Ionic liquid-supported sulfonyl hydrazine: a useful reagent for traceless synthesis of pyrazoles†
Sunita
Choudhary
a,
Manoj Kumar
Muthyala
a,
Keykavous
Parang
b and
Anil
Kumar
*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Rajasthan 333031, India. E-mail: anilkumar@pilani.bits-pilani.ac.in
bChapman University School of Pharmacy, Irvine, California 92618, USA
First published on 20th May 2014
Abstract
Ionic liquid-supported sulfonyl hydrazine has been synthesized and used as a soluble support for the traceless synthesis of highly substituted pyrazoles from two different routes. Various substituted pyrazoles were synthesized in high yields. A key feature of the protocol is an easy and convenient purification process that avoids chromatographic separation of products and thus makes the method eco-friendly and economical.
Introduction
Nitrogen-containing heterocycles are of great chemical and biological significance. In particular, pyrazole is a popular synthetic target for pharmaceutical, material and agrochemical industries.1,2 Numerous compounds containing the pyrazole moiety are known to exhibit biological activities such as analgesic,3 antimicrobial,4 anti-convulsant,5 antidepressant,6 anti-inflammatory,7,8 anti-arthritics,9 cannabinoid-1 (CB1) receptor antagonists,10–12 Iκβ kinase β (IKKβ or IKK-2) inhibition13 and HIV-1 reverse transcriptase inhibition.14 Pyrazole constitutes the basic framework of several drug molecules. Structures of some important pyrazole containing drugs such as Celecoxib (selective COX-2 inhibition, 1), Zaleplon (insomnia, 2), Zoniporide (selective human NHE-1 inhibition, 3), Sildenafil (erectile dysfunction, 4), and Acomplia (selective CB1 receptor blocker, 5) are depicted in Fig. 1.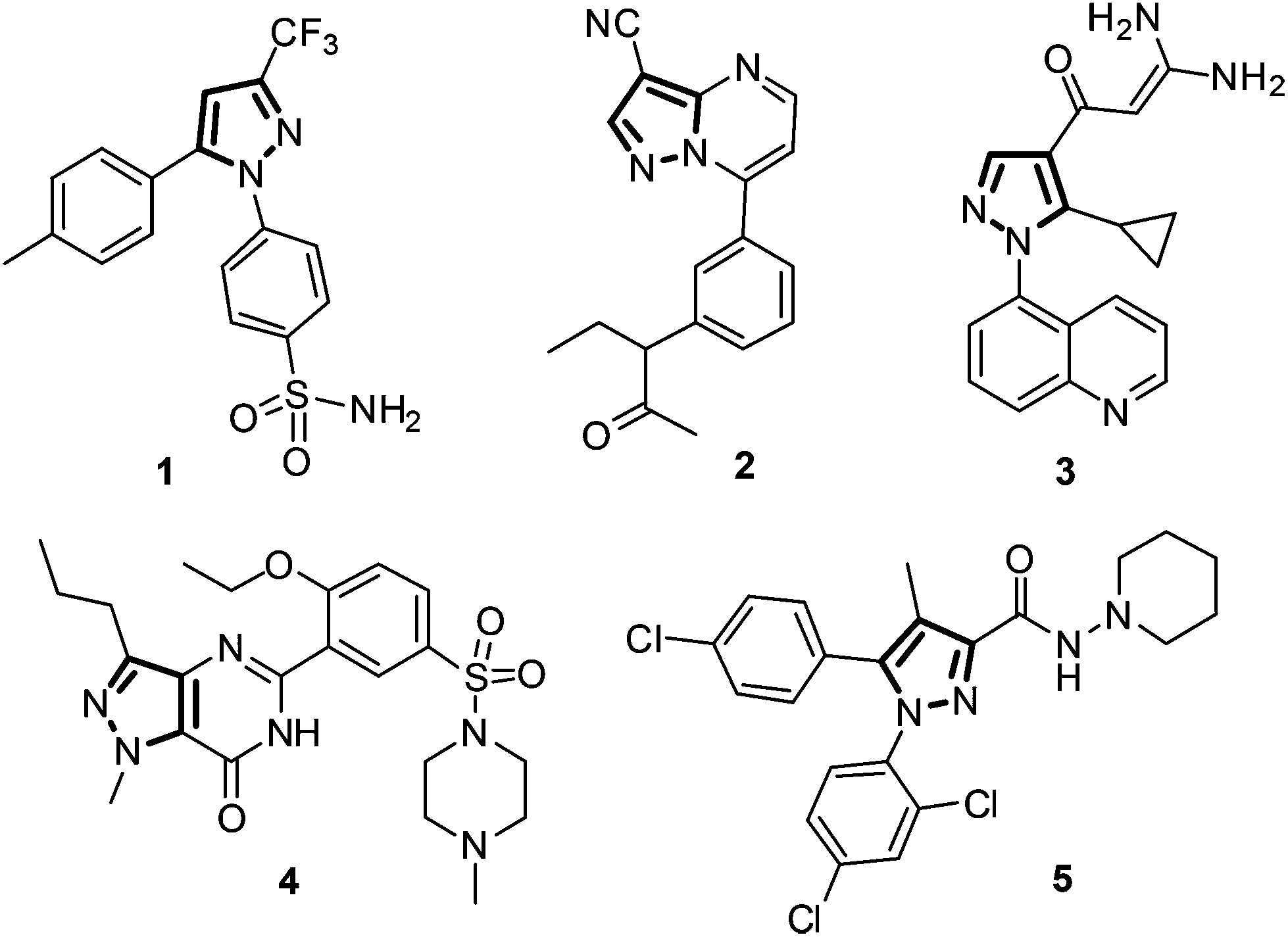 | ||
| Fig. 1 Structure of some drugs containing the pyrazole ring. | ||
Owing to the attractive biological properties of pyrazoles, considerable attention has been focused on developing methods for the preparation of diversely substituted pyrazoles.15–22 The most common methods for preparing pyrazoles are (a) the reaction of 1,3-diketones with hydrazines or its derivatives,23–25 (b) the reaction of α,β-unsaturated aldehydes and ketones with hydrazines followed by oxidation,26 and (c) [3 + 2]-cycloaddition of diazo compounds with alkynes.27 Formation of two regioisomers by the