 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of multivalency as an organization principle for the efficient synthesis of doubly and triply threaded amide rotaxanes†
Lena
Kaufmann
a,
Nora L.
Traulsen
a,
Andreas
Springer
a,
Hendrik V.
Schröder
a,
Toni
Mäkelä
b,
Kari
Rissanen
b and
Christoph A.
Schalley
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195, Berlin, Germany. E-mail: c.schalley@fu-berlin.de
bDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, P.O. Box 35, 40014 Jyväskylä, Finland
First published on 18th April 2014
Abstract
Mono-, di- and trivalent pseudorotaxanes with tetralactam macrocycle hosts and axles containing diamide binding stations as the guests have been synthesised. Their threading behaviour was analyzed in detail by NMR experiments and isothermal titration calorimetry. An X-ray crystal structure of the monovalent pseudorotaxane confirms the binding motif. Double mutant cycle analysis provides the effective molarities and insight into the chelate cooperativity of multivalent binding. While the second binding event in a trivalent pseudorotaxane exhibits a slightly positive cooperativity, the third binding is nearly non-cooperative. Nevertheless, the enhanced binding affinities resulting from the multivalent interaction are the basis for a highly efficient synthesis of di- and trivalent rotaxanes through stoppering the axle termini by “click” chemistry. Evidence for the multiply threaded geometry comes from NMR spectroscopy as well as tandem mass-spectrometric fragmentation experiments of mass-selected rotaxane ions in the gas phase. Furthermore, the trivalent rotaxane can be controlled by external stimuli (chloride addition and removal) which lead to an elevator-type movement of the wheel along the axle.
Introduction
In nature, multivalency is a key principle to establish strong yet reversible Velcro-like interactions. It is therefore of high interest in biology and biochemistry1–15 and describes molecular recognition phenomena between two binding partners, which involve more than one binding site.16 Synthetic multivalent supramolecular complexes have the advantage that they can be changed systematically at will by modifying for example the number of interaction sites or the flexibility and length of spacers connecting them.17–21 This promises great potential for the analysis of multivalent interactions in more detail and thus for a more profound understanding of the different contributions to the overall binding strengths. In contrast to monovalent systems, where only bound or non-bound scenarios exist, more diverse binding situations occur in that partially bound states are possible.Here, we report multivalency as a tool for the efficient synthesis of doubly and triply threaded (pseudo)rotaxanes of the amide type.22–26 Pseudorotaxanes are the precursors of mechanically interlocked molecules like rotaxanes and catenanes, which are fascinating not only because of their topology, but also for playing a major role in the construction of molecular machines.27–37 An intriguing example combining multivalency with a stimuli-controlled molecular shuttling motion is the “molecular elevator” reported by Stoddart et al.,38–40 a trivalent crown-ammonium rotaxane, in which the treatment with acid and base induces a motion of the trivalent wheel component relative to the trivalent axle.
Understanding the binding in multivalent pseudorotaxanes in detail is also pivotal for the synthesis of multiply interlocked molecules as they are their immediate precursors. Their synthesis involves template effects based on weak interactions like metal-coordination,30,41–57 charge transfer interactions58–71 or hydrogen bonding.72–96 When multiply interlocked rotaxanes are to be made, all binding sites need to be interlocked. Multivalency and cooperative binding are a key concept to reach this goal and help avoiding complex mixtures of different partially interlocked rotaxane isomers. In case of multivalent complexes, spacer length and rigidity have a major impact on the binding situation. A recent example shows the spacer adding substantially to the formation of a stable divalent crown-ammonium pseudorotaxane and spacer–spacer interactions to increase the cooperativity of binding significantly.97
Results and discussion
Conceptual considerations and the synthesis of pseudorotaxane components
One class of well-known rotaxane or catenane wheels are tetralactam macrocycles (TLM).81,94 Their four converging amide groups form hydrogen bonds to suitable guest molecules such as dicarbonyl compounds in aprotic and not too strongly competitive solvents like chloroform or dichloromethane.79,88,93,98–110 For the present study, tetralactam macrocycles and diamide axles were chosen as the pseudorotaxane components. As rigid spacers are expected to improve the multivalency-induced binding due to lower entropic losses arising from conformational fixation, these binding motifs were envisaged to be connected by aromatic rings and triple bonds only. At the same time, we aimed at a converging synthesis based on monovalent building blocks that are easy to connect to the same spacer units for hosts (Scheme 1) and guests (Scheme 2) through cross-coupling reactions.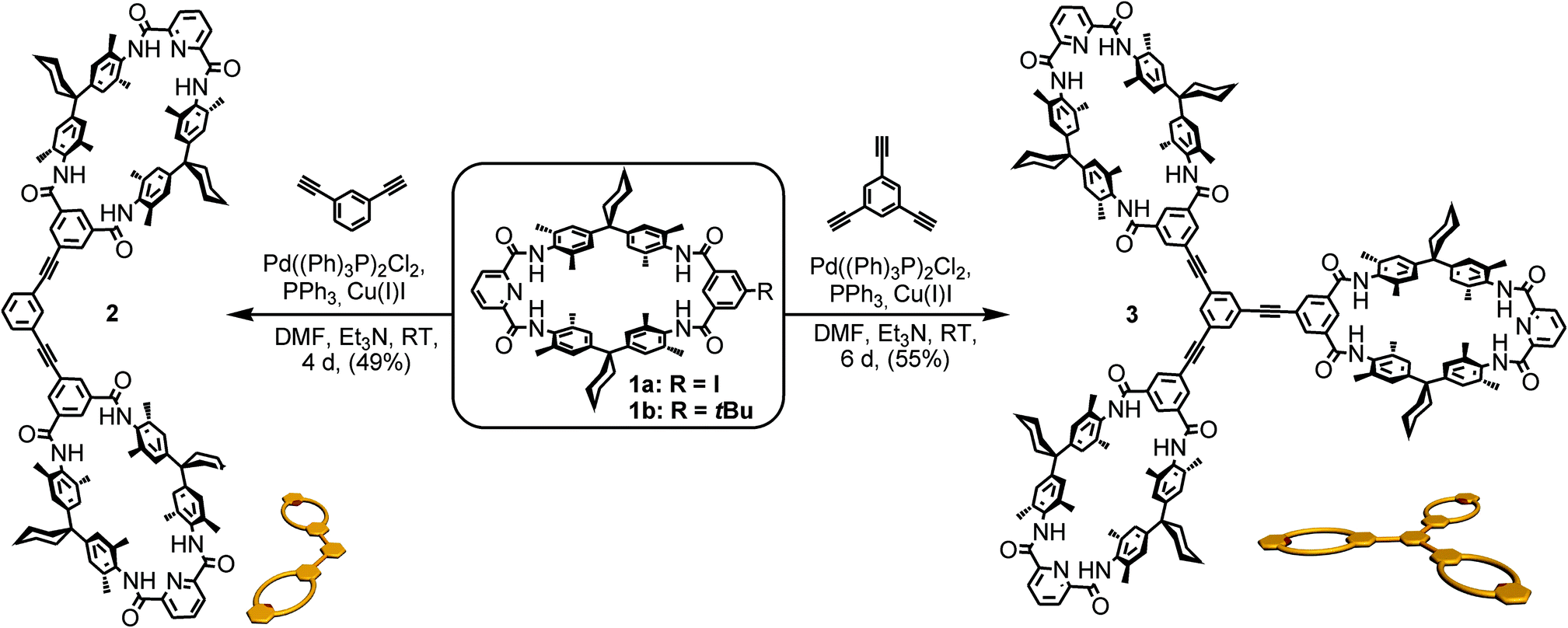 | ||
| Scheme 1 Synthesis of di- and trivalent hosts 2 and 3 from the iodide-substituted tetralactam macrocycle 1a. Macrocycle 1b was used as the monovalent control as the tert-butyl group increases the solubility of the monovalent macrocycle sufficiently for ITC and NMR experiments to be performed. For simplicity the following figures use the cartoons shown below the structures. | ||
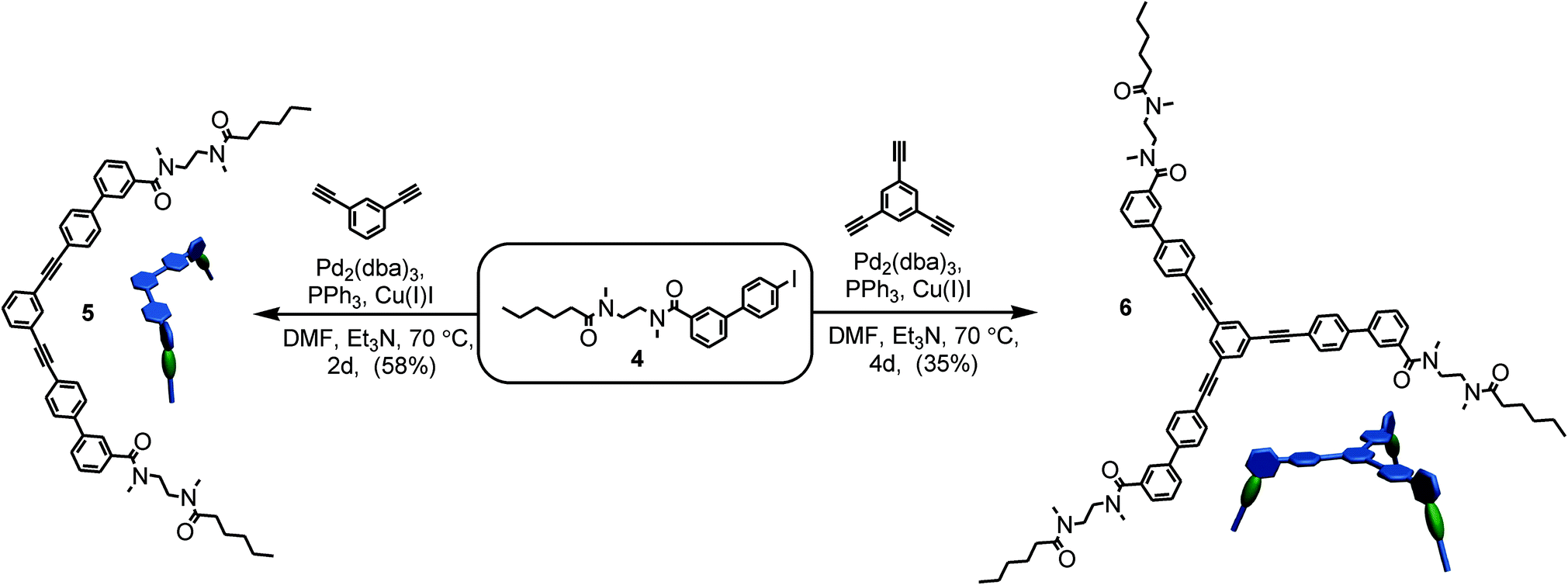 | ||
| Scheme 2 Synthesis of di- and trivalent guests 5 and 6 from the iodide-substituted precursor 4. | ||
TLMs 1a, b were synthesized according to established literature procedures.111 They contain one pyridine-2,6-dicarboxamide unit providing higher yields, increasing solubility, and facilitating NMR spectral analysis.112 Monovalent 1a can easily be converted into di- or trivalent analogues through Sonogashira cross-coupling to suitable spacer molecules (for synthetic details, see ESI†).113 Both multivalent host molecules 2 and 3 are soluble in chloroform, which is decisive for a detailed thermodynamic analysis of the binding situation by isothermal titration calorimetry (ITC).
