrile (2.0 mL), and the reaction substrate (0.2 mmol, 1.0 equiv.). The reaction mixture was then degassed three times by freeze–pump–thaw cycles and stirred at room temperature for 6–48 h. Upon completion, the reaction mixture was poured into diethyl ether (20 mL), filtered, concentrated and purified by silica gel flash column chromatography using diethyl ether–pentane as the eluent.Acknowledgements
Financial support was provided by NIH/NIGMS (R01-GM079554) and the Welch Foundation (I-1596).Notes and references
- (a) L. Que Jr. and W. B. Tolman, Angew. Chem., Int. Ed., 2002, 41, 1114–1137 CrossRef; (b) L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed; (c) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed; (d) M. Bordeaux, A. Galarneau and J. Drone, Angew. Chem., Int. Ed., 2012, 51, 10712–10723 CrossRef CAS PubMed; (e) J. Eames and M. Watkinson, Angew. Chem., Int. Ed., 2001, 40, 3567–3571 CrossRef CAS; (f) T. Punniyamurthy and L. Rout, Coord. Chem. Rev., 2008, 252, 134–154 CrossRef CAS PubMed; (g) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed; see also: (h) Y. Ishihara and P. S. Baran, Synlett, 2010, 1733–1745 CAS; (i) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed.
- (a) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (b) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2011, 45, 936–946 CrossRef PubMed; (c) L. Boisvert and K. I. Goldberg, Acc. Chem. Res., 2011, 45, 899–910 CrossRef PubMed; (d) B. G. Hashiguchi, S. M. Bischof, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2011, 45, 885–898 CrossRef PubMed; (e) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed; (f) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2011, 45, 911–922 CrossRef PubMed; (g) J. F. Hartwig, Acc. Chem. Res., 2011, 45, 864–873 CrossRef PubMed; (h) M. Zhou and R. H. Crabtree, Chem. Soc. Rev., 2011, 40, 1875–1884 RSC; (i) M. C. Haibach, S. Kundu, M. Brookhart and A. S. Goldman, Acc. Chem. Res., 2011, 45, 947–958 CrossRef PubMed.
- (a) K. B. Sharpless and T. R. Verhoeven, Aldrichimica Acta, 1979, 12, 63–74 CAS; (b) C. S. Neumann, D. G. Fujimori and C. T. Walsh, Chem. Biol., 2008, 15, 99–109 CrossRef CAS PubMed; (c) F. H. Vaillancourt, E. Yeh, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef CAS PubMed; (d) A. Butler, M. J. Clague and G. E. Meister, Chem. Rev., 1994, 94, 625–638 CrossRef CAS; (e) A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937–1944 CrossRef CAS.
- (a) T. Hirao, Chem. Rev., 1997, 97, 2707–2724 CrossRef CAS PubMed; (b) V. Conte, F. Di Furia and G. Licini, Appl. Catal., A, 1997, 157, 335–361 CrossRef CAS; (c) A. G. J. Ligtenbarg, R. Hage and B. L. Feringa, Coord. Chem. Rev., 2003, 237, 89–101 CrossRef CAS; (d) J. A. L. da Silva, J. J. R. F. da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2011, 255, 2232–2248 CrossRef CAS PubMed.
- J.-B. Xia, K. W. Cormier and C. Chen, Chem. Sci., 2012, 3, 2240–2245 RSC.
- (a) H. Mimoun, P. Chaumette, M. Mignard and L. Saussine, New J. Chem., 1983, 7, 467–475 CAS; (b) H. Mimoun, L. Saussine, E. Daire, M. Postel, J. Fischer and R. Weiss, J. Am. Chem. Soc., 1983, 105, 3101–3110 CrossRef CAS; (c) M. Bonchio, V. Conte, F. Di Furia, G. Modena and S. Moro, J. Org. Chem., 1994, 59, 6262–6267 CrossRef CAS; (d) E. P. Talsi and K. V. Shalyaev, J. Mol. Catal., 1994, 92, 245–255 CrossRef CAS.
- (a) I. I. Moiseev, A. E. Gekhman and D. I. Shishkin, New J. Chem., 1989, 13, 683–690 CAS; (b) G. B. Shul'pin and G. Süss-Fink, J. Chem. Soc., Perkin Trans. 2, 1995, 1459–1463 RSC; (c) T. F. S. Silva, K. V. L. M. V. Kirillova, M. F. G. da Silva, L. M. D. R. S. Martins and A. J. L. Pombeiro, Adv. Synth. Catal., 2010, 352, 171–187 CrossRef CAS PubMed; (d) K. Kamata, K. Yonehara, Y. Nakagawa, K. Uehara and N. Mizuno, Nat. Chem., 2010, 2, 478–483 CrossRef CAS PubMed.