condensation of 1,3-diketones or unsaturated aldehydes and ketones with hydrazine or arylhydrazines is an inevitable drawback of the first two methods. On the other hand, preparing toxic and potentially explosive diazo compounds and handling reactive 1,3-dipolar compounds limit the synthetic utility of the third method. With the aim to solve the handling problem in diazo compounds, Aggarwal et al. reported a one-pot method for the preparation of pyrazoles by 1,3-dipolar cycloaddition of in situ generated diazo compounds from tosylhydrazones of aldehydes in the presence of a base.28 Wu et al. also synthesized 3,5-disubstituted pyrazoles by the reaction of aldehydes with tosylhydrazine followed by cycloaddition with terminal alkynes.29 Recently, the Tang group applied this protocol for the synthesis of 1,3,5-trisubstituted-1H-pyrazoles from N-alkylated tosylhydrazones to increase the range of substrates and demonstrated excellent tolerance to a variety of substituents, including both electron-donating and electron-withdrawing groups.30 This approach solved some of the traditional problems inherent in the [3 + 2]-cycloaddition of diazo compounds to alkynes for the synthesis of pyrazoles. However, purification of pyrazoles from the co-product tosylsulfonamide remains a challenge and requires extensive chromatographic separation.
We recently synthesized ionic liquid-supported sulfonyl azide and it has proven to be a highly effective and safe diazotransfer reagent for active methylene compounds. The diazo compounds could be easily separated from the co-product sulfonamide without chromatographic separation. We envisioned that the use of ionic liquid-supported sulfonyl hydrazine could solve the problem of separation in the synthesis of pyrazoles and thus designed two routes to synthesize pyrazoles using ionic liquid-supported sulfonyl hydrazine as a soluble support (Scheme 1). The first one is by the condensation of α,β-unsaturated carbonyl compounds to ionic liquid-supported sulfonyl hydrazine 6 followed by cyclization in the presence of a base and the second one by the 1,3-dipolar cycloaddition of alkynes with ionic liquid-supported sulfonyl hydrazones 10, which were obtained by reacting 6 with the corresponding aldehydes. In this article, we report our results on traceless synthesis of pyrazoles using ionic liquid-supported sulfonyl hydrazine.