For the preparation of the corresponding multivalent axles (Scheme 2), monovalent building block 4 was synthesized as the precursor (ESI†). It bears the diamide binding station with a hexanoyl substituent on one side. The alkyl chain increases solubility and the binding constants.102 As all attempts to work with secondary amides failed because of their lower solubility, tertiary amides are used here. Both iodinated precursors can be connected to the same spacers by Sonogashira coupling and according to MM2 force field optimisation (CaChe program package, Fujitsu, Poland) result in complexes with good geometric fits between hosts and guests (Fig. 1).113 In the calculated structures, four N–H⋯O hydrogen bonds are realized between each macrocycle and the corresponding axle station. The spacers are able to interact with optimal stacking distances of 3.54 and 3.57 Å.
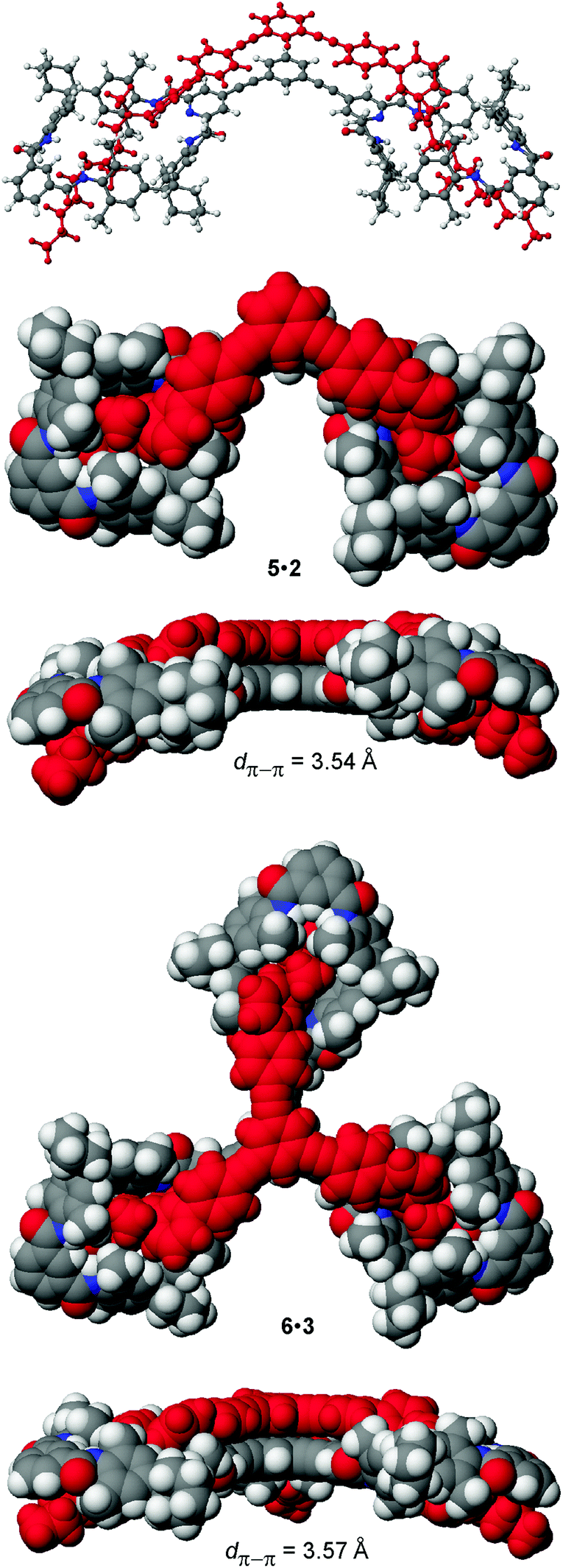 | ||
| Fig. 1 MM2 force field-optimized structures of the divalent and trivalent pseudorotaxanes 5·2 and 6·3. Space-filling representations provide top and side views. | ||
Analysis of the binding behaviour
For monovalent pseudorotaxane 7·1a (Fig. 2, top), a crystal structure (ESI†) provides insight into the binding motif in the solid state. Clearly, axle 7 penetrates the cavity of TLM 1a and is bound by four NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds (Fig. 2, centre). The phenyl part of the axle is more tightly bound to the macrocycle with H⋯O distances of 1.920–1.944 Å and N⋯O distances of 2.824–2.845 Å, while the H⋯O distances for the hexanoyl amide are 2.225–2.237 Å and the N⋯O distances range from 2.995 to 3.019 Å. The N–H⋯O angles are between 159° and 167° and thus the H-bonding interactions are within the expected range compared to other amide rotaxanes and catenanes.101,102,106–108 The packing of the pseudorotaxanes in the crystal exhibits intermolecular NCH3⋯OC interactions from one of the axle's NCH3 H-atoms to the carbonyl oxygen of the adjacent macrocycle forming a weakly H-bonded dimer (Fig. 2, bottom). Due to the tight packing of these dimers, the crystal lattice does not contain any solvent molecules.
C hydrogen bonds (Fig. 2, centre). The phenyl part of the axle is more tightly bound to the macrocycle with H⋯O distances of 1.920–1.944 Å and N⋯O distances of 2.824–2.845 Å, while the H⋯O distances for the hexanoyl amide are 2.225–2.237 Å and the N⋯O distances range from 2.995 to 3.019 Å. The N–H⋯O angles are between 159° and 167° and thus the H-bonding interactions are within the expected range compared to other amide rotaxanes and catenanes.101,102,106–108 The packing of the pseudorotaxanes in the crystal exhibits intermolecular NCH3⋯OC interactions from one of the axle's NCH3 H-atoms to the carbonyl oxygen of the adjacent macrocycle forming a weakly H-bonded dimer (Fig. 2, bottom). Due to the tight packing of these dimers, the crystal lattice does not contain any solvent molecules.