- (a) H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed; (b) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (d) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- (a) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661–1664 CrossRef CAS PubMed; (b) E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639–642 CrossRef CAS PubMed; (c) V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681–1684 CrossRef CAS PubMed; (d) J. J. Topczewski, T. J. Tewson and H. M. Nguyen, J. Am. Chem. Soc., 2011, 133, 19318–19321 CrossRef CAS PubMed; (e) M. Rueda-Becerril, C. C. Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS PubMed; (f) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS PubMed; (g) Z. Li, L. Song and C. Li, J. Am. Chem. Soc., 2013, 135, 4640–4643 CrossRef CAS PubMed; (h) C. Zhang, Z. Li, L. Zhu, L. Yu, Z. Wang and C. Li, J. Am. Chem. Soc., 2013, 135, 14082–14085 CrossRef CAS PubMed; (i) Z. Li, C. Zhang, L. Zhu, C. Liu and C. Li, Org. Chem. Front., 2014, 1, 100–104 RSC; (j) T. J. Barker and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13588–13591 CAS; (k) S. Suzuki, Y. Kitamura, S. Lectard, Y. Hamashima and M. Sodeoka, Angew. Chem., Int. Ed., 2012, 51, 4581–4585 CrossRef CAS PubMed; (l) W. Kong, P. Feige, T. d. Haro and C. Nevado, Angew. Chem., Int. Ed., 2013, 52, 2469–2473 CrossRef CAS PubMed; (m) J. R. Wolstenhulme, J. Rosenqvist, O. Lozano, J. Ilupeju, N. Wurz, K. M. Engle, G. W. Pidgeon, P. R. Moore, G. Sandford and V. Gouverneur, Angew. Chem., Int. Ed., 2013, 52, 9796–9800 CrossRef CAS PubMed; (n) N. A. Cochrane, H. Nguyen and M. R. Gagne, J. Am. Chem. Soc., 2013, 135, 628–631 CrossRef CAS PubMed; (o) X. Mu, H. Zhang, P. Chen and G. Liu, Chem. Sci., 2014, 5, 275–280 RSC.
- (a) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134–7135 CrossRef CAS PubMed; (b) K. B. McMurtrey, J. M. Racowski and M. S. Sanford, Org. Lett., 2012, 14, 4094–4097 CrossRef CAS PubMed; (c) J. M. Racowski, J. B. Gary and M. S. Sanford, Angew. Chem., Int. Ed., 2012, 51, 3414–3417 CrossRef CAS PubMed.
- (a) X. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520–7521 CrossRef CAS PubMed; (b) K. S. L. Chan, M. Wasa, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 9081–9084 CrossRef CAS PubMed.
- (a) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard III and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed; (b) W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024–6027 CrossRef CAS PubMed.
- (a) S. Bloom, C. R. Pitts, D. C. Miller, N. Haselton, M. G. Holl, E. Urheim and T. Lectka, Angew. Chem., Int. Ed., 2012, 51, 10580–10583 CrossRef CAS PubMed; (b) S. Bloom, C. R. Pitts, R. Woltornist, A. Griswold, M. G. Holl and T. Lectka, Org. Lett., 2013, 15, 1722–1724 CrossRef CAS PubMed; (c) S. Bloom, J. L. Knippel and T. Lectka, Chem. Sci., 2014, 5, 1175–1178 RSC.
- J.-B. Xia, C. Zhu and C. Chen, J. Am. Chem. Soc., 2013, 135, 17494–17500 CrossRef CAS PubMed.
- Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160–2163 CrossRef CAS PubMed.
- M.-G. Braun and A. G. Doyle, J. Am. Chem. Soc., 2013, 135, 12990–12993 CrossRef CAS PubMed.
- P. S. Fier and J. F. Hartwig, Science, 2013, 342, 956–960 CrossRef CAS PubMed.
- (a) R. D. Chambers, M. Parsons, G. Sandford and R. Bowden, Chem. Commun., 2000, 959–960 RSC; (b) R. D. Chambers, A. M. Kenwright, M. Parsons, G. Sandford and J. S. Moilliet, J. Chem. Soc., Perkin Trans. 1, 2002, 2190–2197 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00057a |
| This journal is © the Partner Organisations 2014 |