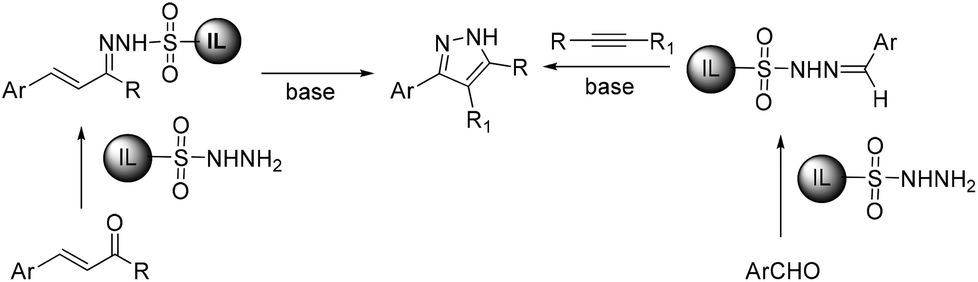 | ||
| Scheme 1 Synthetic approach for the synthesis of pyrazoles from ionic liquid-supported sulfonyl hydrazine. | ||
Results and discussion
Our investigations commenced with the synthesis of ionic liquid-supported sulfonyl hydrazones 9 and 10 by the reaction of ionic liquid-supported sulfonyl hydrazine 6 with α,β-unsaturated carbonyl compounds 7 and aryl aldehydes 8, respectively (Scheme 2). The reaction of 6 with 1-phenylbut-2-en-1-one 7a in ethanol for 5 h at 40 °C followed by evaporation of ethanol, washing of the ionic liquid layer with an ethyl acetate–hexane mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and drying under vacuum gave 9a in excellent yield (86%) with high purity. The structure and purity of ionic liquid-supported hydrazones 9a were confirmed by 1H and 13C NMR data. In 1H NMR of 9a the characteristic peak for CH3–C
:1 v/v) and drying under vacuum gave 9a in excellent yield (86%) with high purity. The structure and purity of ionic liquid-supported hydrazones 9a were confirmed by 1H and 13C NMR data. In 1H NMR of 9a the characteristic peak for CH3–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N proton appeared at 2.08 ppm as a singlet and CN carbon appeared at around 154.49 ppm in the 13C NMR spectrum.
N proton appeared at 2.08 ppm as a singlet and CN carbon appeared at around 154.49 ppm in the 13C NMR spectrum.
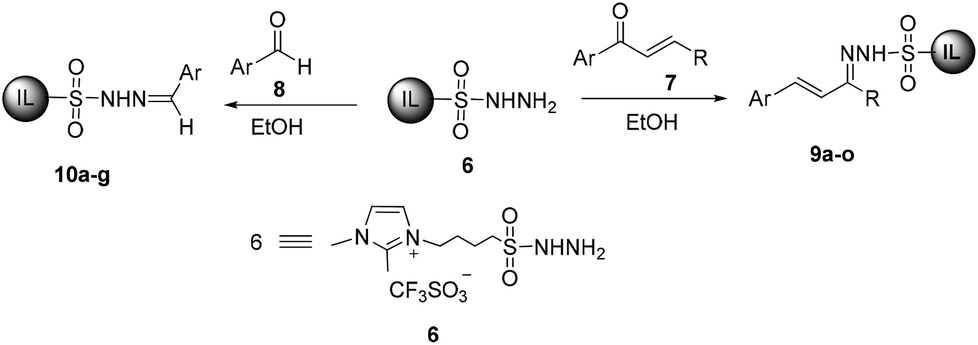 | ||
| Scheme 2 Synthesis of ionic liquid-supported sulfonyl hydrazones 9 and 10. | ||
As can be seen from Table 1, the reaction has wide substrate scope. Reaction of 6 with 7 or 8 bearing both electron-withdrawing and/or electron donating substituents resulted in the corresponding ionic liquid-supported sulfonyl hydrazones 9a–o & 10a–g in good to excellent yields (54–92%). The 1H and 13C NMR spectra of all the sulfonyl hydrazones 9 & 10 were in agreement with the structure (ESI).