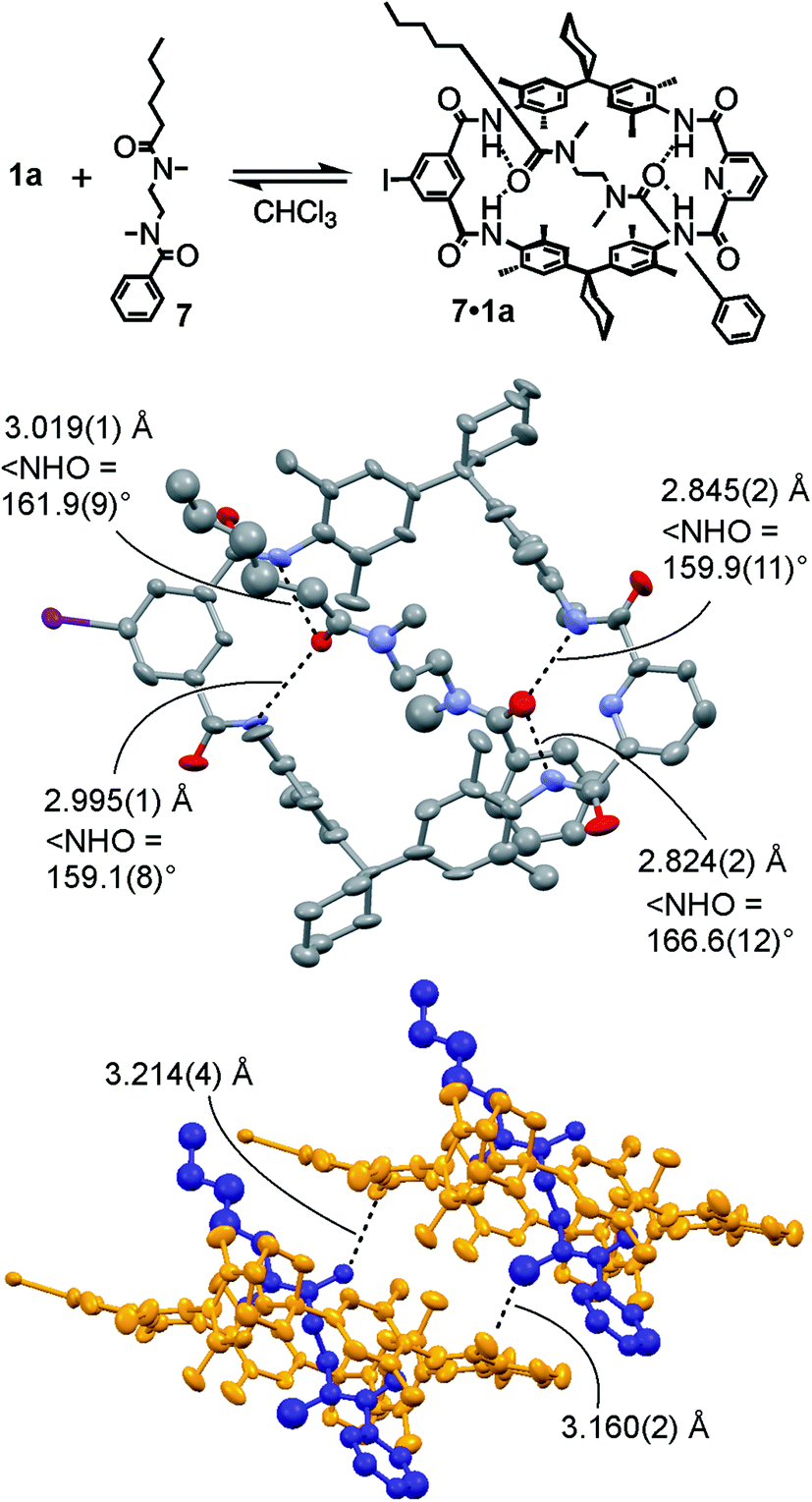 | ||
| Fig. 2 Top: Chemical structure of monovalent pseudorotaxane 7·1a. Centre: Mercury114 plot with anisotropic thermal ellipsoids (50% probability level) of its solid-state structure showing the N–H⋯OC H-bonds (dashed lines) connecting axle and wheel (H-atoms removed for clarity). Bottom: Packing of two 7·1a pseudorotaxanes showing the intermolecular C–H⋯OC interactions. For more crystallographic details, see ESI.† | ||
The formation of the monovalent pseudorotaxane in chloroform is verified by 1H NMR spectroscopy (Fig. 3, right). The typical downfield shifts of the amide signals (>1 ppm) indicate hydrogen bond formation between axle and macrocycle. Also, wheel proton 5 shifts downfield due to the influence of the penetrating axle. The signals of the axle NCH3 and NCH2 protons shift to higher field, indicating that they are positioned in the centre of the macrocycle cavity. One drawback of the tertiary axle amides is the slow interconversion of trans/trans-, cis/trans-, and cis/cis-isomers of the tertiary axle diamides that renders the interpretation of this spectral region somewhat difficult. The axle exchange is fast on the NMR timescale for the monovalent complex and a 1H NMR titration provided a binding constant of K = 3000 M−1.115,116 ITC experiments confirm this result (ESI†). The multivalent complexes exhibit the same typical shifts (trivalent pseudorotaxane: Fig. 3, left; divalent analogue: ESI†) revealing complex formation. In contrast to the monovalent pseudorotaxane, a slow exchange is observed for the di- and trivalent pseudorotaxanes: a second, separate increasing set of signals for the pseudorotaxane is found upon stepwise addition of the axle to the wheel rather than a gradual shift. The second set of signals indicates the pseudorotaxane to have the same symmetry as the two components and thus points to the formation of fully threaded 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes of axle and wheel. The positions of the new signals for the multivalent complexes are even more shifted than those of the monovalent pseudorotaxanes. They are in the same order of magnitude as those of strongly binding axles such as diketopiperazines or squaraines.99,102,117,118
:1 complexes of axle and wheel. The positions of the new signals for the multivalent complexes are even more shifted than those of the monovalent pseudorotaxanes. They are in the same order of magnitude as those of strongly binding axles such as diketopiperazines or squaraines.99,102,117,118
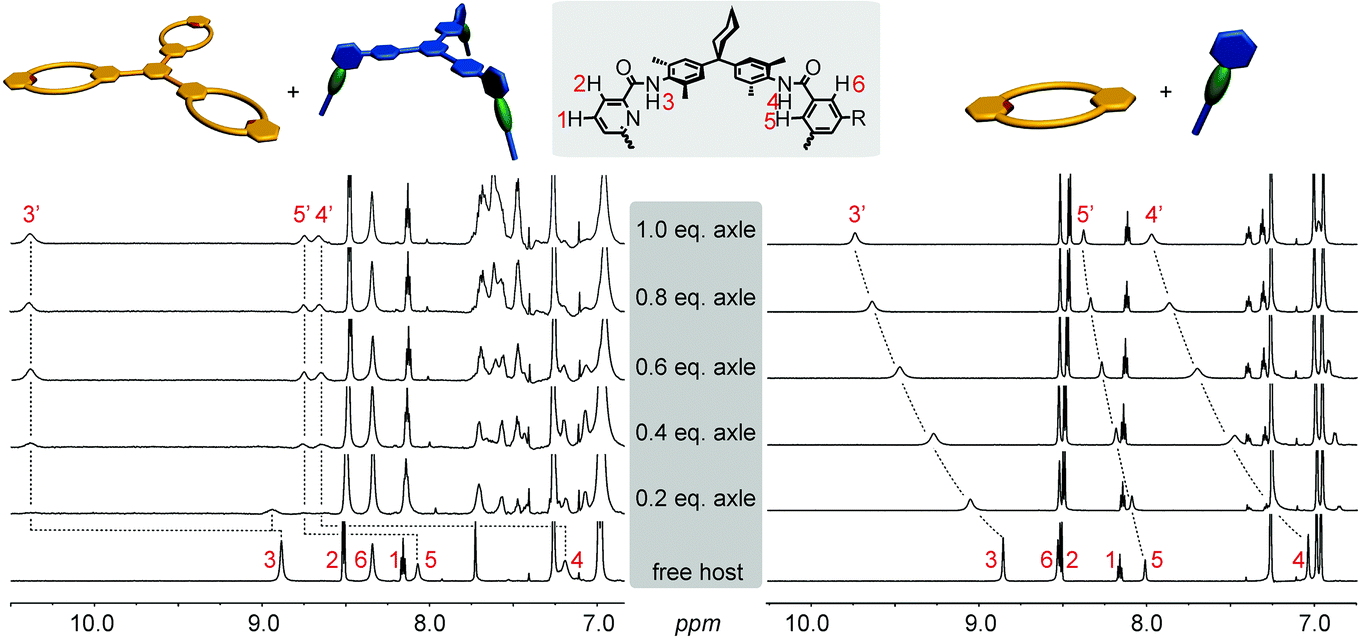 | ||
| Fig. 3 1H NMR titrations (chloroform, 298 K, 700 MHz): trivalent pseudorotaxane 6·3 (left), monovalent 7·1a (right). | ||
While UV/Vis spectra of the trivalent pseudorotaxane are more or less a superposition of the spectra of its components (ESI†) and are thus not very conclusive, the fluorescence of the trivalent axle is significantly affected, when the pseudorotaxane forms. The fluorescence titration curve provides evidence for a 1:1 complex stoichiometry.
Thermodynamic analysis of multivalent binding
For a detailed thermodynamic evaluation of the di- and trivalent binding interactions, an analysis of chelate cooperativity is required. One important factor, in which cooperative binding is expressed, is the effective molarity (EM) that describes the critical concentration above which oligomerisation is preferred over the formation of the closed multivalent complex. If the dimensionless quantity KmonoEM > 1, binding can be regarded as positively cooperative, while KmonoEM < 1 indicates the system to prefer partially bound states over the fully bound one.119 To determine the effective molarities we followed the double mutant cycle (DMC) analysis as proposed by Fersht et al.120 and established for other systems by Anderson et al.,121 Hunter et al.119,122–126 and others. Although there is still a controversy about the best method to assess chelate cooperativity and other ways have been suggested by Ercolani et al.,127–130 the DMC concept represents a useful method for quantification of non-covalent interactions for example in proteins. It waives all effects that are not caused by chelate cooperativity.120,126The DMC concept is depicted for the trivalent pseudorotaxane in Fig. 4 (for the divalent pseudorotaxane, see ESI†). One can consider the DMC as an equilibrium between the four situations A, B, C and D as given in eqn (1). The free enthalpy change ΔΔG for this equilibrium can be described as the difference of the contributions of the individual complexes that are involved (eqn (2)). The equilibrium constant K for this equilibrium can be calculated from the single constants KA, KB, KC and KD (eqn (3)).
| B + C ⇄ A + D | (1) |
| ΔΔG = ΔGA + ΔGD − ΔGB − ΔGC | (2) |
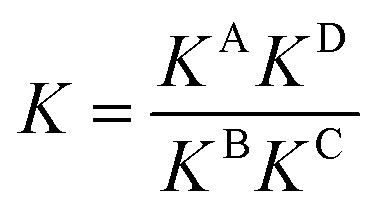 | (3) |
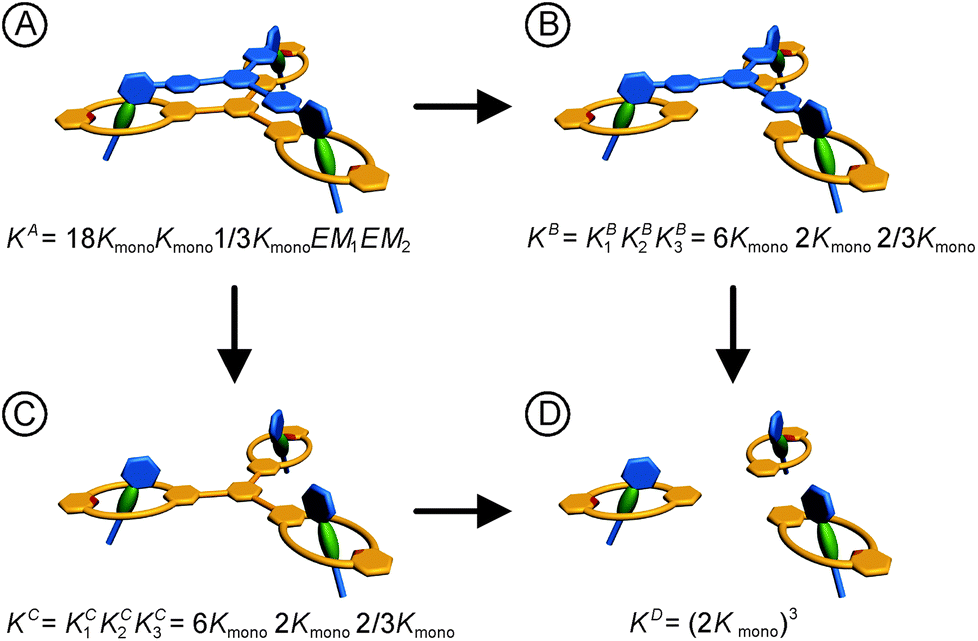 | ||
| Fig. 4 Double mutant cycle analysis for the trivalent pseudorotaxane. The statistical factors are obtained as discussed in the ESI† in detail. For the analogous analysis of the divalent pseudorotaxane, also see the ESI.† | ||
The individual binding constants KA − KD can on one hand be experimentally determined separately. On the other hand, they can be expressed by combinations of the appropriate statistical factors, the monovalent binding constant Kmono and – in the trivalent case – the effective molarities EM1 and EM2 for the two ring closure steps connected to the second and third threading event (Fig. 4 and eqn (4)). The same analysis (ESI†) leads to eqn (5) for the divalent pseudorotaxane.