| Entry | Ar | R | Product 9 or 10 | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: 6 (1 mmol), 7 or 8 (1.1 mmol), EtOH (10 mL), 40 °C for 5 h. b Isolated yield. | ||||
| 1 | C6H5 | CH3 | 9a | 86 |
| 2 | 2-FC6H4 | CH3 | 9b | 83 |
| 3 | 2-ClC6H4 | CH3 | 9c | 81 |
| 4 | 3-ClC6H4 | CH3 | 9d | 84 |
| 5 | 3-NO2C6H4 | CH3 | 9e | 79 |
| 6 | 4-ClC6H4 | CH3 | 9f | 74 |
| 7 | 4-BrC6H4 | CH3 | 9g | 87 |
| 8 | 2,6-(Cl)2C6H3 | CH3 | 9h | 80 |
| 9 | 3-OCH3C6H4 | CH3 | 9i | 54 |
| 10 | 4-CH3C6H4 | CH3 | 9j | 67 |
| 11 | 4-OCH3C6H4 | CH3 | 9k | 92 |
| 12 | 3,4-(OCH3)2C6H3 | CH3 | 9l | 71 |
| 13 | C4H3S | CH3 | 9m | 89 |
| 14 | C4H3O | CH3 | 9n | 88 |
| 15 | C6H5 | H | 9o | 59 |
| 16 | C6H5 | — | 10a | 88 |
| 17 | 3-ClC6H4 | — | 10b | 76 |
| 18 | 4-ClC6H4 | — | 10c | 72 |
| 19 | 4-BrC6H4 | — | 10d | 79 |
| 20 | 2,6-(Cl)2C6H3 | — | 10e | 77 |
| 21 | 4-CH3C6H4 | — | 10f | 89 |
| 22 | C4H3S | — | 10g | 88 |
In an initial experiment for the synthesis of pyrazoles, we performed the cyclization of 9g in the presence of K2CO3 at 100 °C in water. The reaction afforded pyrazole 11g in 72% yield after 24 h. After screening various bases viz. K2CO3, Cs2CO3, KOH and NaOH, and solvents it was found that the best yield (88%) of 11g was achieved by employing 3 N NaOH in water (Table 2, entry 5). Almost similar yields of 11g were obtained in DMF in the presence of 3 N NaOH at 150 °C and water using 5 equiv. Cs2CO3 at 100 °C (Table 2, entries 7 & 13).
|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | T (°C) | Yielda (%) |
| a Isolated yields. b No product isolated. | ||||
| 1 | NaOH (1 N) | Water | 100 | 71 |
| 2 | NaOH (2 N) | Water | 100 | 76 |
| 3 | KOH (2 N) | Water | 100 | 66 |
| 4 | NaOH (3 N) | Water | 30 | —b |
| 5 | NaOH (3 N) | Water | 100 | 88 |
| 6 | NaOH (3 N) | DMSO | 150 | 47 |
| 7 | NaOH (3 N) | DMF | 150 | 85 |
| 8 | NaOH (3 N) | CH3CN | 80 | 52 |
| 9 | NaOH (3 N) | CH3OH | 60 | 30 |
| 10 | NaOH (5 N) | Water | 100 | 85 |
| 11 | K2CO3 (1 eq) | Water | 100 | 72 |
| 12 | K2CO3 (2 eq) | Water | 100 | 67 |
| 13 | Cs2CO3 (5 eq) | Water | 100 | 87 |
| 14 | Cs2CO3 (3 eq) | Water | 100 | 54 |
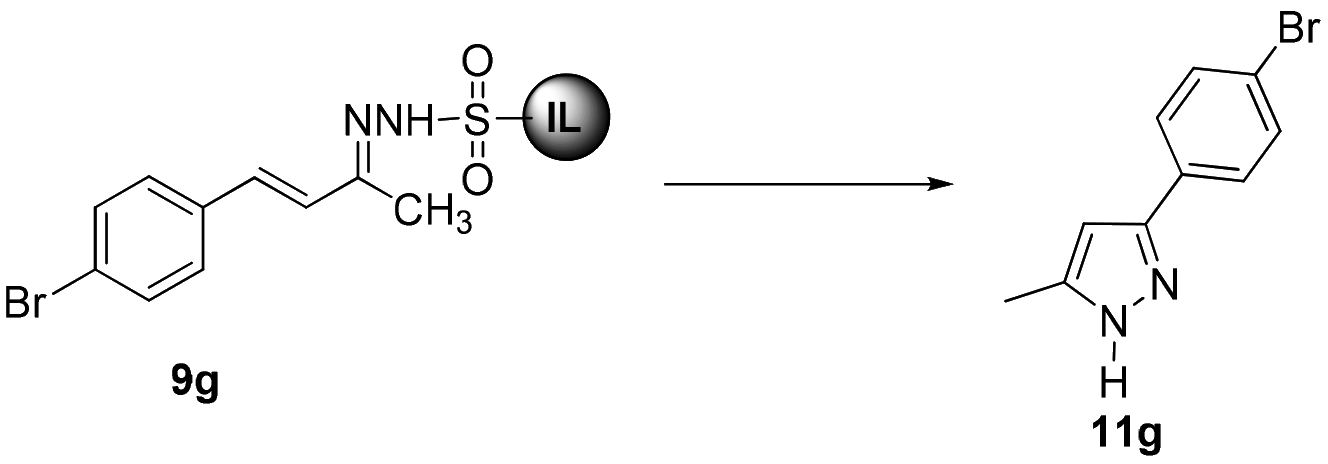