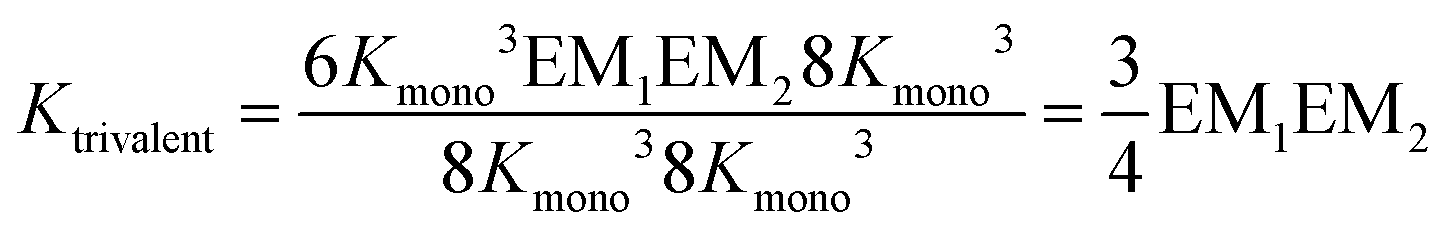 | (4) |
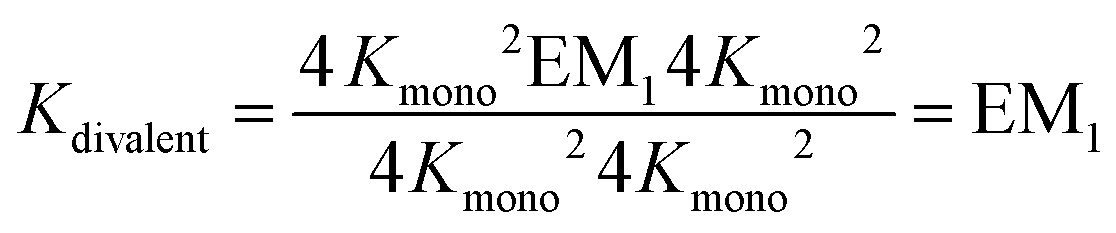 | (5) |
Assuming that EM1 is the same effective molarity in the divalent as well as the trivalent pseudorotaxane, one can differentiate the effective molarities for the second and third binding step in the trivalent pseudorotaxane by separately determining EM1 for the divalent case. Consequently, from a DMC analysis of both, the divalent and trivalent pseudorotaxanes 5·2 and 6·3, one can draw conclusions on the chelate cooperativity operative during the second as well as third binding step separately.
Isothermal titration calorimetry (ITC)131–135 has been used as a very sensitive method to analyse the binding situation of all complexes in greater detail (Table 1 and ESI†). Due to its better solubility, TLM 1b was used as the monovalent host instead of TLM 1a. In a typical ITC experiment, a solution of the axle (5, 6, or 7) was added to a solution of the corresponding wheel (1b, 2, or 3). All experiments were conducted at 298 K in dry CHCl3. For curve fitting, we used 1:1, 2:1 and 3:1 binding models, respectively, for the different combinations of mono- and multivalent axles and wheels. The curve shapes provide the binding constants (K) with errors of about ±10% resulting in errors of ΔG in the order of ±3%. The accuracy of the enthalpy (ΔH) and the resulting entropy (ΔS) values is certainly somewhat lower.97
| K a [M−1] | ΔG [kJ mol−1] | ΔH [kJ mol−1] | −TΔS [kJ mol−1] | EM [mM] | EM Kmono | ||
|---|---|---|---|---|---|---|---|
| a It should be briefly noted that allosteric cooperativity is negligible. This is indicated by the K1 − K3 values obtained for the two/three consecutive binding events occurring for 5·1b2 and 72·2 in the divalent case and 6·1b3 and 73·3 in the trivalent case. As expected from the statistical factors, one expects K1 = 4Kmono, K2 = Kmono for the divalent and K1 = 6Kmono, K2 = 2Kmono, K1 = 2/3Kmono for the trivalent case. These values are close to the experimentally determined ones indicating allosteric cooperativity not to play a significant role here. b For the combination of trivalent axle 6 and monovalent wheel 1b, the titration curve does not yield unambiguous values for ΔH, so that the −TΔS value cannot be calculated precisely from the titration data. | |||||||
| 7·1b | 2640 ± 250 | −19.5 ± 0.3 | −12.2 | −7.2 | |||
| 5·2 | 11800 ± 1200 |
−23.2 ± 0.3 | −41.2 | +18.0 | 0.65 (EM1) | 1.73 | |
| 5·1b2 | K 1 | 10100 ± 1000 |
−22.9 ± 0.3 | −7.6 | −15.3 | ||
| K 2 | 2260 ± 220 | −19.1 ± 0.2 | −2.4 | −16.7 | |||
| 72·2 | K 1 | 10500 ± 1000 |
−23.0 ± 0.3 | −23.7 | +0.7 | ||
| K 2 | 2100 ± 200 | −19.0 ± 0.2 | −4.5 | −14.5 | |||
| 6·3 | 19000 ± 1900 |
−24.4 ± 0.3 | −51.2 | +26.8 | 0.65 (EM1) | 1.73 | |
| 0.32 (EM2) | 0.84 | ||||||
| 6·1b3 | K 1 | 15900 ± 160 |
−23.9 ± 0.3 | ||||
| K 2 | 5280 ± 530 | −21.2 ± 0.3 | |||||
| K 3 | 1760 ± 180 | −18.5 ± 0.2 | |||||
| 73·3 | K 1 | 16400 ± 1600 |
−24.1 ± 0.3 | −13.0 | −11.1 | ||
| K 2 | 4390 ± 440 | −20.8 ± 0.3 | −6.7 | −14.1 | |||
| K 3 | 1690 ± 170 | −18.4 ± 0.2 | −10.7 | −7.7 | |||
From the data in Table 1, it is clear that the binding constants and the free binding enthalpies ΔG increase slightly from the mono- (ΔG = −19.5 kJ mol−1) to the di- (ΔG = −23.2 kJ mol−1) and trivalent (ΔG = −24.4 kJ mol−1) pseudorotaxane. The binding enthalpies ΔH instead exhibit a strong increase with the number of binding sites from −12.2 kJ mol−1 (monovalent) to −41.2 kJ mol−1 (divalent) and −51.2 kJ mol−1 (trivalent). The binding entropies indicate an interesting difference between mono- and multivalent binding: the complexes containing the monovalent axle or the monovalent wheel all exhibit positive binding entropies that thus support binding. This effect is likely caused by solvent molecules in the macrocycle cavity that are replaced by the axle. The overall particle number thus increases in the binding event.102,136 In contrast, the di- and trivalent pseudorotaxanes reveal negative entropies, which then counterbalance part of the ΔH. The axle–wheel binding in the di- and trivalent pseudorotaxanes suffers from conformational fixation, once the second and third binding events occur. This effect leads to an overcompensation of the solvent effect and thus turns the binding entropy negative.
For the divalent pseudorotaxane, an effective molarity of EM1 = 0.65 mM is obtained, which translates into KmonoEM1 = 1.73. Consequently, the formation of the divalent pseudorotaxane is only very slightly positively cooperative. Using this effective molarity for the second binding interaction in the trivalent pseudorotaxane, the third binding event occurs with an EM2 = 0.32 mM and a KmonoEM2 = 0.84. In conclusion, the third binding event occurs in a more or less non-cooperative way.
This finding is also reflected in the binding enthalpies: the large step in binding enthalpy occurs from the mono- to the divalent pseudorotaxane, while the step to the trivalent system is comparably small. Despite of the apparently good geometric fit found in the force-field-optimized geometries shown in Fig. 1, these results indicate some strain to build up due to a non-perfect fit of the two spacer units in the trivalent case, while the divalent pseudorotaxane can likely adopt a more favourable geometry. The strain generated during the third binding event nevertheless does not overcompensate the additional binding contribution of the third site and triply bound pseudorotaxanes are still obtained almost exclusively as indicated by the NMR data in Fig. 3.