With the optimized conditions in hand, we next investigated the scope and generality of the cyclization, and the results are summarized in Table 3. Various electron-withdrawing and electron-donating groups on the aryl ring of ionic liquid-supported hydrazones 9 were well tolerated in this reaction. It was observed that compound 9 with electron-withdrawing groups (Table 3, entries 1–4 and 6–8) furnished the target pyrazoles in slightly better yields compared to electron-rich groups (Table 3, entries 9–12). Remarkably, thiophene and furan-substituted pyrazoles 9m & 9n could also be obtained under the given conditions in good to moderate yields (Table 3, entries 13 and 14). Ionic liquid-supported sulfonyl hydrazone of cinnamaldehyde 9o resulted in the corresponding pyrazole 11o in moderate yields (Table 3, entry 15). In all cases the product was isolated by simple extraction with an ethyl acetate–hexane mixture (1:1 v/v). It is worth mentioning that the pyrazoles 11 were highly pure and no further chromatographic separation was required. The chemical structures of 11 were determined by 1H and 13C NMR and data for known pyrazoles were in agreement with the reported values. In the 1H NMR spectra of 11a, a characteristic singlet peak appeared around 6.3 ppm for C4-protons of the pyrazole ring along with other protons and a peak corresponding to C4-carbon of the pyrazole ring appeared around 102 ppm in the 13C NMR. However, signals for quaternary C3- and C5-carbons of some 3,5-disubsituted pyrazoles could not be observed due to their longer relaxation time, which is a consequence of the neighboring nitrogen atoms present in the highly conjugated pyrazole ring.28,31
|
|
|||||
|---|---|---|---|---|---|
| Entry | 9 | Ar | R | Product | Yieldb (%) |
| a Reaction conditions: 9 (1 mmol), 3 N NaOH (2 mL), 100 °C for 24 h. b Isolated yield. | |||||
| 1 | 9a | C6H5 | CH3 | 11a | 70 |
| 2 | 9b | 2-FC6H4 | CH3 | 11b | 77 |
| 3 | 9c | 2-ClC6H4 | CH3 | 11c | 70 |
| 4 | 9d | 3-ClC6H4 | CH3 | 11d | 76 |
| 5 | 9e | 3-NO2C6H4 | CH3 | 11e | 33 |
| 6 | 9f | 4-ClC6H4 | CH3 | 11f | 83 |
| 7 | 9g | 4-BrC6H4 | CH3 | 11g | 88 |
| 8 | 9h | 2,6-(Cl)2C6H3 | CH3 | 11h | 93 |
| 9 | 9i | 3-OCH3C6H4 | CH3 | 11i | 57 |
| 10 | 9j | 4-CH3C6H4 | CH3 | 11j | 62 |
| 11 | 9k | 4-OCH3C6H4 | CH3 | 11k | 60 |
| 12 | 9l | 3,4-(OCH3)2C6H3 | CH3 | 11l | 27 |
| 13 | 9m | C4H3S | CH3 | 11m | 80 |
| 14 | 9n | C4H3O | CH3 | 11n | 57 |
| 15 | 9o | C6H5 | H | 11o | 65 |
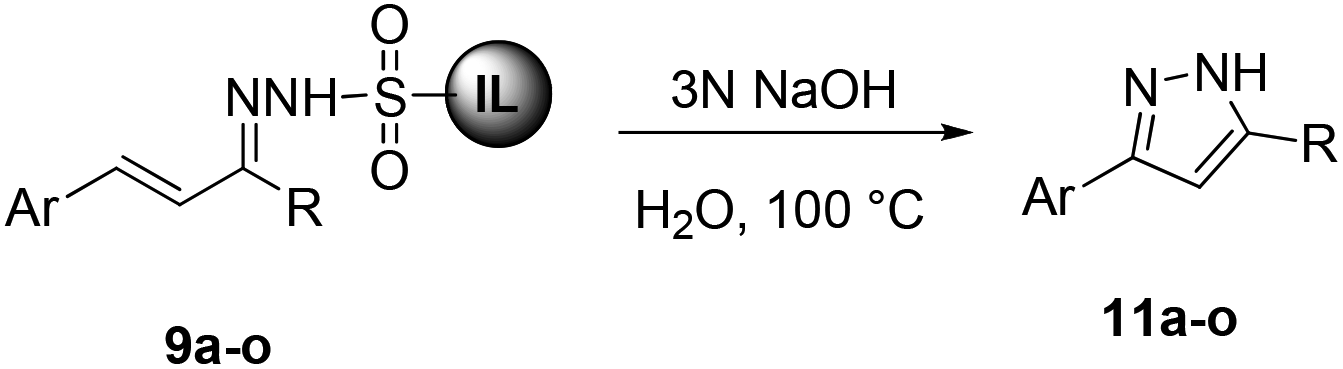
These results encouraged us to further explore an alternative protocol for the synthesis of 3,5-disubstituted pyrazoles by 1,3-dipolar cycloaddition of ionic liquid-supported tosylhydrazones 10 to alkynes 12. To our delight the reaction of tosylhydrazone 10d with phenylacetylene 12a in the presence of 5 N NaOH at 60 °C afforded 3-(4-bromophenyl)-5-phenyl-1H-pyrazole 13da in 85% yield. It is important to mention that the yield of 13da was low when 3 N NaOH and other bases were used in this reaction.