Synthesis of doubly and triply threaded rotaxanes
Based on these binding data, doubly and triply threaded, di- and trivalent rotaxanes have been synthesised. Multivalency-enhanced binding is expected to result in a highly efficient rotaxane synthesis. For this purpose, we modified the multivalent axle by introducing two and three terminal 5-hexynoyl amide groups instead of the hexanoyl amides, respectively (ESI†). The terminal alkynes serve as groups suitable for the attachment of bulky azide-functionalized trityl phenyl stopper groups in a Cu-catalysed Huisgen–Sharpless–Meldal 1,3-dipolar cycloaddition (Fig. 5).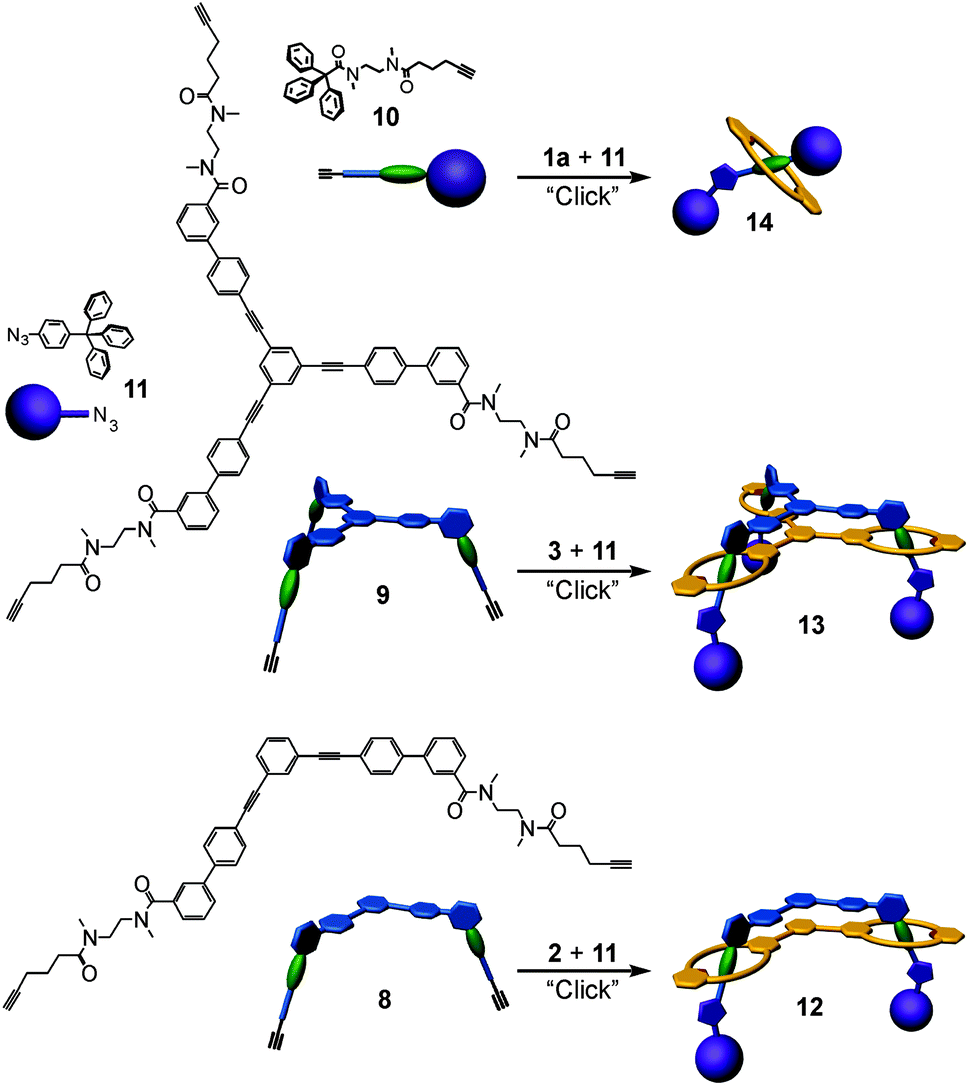 | ||
| Fig. 5 Synthesis of the di-, tri- and monovalent rotaxanes 12, 13, and 14 by “click” reactions of azide-functionalized stopper 11 and the pseudorotaxanes with alkyne-terminated axle 8·2, 9·3, and 10·1a, respectively. The reactions were catalysed by bromotris(triphenylphosphine)copper with triethylamine as the base. | ||
After two weeks of stirring, the di- or trivalent macrocycle, the corresponding axle, the azide-functionalized trityl phenyl stopper and (PPh3)3CuBr as the catalyst in dichloromethane in a sealed tube at 40 °C, the TLC shows the almost quantitative formation of the di- and trivalent rotaxanes 12 and 13, respectively, which can be isolated with 94% and 74% yield from the reaction mixture. Neither free macrocycle nor free axle was detected in the raw product. This is quite remarkable, as a mixture of the rotaxane, the free tetralactam macrocycle and the free stoppered axle is always obtained for monovalent cases such as 14. The course of the reaction can be roughly followed by ESI mass spectrometry. Over time, rotaxanes with one and then with two stopper groups form and vanish as reaction intermediates finally yielding the doubly and triply stoppered end products of the di- and trivalent systems.
Clearly, the synthesis benefits from the di- and trivalent binding situation. Two pieces of evidence confirm the triply threaded structure of trivalent rotaxane 13: (i) although the 1H NMR spectra are quite complex due to the superposition with the stopper signals, the typical amide NH shifts clearly reveal all macrocycles to be involved in hydrogen bonding and indicate the threefold symmetry of the trivalent rotaxane. (ii) An additional confirmation is provided by tandem MS experiments (Fig. 6 and ESI†): collision-induced dissociation of the mass-selected trivalent rotaxane trication at m/z 1726 (Fig. 6) shows fragments that lose one, then two stoppers without a competing separation of axle and wheel. For a singly or doubly threaded rotaxane, one would instead expect that loss of one or two stoppers, respectively, would result in a non-covalently bound and thus easy-to-dissociate complex. In such a case, an axle fragment would appear in the spectra, which bears at least one residual stopper at the previously non-threaded axle part. This is clearly not the case. The only axle fragment without the wheel appears at m/z 918 and corresponds to the axle which has lost all three stopper groups. Consequently, we can safely conclude that three mechanical bonds need to be cleaved before axle and wheel can dissociate from each other. Rotaxane 13 is thus triply threaded.
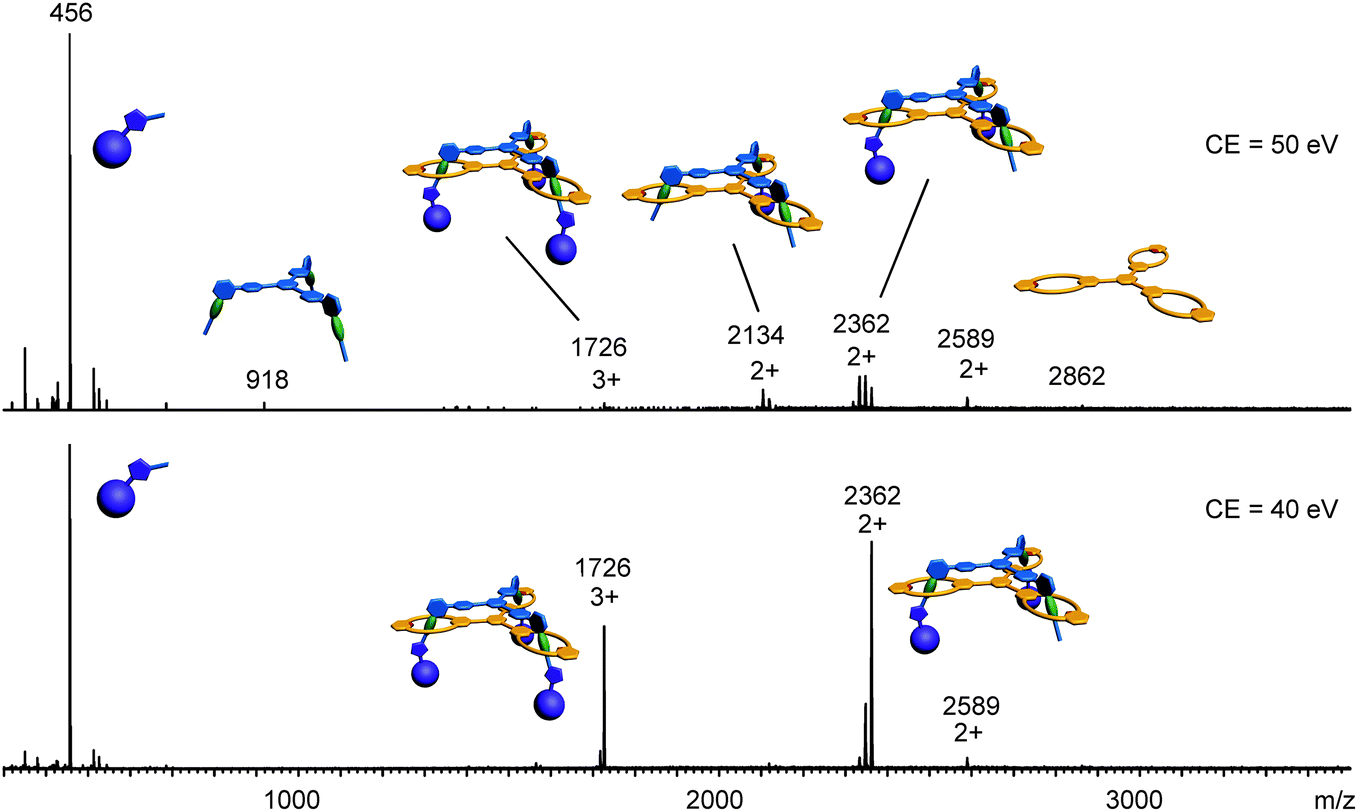 | ||
| Fig. 6 ESI-MS/MS: (da sonst Doppelung) collision-induced dissociation of the mass-selected triply protonated trivalent rotaxane at two different collision energies. Clearly, the separation of the wheel and axle component does not occur prior to the loss of all three stopper groups as indicated by the fragment at m/z 918, which corresponds to the axle lacking all three stoppers, while no such fragment is found that bears one, two or even three stopper groups. The signal for the singly charged stopper at m/z 456 is cut at half its intensity in order to make the other signals more easily visible. Losses of neutral fragments with a molecular mass of 28 can be assigned to N2 losses originating from the triazole rings. | ||
Towards amide-rotaxane-based molecular elevators
Earlier monovalent rotaxanes bearing diamide stations and triazoles in their axles exhibited chloride-induced molecular motion.99,118 The anion binds inside the cavity by hydrogen-bonding to two amide NH groups. The diamide is then pushed out of the cavity and the triazole slips in forming a C–H⋯Cl hydrogen bond to the chloride.The addition of chloride ions to a solution of trivalent rotaxane 13 may thus result in an elevator-type motion of the host relative to the axle by a similar shift from the diamide to the triazole stations (Fig. 7). As indicated by the 1H NMR chemical shifts discussed above, the diamide station is preferred, when no chloride is present. Upon addition of 3 eq. of tetrabutyl ammonium chloride, a significant downfield shift of the triazole C–H signal in the 1H NMR spectrum is observed (Fig. 7). Also, the wheel amide NH protons are significantly affected indicating the expected elevator motion to occur. Chloride binding can be reversed by adding NaBPh4 which precipitates NaCl from the solution and lets the host slide back to the diamide station. The 1H NMR spectrum after the addition of NaBPh4 is identical with that prior to chloride addition with the exception of the additional BPh4− signals.
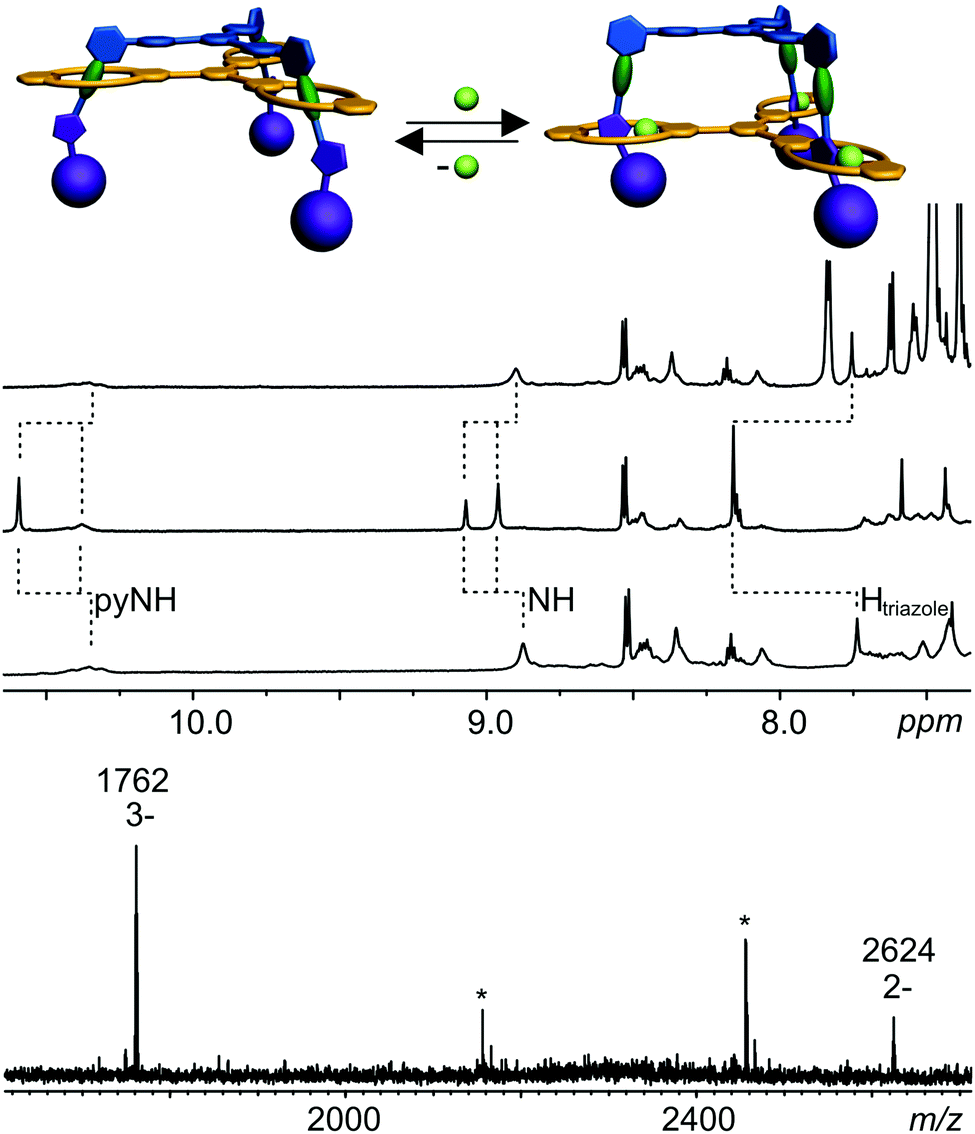 | ||
| Fig. 7 Switching the triply charged rotaxane by adding/removing chloride ions. Top: 1H NMR spectra (CDCl3, 298 K, 700 MHz) of the rotaxane prior to chloride addition (bottom spectrum), after addition of NBu4Cl (centre), and after removing the chloride ions by NaBPh4 (top spectrum). The cartoons illustrate the switching process. Bottom: Mass spectrum of the rotaxane after adding one equivalent of NBu4Cl shows as most prominent peak the triply charged rotaxane containing three chloride ions at m/z 1762 and a minor signal for the doubly charged rotaxane with two chloride ions at m/z 2624. No peak for the rotaxane containing just one chloride ion could be observed. The peaks labelled with asterisks are due to internal calibration. | ||
In the switched state after chloride addition, the isophthaloyl amide NH and the pyridine dicarboxylic amide NH protons both appear as two signals that integrate in a 1:2 ratio. The reason for this signal splitting could have two different reasons: (i) only part of the rotaxanes is switched and different isomers coexist. In this case, the signals that are close to the position before chloride addition would correspond to non-moved binding stations, the signals that are shifted further away from that position would correspond to the binding stations that have moved. However, this scenario is not in agreement with the fact that all triazole C–H protons give rise to one shifted signal. A second observation also rules out this scenario: if the two sets of signals would correspond to the switched and non-switched rotaxanes, one would expect that the integration ratio would be the same for the amide protons on the pyridine and the isophthalic acid hemisphere of the wheels. Instead, one observes a 2:1 ratio for the pyridine and a 1:2 ratio for the isophthalic acid hemisphere. (ii) The second scenario is the formation of a fully shifted, but unsymmetrical complex bearing three chloride ions. If one wheel is different from the other two, the 1:2 integration ratio can easily be understood. This scenario is also consistent with a uniform shift for all triazole C–H protons, which then all bind to chloride ions. If we assume that the amide protons that bind to the anion experience a larger complexation-induced shift as compared to those more remote from the chloride, also the different integration ratios for the two hemispheres become clear. A 2:1 ratio for the two amide signals of the pyridine hemisphere is in agreement with two of the three wheels binding the chloride close to this “outer” hemisphere of the wheel. The triazole inside the cavity of the third wheel is then turned around with the chloride located at the inner hemisphere. Therefore, two of the six pyridine amide protons experience only a smaller shift. In turn, two of the isophthaloyl diamide groups are more remote from the chloride and experience only a weaker shift, while the chloride is binding to these amides in the third wheel. Consequently, the integration ratio must be reversed here in line with the observed 1:2 ratio. From these considerations, one arrives at a clear-cut conclusion: two chlorides bind at the periphery of two of the wheels, the third one binds at the “inside” diamide just as depicted in the cartoon in Fig. 7. One question remains: if the binding site does not feel the structure of the adjacent two, the switched rotaxane could be a mixture of all out3, out2–in, out–in2, and in3 isomers. However, the fact that the integration ratio is almost exactly 2:1 and 1:2, respectively, makes us confident that we deal with a uniform out2–in structure. This interesting positional selectivity is likely caused by small differences in strain for the different isomers.
When discussing cooperativity, it would be a highly interesting aspect to assess, whether the elevator motion occurs in a concerted way. Would the addition of a substoichiometric amount – e.g. 1 eq. – of chloride lead to the exclusive formation of a mixture of completely switched and non-switched rotaxane? Or is a mixture of different switching states formed? Unfortunately, the rather complicated NMR spectra of a 1:1 mixture of the chloride salt and the trivalent rotaxane do not provide straightforward insight here due to more pronounced peak broadening. The only hint that a concerted switching process might indeed occur is the finding that the ESI mass spectra (Fig. 7, bottom) exhibit a strong signal for a 3:1 complex of chloride and rotaxane even when only one equivalent of chloride is added. Just a minor signal for a rotaxane with two chloride ions is observed and the signal for the 1:1 complex is absent. However, this is merely an indication and for the time being we unfortunately cannot determine with certainty, whether the switching process is occurring cooperatively.
Conclusions
Multivalency was used here to increase the efficiency of the synthesis of multiply threaded amide rotaxanes. Diamide stations incorporated in the axles form hydrogen bonds to tetralactam macrocycles that are used as the wheels. The building blocks were convergently prepared by applying the same spacer units for the wheel and the axle components. Spacer rigidity is achieved through a Sonogashira cross-coupling protocol.Divalent and trivalent pseudorotaxanes form, when axle and wheel components are mixed in non-competitive solvents. Their kinetic properties differ significantly from those of the monovalent analogue in that axle binding is slow on the NMR time scale for the di- and trivalent case, while the monovalent axle quickly exchanges. A detailed double mutant cycle thermodynamic analysis based on NMR and ITC binding data revealed the second binding step to exhibit positive cooperativity. Due to unfavourable strain in the trivalent pseudorotaxane, the third binding step is more or less non-cooperative. Nevertheless, the preorganization of the two components achieved in the second step ensures formation of the completely threaded trivalent pseudorotaxane.
Our results open several routes to an optimization of the rotaxanes under study: replacing the diamide stations by, for example diketopiperazines, will not only increase the individual binding interaction Kmono, but will also avoid the cis/trans-amide isomer problem which complicates the NMR spectral analysis. Also, the spacers are not perfectly designed yet and a somewhat higher flexibility may help avoiding the strained situation that is counterproductive in terms of chelate cooperativity.
When terminal alkynes are incorporated in the axle, the di- and trivalent pseudorotaxanes can be equipped with bulky stopper groups through a copper-catalysed 1,3-dipolar cycloaddition with an azide-functionalized tritylphenyl stopper. The corresponding rotaxanes form with significantly higher isolated yields (divalent: 94%, trivalent: 74%) than the monovalent rotaxane (37%). NMR as well as tandem mass spectrometric experiments confirm the completely threaded structure.
Finally, chloride addition is capable of shifting binding stations in all three axles of the trivalent rotaxane. The diamide is the preferred binding station in the absence of chloride. In its presence, however, a network of hydrogen bonds connects the triazole units in the axle with the macrocycles mediated by the chloride. The trivalent rotaxane undergoes an elevator-type motion. The switching behaviour is expressed in quite significant NMR shift changes, which can be completely reversed when the chloride is precipitated by addition of NaBPh4.