The scope of the reaction was evaluated with regard to the structure of aldehydes and alkynes. As shown in Table 4, various substituted aldehydes worked well with alkynes to provide the corresponding 3,5-disubstituted-1H-pyrazoles 13 in moderate to excellent yields. The reaction could tolerate alkynes and aldehydes with various steric and electronic properties. In general, the electronic effect on the aldehydes had little influence on the reaction; both electron-withdrawing and electron-donating substituents gave good to excellent yields with simple phenylactylene 12a and 4′-methylphenylacetylene 12b. We also found that the reaction could tolerate heteroaryl motifs (Table 4, entry 11). However, for the disubstituted alkynes the yields of the corresponding pyrazole were poor (Table 4, entries 2 and 6). The chemical structures of the synthesized pyrazoles 13 were elucidated using 1H and 13C NMR data (ESI).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 10 | Ar | 12 | R | R1 | Product | Yieldb (%) |
| a Reaction conditions: 10 (1 mmol), 12 (1.5 mmol), 5 N NaOH (2 mL), 60 °C for 24 h. b Isolated yield. | |||||||
| 1 | 10a | C6H5 | 12a | C6H5 | H | 13aa | 90 |
| 2 | 10a | C6H5 | 12a′ | C6H5 | CH3 | 13aa′ | 15 |
| 3 | 10b | 3-ClC6H4 | 12a | C6H5 | H | 13ba | 87 |
| 4 | 10c | 4-ClC6H4 | 12a | C6H5 | H | 13ca | 87 |
| 5 | 10d | 4-BrC6H4 | 12a | C6H5 | H | 13da | 85 |
| 6 | 10d | 4-BrC6H4 | 12a′ | C6H5 | CH3 | 13da′ | 14 |
| 7 | 10e | 2,6-(Cl)2C6H3 | 12a | C6H5 | H | 13ea | 78 |
| 8 | 10e | 2,6-(Cl)2C6H3 | 12b | 4-CH3C6H4 | H | 13eb | 74 |
| 9 | 10f | 4-CH3C6H4 | 12a | C6H5 | H | 13fa | 80 |
| 10 | 10f | 4-CH3C6H4 | 12b | 4-CH3C6H4 | H | 13fb | 83 |
| 11 | 10g | C4H3S | 12a | C6H5 | H | 13ga | 41 |
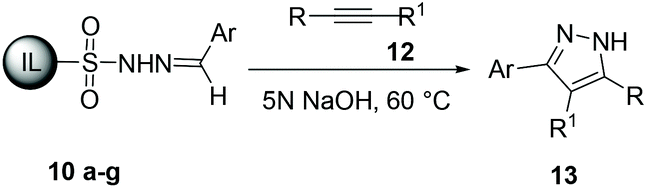
In summary, a facile and economical approach for the traceless synthesis of highly substituted pyrazoles has been developed using ionic liquid-supported sulfonyl hydrazine. A key feature of the protocol is an easy and convenient purification process that avoids chromatographic separation of products and thus makes the method ecofriendly and economical.
Experimental
General information