Acknowledgements
We are grateful to the Deutsche Forschungsgemeinschaft (C. A. S.: collaborative research centre 765 “multivalency”) and the Academy of Finland (K. R.: project no. 122350, 140718, 265328 and 263256) for funding this project. L. K. thanks the Studienstiftung des Deutschen Volkes (German National Academic Foundation) for a doctoral fellowship and the Centre for International Cooperation (FU Berlin) for a travel grant to Finland. We thank Niklas König, B.Sc., for help in the synthesis. T. M. thanks the Department of Chemistry, University of Jyväskylä for a postgraduate fellowship.Notes and references
- R. H. Kramer and J. W. Karpen, Nature, 1998, 395, 710 CrossRef CAS PubMed.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754 CrossRef.
- H. Herzner, T. Reipen, M. Schultz and H. Kunz, Chem. Rev., 2000, 100, 4495 CrossRef CAS PubMed.
- P. I. Kitov, J. M. Sadowska, G. Mulvey, G. D. Armstrong, H. Ling, N. S. Pannu, R. J. Read and D. R. Bundle, Nature, 2000, 403, 669 CrossRef CAS PubMed.
- C. W. Cairo, J. E. Gestwicki, M. Kanai and L. L. Kiessling, J. Am. Chem. Soc., 2002, 124, 1615 CrossRef CAS PubMed.
- B. Houseman and M. Mrksich, Top. Curr. Chem., 2002, 218, 1 CrossRef CAS.
- T. Lindhorst, Top. Curr. Chem., 2002, 218, 201 CrossRef CAS.
- P. I. Kitov and D. R. Bundle, J. Am. Chem. Soc., 2003, 125, 16271 CrossRef CAS PubMed.
- M. Hartmann and T. K. Lindhorst, Eur. J. Org. Chem., 2011, 3583 CrossRef CAS.
- J. Rao, J. Lahiri, L. Isaacs, R. M. Weis and G. M. Whitesides, Science, 1998, 280, 708 CrossRef CAS.
- N. Röckendorf and T. Lindhorst, Top. Curr. Chem., 2001, 217, 201 CrossRef.
- J. Rojo, Top. Curr. Chem., 2002, 218, 45 CrossRef CAS.
- R. Roy, Top. Curr. Chem., 1997, 187, 241 CrossRef CAS.
- D. Schwefel, C. Maierhofer, J. G. Beck, S. Seeberger, K. Diederichs, H. M. Möller, W. Welte and V. Wittmann, J. Am. Chem. Soc., 2010, 132, 8704 CrossRef CAS PubMed.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754 CrossRef.
- S.-K. Choi, Synthetic Multivalent Molecules, Wiley, Hoboken, 2004 Search PubMed.
- X. Chi, A. J. Guerin, R. A. Haycock, C. A. Hunter and L. D. Sarson, J. Chem. Soc., Chem. Commun., 1995, 2563 RSC.
- A. Mulder, J. Huskens and D. N. Reinhoudt, Org. Biomol. Chem., 2004, 2, 3409 CAS.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472 CrossRef CAS PubMed.
- J. D. Badjić, A. Nelson, S. J. Cantrill, W. B. Turnbull and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 723 CrossRef PubMed.
- C.-F. Chen, Chem. Commun., 2011, 47, 1674 RSC.
- R. Jäger and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1997, 36, 930 CrossRef.
- C. Schalley, T. Weilandt, J. Brüggemann and F. Vögtle, Top. Curr. Chem., 2004, 248, 141 CrossRef.
- J. Berná, G. Bottari, D. A. Leigh and E. M. Pérez, Pure Appl. Chem., 2007, 79, 39 CrossRef.
- C. M. Keaveney and D. A. Leigh, Angew. Chem., Int. Ed., 2004, 43, 1222 CrossRef CAS PubMed.
- D. A. Leigh, A. Venturini, A. J. Wilson, J. K. Y. Wong and F. Zerbetto, Chem. – Eur. J., 2004, 10, 4960 CrossRef CAS PubMed.
- R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi and M. Venturi, Acc. Chem. Res., 2001, 34, 445 CrossRef CAS PubMed.
- V. Balzani, A. Credi, F. M. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348 CrossRef CAS.
- V. Balzani, A. Credi, S. Silvi and M. Venturi, Chem. Soc. Rev., 2006, 35, 1135 RSC.
- M.-J. Blanco, M. Consuelo Jimenez, J.-C. Chambron, V. Heitz, M. Linke and J.-P. Sauvage, Chem. Soc. Rev., 1999, 28, 293 RSC.
- P. Bodis, M. R. Panman, B. H. Bakker, A. Mateo-Alonso, M. Prato, W. J. Buma, A. M. Brouwer, E. R. Kay, D. A. Leigh and S. Woutersen, Acc. Chem. Res., 2009, 42, 1462 CrossRef CAS PubMed.
- M. Clemente-León, A. Credi, M. V. Martínez-Díaz, C. Mingotaud and J. F. Stoddart, Adv. Mater., 2006, 18, 1291 CrossRef.
- A. Harada, Acc. Chem. Res., 2001, 34, 456 CrossRef CAS PubMed.
- M. R. Panman, P. Bodis, D. J. Shaw, B. H. Bakker, A. C. Newton, E. R. Kay, A. M. Brouwer, W. J. Buma, D. A. Leigh and S. Woutersen, Science, 2010, 328, 1255 CrossRef CAS PubMed.
- J.-P. Sauvage, Acc. Chem. Res., 1998, 31, 611 CrossRef CAS.
- C. A. Schalley, K. Beizai and F. Vögtle, Acc. Chem. Res., 2001, 34, 465 CrossRef CAS PubMed.
- S. Silvi, M. Venturi and A. Credi, J. Mater. Chem., 2009, 19, 2279 RSC.
- J. D. Badjić, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845 CrossRef PubMed.
- J. D. Badjić, C. M. Ronconi, J. F. Stoddart, V. Balzani, S. Silvi and A. Credi, J. Am. Chem. Soc., 2006, 128, 1489 CrossRef PubMed.
- J. D. Badjić, S. J. Cantrill and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 2288 CrossRef PubMed.
- M. Ammann, A. Rang, C. A. Schalley and P. Bäuerle, Eur. J. Org. Chem., 2006, 1940 CrossRef CAS.
- S. Anderson, H. L. Anderson and J. K. M. Sanders, Acc. Chem. Res., 1993, 26, 469 CrossRef CAS.
- P. Bäuerle, M. Ammann, M. Wilde, G. Götz, E. Mena-Osteritz, A. Rang and C. A. Schalley, Angew. Chem., Int. Ed., 2007, 46, 363 CrossRef PubMed.
- S. Bonnet and J.-P. Collin, Chem. Soc. Rev., 2008, 37, 1207 RSC.
- J.-C. Chambron, J.-P. Collin, V. Heitz, D. Jouvenot, J.-M. Kern, P. Mobian, D. Pomeranc and J.-P. Sauvage, Eur. J. Org. Chem., 2004, 1627 CrossRef CAS.
- G. H. Clever and M. Shionoya, Chem. – Eur. J., 2010, 16, 11792 CrossRef CAS PubMed.
- J.-P. Collin, F. Durola, V. Heitz, F. Reviriego, J.-P. Sauvage and Y. Trolez, Angew. Chem., Int. Ed., 2010, 49, 10172 CrossRef CAS PubMed.
- J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530 RSC.
- C. O. Dietrich-Buchecker and J. P. Sauvage, Chem. Rev., 1987, 87, 795 CrossRef CAS.
- J. A. Faiz, V. Heitz and J.-P. Sauvage, Chem. Soc. Rev., 2009, 38, 422 RSC.
- P. E. Glen, J. A. T. O'Neill and A.-L. Lee, Tetrahedron, 2013, 69, 57 CrossRef CAS PubMed.
- J. Guo, P. C. Mayers, G. A. Breault and C. A. Hunter, Nat. Chem., 2010, 2, 218 CrossRef CAS PubMed.
- K. D. Hanni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240 RSC.
- M. Hutin, C. A. Schalley, G. Bernardinelli and J. R. Nitschke, Chem. – Eur. J., 2006, 12, 4069 CrossRef CAS PubMed.
- J. D. Megiatto, D. I. Schuster, S. Abwandner, G. de Miguel and D. M. Guldi, J. Am. Chem. Soc., 2010, 132, 3847 CrossRef CAS PubMed.
- C. D. Meyer, C. S. Joiner and J. F. Stoddart, Chem. Soc. Rev., 2007, 36, 1705 RSC.
- K.-i. Yamashita, M. Kawano and M. Fujita, J. Am. Chem. Soc., 2007, 129, 1850 CrossRef CAS PubMed.
- D. B. Amabilino and J. F. Stoddart, Chem. Rev., 1995, 95, 2725 CrossRef CAS.
- D. B. Amabilino, J. F. Stoddart and D. J. Williams, Chem. Mater., 1994, 6, 1159 CrossRef CAS.
- E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251 CrossRef CAS PubMed.
- H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2010, 49, 5331 CrossRef CAS PubMed.
- F. Biedermann, U. Rauwald, J. M. Zayed and O. A. Scherman, Chem. Sci., 2011, 2, 279 RSC.
- F. B. L. Cougnon, H. Y. Au-Yeung, G. D. Pantoş and J. K. M. Sanders, J. Am. Chem. Soc., 2011, 133, 3198 CrossRef CAS PubMed.
- W. R. Dichtel, O. S. Miljanić, W. Zhang, J. M. Spruell, K. Patel, I. Aprahamian, J. R. Heath and J. F. Stoddart, Acc. Chem. Res., 2008, 41, 1750 CrossRef CAS PubMed.
- L. Fang, S. Basu, C.-H. Sue, A. C. Fahrenbach and J. F. Stoddart, J. Am. Chem. Soc., 2010, 133, 396 CrossRef PubMed.
- L. Isaacs, Chem. Commun., 2009, 619 RSC.