The NMR spectra were recorded on 300 MHz, 400 MHz and 500 MHz spectrometers using CDCl3 and DMSO-d6 as solvents. The chemical shifts were expressed in ppm. Reactions were monitored by thin-layer chromatography (TLC) carried out on silica-coated aluminum plates (60F-254) using UV light as a visualizing agent. Melting points were determined on an open capillary tube on automated melting point apparatus and are uncorrected. All the chemicals and reagents were purchased at the highest commercial quality and were used without further purification, unless otherwise stated. Ionic liquid-supported sulfonyl hydrazine was prepared from ionic liquid-supported sulfonic acid which was prepared by the reaction of 1,4-butanesultone with 1,2-dimethylimidazole followed by acidification with trifluoromethane sulfonic acid.32General procedure for the synthesis of ionic liquid-supported sulfonyl hydrazones 9a–o & 10a–g
To the ionic liquid-supported sulfonyl hydrazine (6, 1 mmol) in ethanol, α,β-unsaturated carbonyl compounds (7, 1.1 mmol) or aldehydes (8, 1.1 mmol) were added and the resulting solution was stirred at room temperature vigorously for 5 h. After completion of the reaction, ethanol was removed and the resulting mixture was washed with an ethyl acetate–hexane mixture (1:1 v/v) and dried under reduced pressure to obtain the pure product.
General procedure for the synthesis of pyrazoles 11a–o from ionic liquid-supported sulfonyl hydrazones 9a–o
To the ionic liquid-supported sulfonyl hydrazones 9 (1 mmol) 2 mL of 3 N NaOH was added and the reaction mixture was heated at 100 °C for 24 h. The progress of the reaction was monitored by thin-layer chromatography. After completion of the reaction, the product was extracted in ethyl acetate (2 × 10 mL) and dried over sodium sulfate. The combined organic layers were evaporated under reduced pressure to obtain pure compound 11.General procedure for the synthesis of pyrazole 13 from ionic liquid-supported sulfonyl hydrazones 10a–g
Alkyne (1.5 mmol) and 2 mL of 5 N NaOH were added to the ionic liquid-supported sulfonyl hydrazone 10 (1.0 mmol) at room temperature. The resulting solution was stirred for 24 h at 60 °C and the progress of the reaction was monitored by thin-layer chromatography. After completion of the reaction, the product was extracted in ethyl acetate (2 × 10 mL) and dried over sodium sulfate. The combined organic layers were evaporated under reduced pressure to obtain a pure compound.Acknowledgements
We gratefully acknowledge the Council for Scientific and Industrial Research (CSIR), New Delhi for the financial support (02(0115)/13/EMR-II) to carry out this work. SC and MKM thank CSIR, New Delhi for a senior research fellowship (SRF).Notes and references
- A. Schmidt and A. Dreger, Curr. Org. Chem., 2011, 15, 1423 CrossRef CAS.