- C. Ke, R. A. Smaldone, T. Kikuchi, H. Li, A. P. Davis and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 381 CrossRef CAS PubMed.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267 RSC.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621 CrossRef CAS PubMed.
- S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1959 CrossRef CAS PubMed.
- Z. Niu, F. Huang and H. W. Gibson, J. Am. Chem. Soc., 2011, 133, 2836 CrossRef CAS PubMed.
- P. D. Beer, M. R. Sambrook and D. Curiel, Chem. Commun., 2006, 2105 RSC.
- M. J. Chmielewski, J. J. Davis and P. D. Beer, Org. Biomol. Chem., 2009, 7, 415 CAS.
- N. H. Evans, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2011, 50, 2507 CrossRef CAS PubMed.
- L. Fang, M. A. Olson, D. Benitez, E. Tkatchouk, W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17 RSC.
- T. B. Gasa, C. Valente and J. F. Stoddart, Chem. Soc. Rev., 2011, 40, 57 RSC.
- P. Ghosh, O. Mermagen and C. A. Schalley, Chem. Commun., 2002, 2628 RSC.
- H.-Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406 CrossRef CAS PubMed.
- G. M. Hübner, J. Gläser, C. Seel and F. Vögtle, Angew. Chem., Int. Ed., 1999, 38, 383 CrossRef.
- C. A. Hunter, J. Chem. Soc., Chem. Commun., 1991, 749 RSC.
- C. A. Hunter, J. Am. Chem. Soc., 1992, 114, 5303 CrossRef CAS.
- W. Jiang, A. Schäfer, P. C. Mohr and C. A. Schalley, J. Am. Chem. Soc., 2010, 132, 2309 CrossRef CAS PubMed.
- K. Kim, Chem. Soc. Rev., 2002, 31, 96 RSC.
- M. D. Lankshear and P. D. Beer, Acc. Chem. Res., 2007, 40, 657 CrossRef CAS PubMed.
- C.-F. Lee, D. A. Leigh, R. G. Pritchard, D. Schultz, S. J. Teat, G. A. Timco and R. E. P. Winpenny, Nature, 2009, 458, 314 CrossRef CAS PubMed.
- X. Ma and H. Tian, Chem. Soc. Rev., 2010, 39, 70 RSC.
- K. M. Mullen and P. D. Beer, Chem. Soc. Rev., 2009, 38, 1701 RSC.
- S. Ottens-Hildebrandt, S. Meier, W. Schmidt and F. Vögtle, Angew. Chem., Int. Ed. Engl., 1994, 33, 1767 CrossRef.
- C. J. Serpell, J. Cookson, A. L. Thompson and P. D. Beer, Chem. Sci., 2011, 2, 494 RSC.
- G. T. Spence, N. G. White and P. D. Beer, Org. Biomol. Chem., 2012, 10, 7282 CAS.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802 RSC.
- M. S. Vickers and P. D. Beer, Chem. Soc. Rev., 2007, 36, 211 RSC.
- F. Vögtle, T. Dünnwald and T. Schmidt, Acc. Chem. Res., 1996, 29, 451 CrossRef.
- F. Vögtle, S. Meier and R. Hoss, Angew. Chem., Int. Ed. Engl., 1992, 31, 1619 CrossRef.
- N. G. White and P. D. Beer, Org. Biomol. Chem., 2013, 11, 1326 CAS.
- K. Zhu, V. N. Vukotic, N. Noujeim and S. J. Loeb, Chem. Sci., 2012, 3, 3265 RSC.
- W. Jiang, K. Nowosinski, N. L. Löw, E. V. Dzyuba, F. Klautzsch, A. Schäfer, J. Huuskonen, K. Rissanen and C. A. Schalley, J. Am. Chem. Soc., 2011, 134, 1860 CrossRef PubMed.
- S.-Y. Chang, H. S. Kim, K.-J. Chang and K.-S. Jeong, Org. Lett., 2003, 6, 181 CrossRef PubMed.
- E. V. Dzyuba, L. Kaufmann, N. L. Löw, A. K. Meyer, H. D. F. Winkler, K. Rissanen and C. A. Schalley, Org. Lett., 2011, 13, 4838 CrossRef CAS PubMed.
- T. Felder and C. A. Schalley, Angew. Chem., Int. Ed., 2003, 42, 2258 CrossRef CAS PubMed.
- P. Ghosh, G. Federwisch, M. Kogej, C. A. Schalley, D. Haase, W. Saak, A. Lützen and R. M. Gschwind, Org. Biomol. Chem., 2005, 3, 2691 CAS.
- L. Kaufmann, E. V. Dzyuba, F. Malberg, N. L. Low, M. Groschke, B. Brusilowskij, J. Huuskonen, K. Rissanen, B. Kirchner and C. A. Schalley, Org. Biomol. Chem., 2012, 10, 5954 CAS.
- B. Kirchner, C. Spickermann, W. Reckien and C. A. Schalley, J. Am. Chem. Soc., 2009, 132, 484 CrossRef PubMed.
- I. Kossev, W. Reckien, T. Felder, M. R. Kishan, C. A. Schalley and M. Sokolowski, J. Phys. Chem. C, 2009, 113, 12870 CAS.
- X.-y. Li, J. Illigen, M. Nieger, S. Michel and C. A. Schalley, Chem. – Eur. J., 2003, 9, 1332 CrossRef CAS PubMed.
- A. Mohry, F. Vögtle, M. Nieger and H. Hupfer, Chirality, 2000, 12, 76 CrossRef CAS.
- S. Ottens-Hildebrandt, M. Nieger, K. Rissanen, J. Rouvinen, S. Meier, G. Harder and F. Vögtle, J. Chem. Soc., Chem. Commun., 1995, 777 RSC.
- C. Reuter, C. Seel, M. Nieger and F. Vögtle, Helv. Chim. Acta, 2000, 83, 630 CrossRef CAS.
- C. Reuter and F. Vögtle, Org. Lett., 2000, 2, 593 CrossRef CAS PubMed.
- C. Seel, A. H. Parham, O. Safarowsky, G. M. Hübner and F. Vögtle, J. Org. Chem., 1999, 64, 7236 CrossRef CAS.
- O. Braun, A. Hünten and F. Vögtle, J. Prakt. Chem., 1999, 341, 542 CrossRef CAS.
- F. J. Carver, C. A. Hunter and R. J. Shannon, J. Chem. Soc., Chem. Commun., 1994, 1277 RSC.
- N. L. Löw, E. V. Dzyuba, B. Brusilowskij, L. Kaufmann, E. Franzmann, W. Maison, E. Brandt, D. Aicher, A. Wiehe and C. A. Schalley, Beilstein J. Org. Chem., 2012, 8, 234 CrossRef PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
- K. Hirose, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 193 CrossRef CAS.
- K. Hirose, Quantitative Analysis of Binding Properties, in Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Wiley-VCH, Weinheim, 2012, vol. 1, p. 27 Search PubMed.
- J. J. Gassensmith, L. Barr, J. M. Baumes, A. Paek, A. Nguyen and B. D. Smith, Org. Lett., 2008, 10, 3343 CrossRef CAS PubMed.
- J. J. Gassensmith, S. Matthys, J.-J. Lee, A. Wojcik, P. V. Kamat and B. D. Smith, Chem. – Eur. J., 2010, 16, 2916 CrossRef CAS PubMed.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488 CrossRef CAS PubMed.
- P. J. Carter, G. Winter, A. J. Wilkinson and A. R. Fersht, Cell, 1984, 38, 835 CrossRef CAS.
- P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538 CrossRef CAS.
- C. A. Hunter, N. Ihekwaba, M. C. Misuraca, M. D. Segarra-Maset and S. M. Turega, Chem. Commun., 2009, 3964 RSC.
- C. A. Hunter, P. S. Jones, P. Tiger and S. Tomas, Chem. – Eur. J., 2002, 8, 5435 CrossRef CAS.
- C. A. Hunter, M. C. Misuraca and S. M. Turega, J. Am. Chem. Soc., 2010, 133, 582 CrossRef PubMed.
- A. Camara-Campos, D. Musumeci, C. A. Hunter and S. Turega, J. Am. Chem. Soc., 2009, 131, 18518 CrossRef CAS PubMed.
- S. L. Cockroft and C. A. Hunter, Chem. Soc. Rev., 2007, 36, 172 RSC.
- G. Ercolani, J. Am. Chem. Soc., 2003, 125, 16097 CrossRef CAS PubMed.
- G. Ercolani, J. Phys. Chem. B, 2003, 107, 5052 CrossRef CAS.
- G. Ercolani, C. Piguet, M. Borkovec and J. Hamacek, J. Phys. Chem. B, 2007, 111, 12195 CrossRef CAS PubMed.
- G. Ercolani and L. Schiaffino, Angew. Chem., Int. Ed., 2011, 50, 1762 CrossRef CAS PubMed.
- T. K. Dam and C. F. Brewer, Chem. Rev., 2002, 102, 387 CrossRef CAS PubMed.
- J. E. Ladbury and B. Z. Chowdhry, Chem. Biol., 1996, 3, 791 CrossRef CAS.
- F. P. Schmidtchen, Isothermal Titration Calorimetry in Supramolecular Chemistry, in Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Wiley-VCH, Weinheim, 2012, vol. 1, p. 67 Search PubMed.
- T. Weilandt, N. L. Löw, G. Schnakenburg, J. Daniels, M. Nieger, C. A. Schalley and A. Lützen, Chem. – Eur. J., 2012, 18, 16665 CrossRef CAS PubMed.
- T. Wiseman, S. Williston, J. F. Brandts and L.-N. Lin, Anal. Biochem., 1989, 179, 131 CrossRef CAS.
- C. Spickermann, T. Felder, C. A. Schalley and B. Kirchner, Chem. – Eur. J., 2008, 14, 1216 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterisation data and original 1H and 13C NMR spectra of new compounds; crystallographic data; additional NMR and UV/Vis spectroscopic and (tandem) mass spectrometric data; ITC titration experiments; analysis of statistical factors and double mutant cycle analysis for the divalent pseudorotaxane. CCDC 938974. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00077c |
| This journal is © the Partner Organisations 2014 |