- D. Kumar and S. P. Singh, Heterocycles, 2004, 63, 145 CrossRef CAS PubMed.
- H. A. Saad, N. A. Osman and A. H. Moustafa, Molecules, 2011, 16, 10187 CrossRef CAS PubMed.
- R. N. Mahajan, F. H. Havaldar and P. S. Fernandes, J. Indian Chem. Soc., 1991, 68, 245 CAS.
- Z. Özdemir, H. B. Kandilci, B. Gümüşel, Ü. Çalış and A. A. Bilgin, Eur. J. Med. Chem., 2007, 42, 373 CrossRef PubMed.
- H. A. DeWald, S. Lobbestael and B. P. H. Poschel, J. Med. Chem., 1981, 24, 982 CrossRef CAS.
- E. Nassar, H. A. Abdel-Aziz, H. S. Ibrahim and A. M. Mansour, Sci. Pharm., 2011, 79, 507 CrossRef CAS PubMed.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef CAS PubMed.
- M. Ezawa, D. S. Garvey, D. R. Janero, S. P. Khanapure, L. G. Letts, A. Martino, R. R. Ranatunge, D. J. Schwalb and D. V. Young, Lett. Drug Des. Discov., 2005, 2, 40 CAS.
- S. R. Donohue, R. F. Dannals, C. Halldin and V. W. Pike, J. Med. Chem., 2011, 54, 2961 CrossRef CAS PubMed.
- M. Gao, M. Wang and Q.-H. Zheng, Bioorg. Med. Chem. Lett., 2012, 22, 3704 CrossRef CAS PubMed.
- S. B. Kumar, S. Darbu, S. Kumar and R. H. Gupta, Recent Pat. Anti-Infective Drug Discov., 2009, 4, 154 CrossRef CAS.
- J. Xie, G. I. Poda, Y. Hu, N. X. Chen, R. F. Heier, S. G. Wolfson, M. T. Reding, P. J. Lennon, R. G. Kurumbail, S. R. Selness, X. Li, N. N. Kishore, C. D. Sommers, L. Christine, S. L. Bonar, N. Venkatraman, S. Mathialagan, S. J. Brustkern and H.-C. Huang, Bioorg. Med. Chem., 2011, 19, 1242 CrossRef CAS PubMed.
- M. J. Genin, C. Biles, B. J. Keiser, S. M. Poppe, S. M. Swaney, W. G. Tarpley, Y. Yagi and D. L. Romero, J. Med. Chem., 2000, 43, 1034 CrossRef CAS PubMed.
- S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984 CrossRef CAS PubMed.
- S. Kumar, H. Ila and H. Junjappa, J. Org. Chem., 2009, 74, 7046 CrossRef CAS PubMed.
- Y. L. Janin, Chem. Rev., 2012, 112, 3924 CrossRef CAS PubMed.
- K. Makino, H. S. Kim and Y. Kurasawa, J. Heterocycl. Chem., 1998, 35, 489 CrossRef CAS.
- X. Deng and N. S. Mani, Org. Lett., 2008, 10, 1307 CrossRef CAS PubMed.
- X. Deng and N. S. Mani, J. Org. Chem., 2008, 73, 2412 CrossRef CAS PubMed.
- X. Deng and N. S. Mani, Org. Lett., 2006, 8, 3505 CrossRef CAS PubMed.
- S. V. Kumar, S. K. Yadav, B. Raghava, B. Saraiah, H. Ila, K. S. Rangappa and A. Hazra, J. Org. Chem., 2013, 78, 4960 CrossRef CAS PubMed.
- F. Gosselin, P. D. O'Shea, R. A. Webster, R. A. Reamer, R. D. Tillyer and E. J. J. Grabowski, Synlett, 2006, 3267 CrossRef CAS PubMed.
- Z.-X. Wang and H.-L. Qin, Green Chem., 2004, 6, 90 RSC.
- S. Fustero, R. Román, J. F. Sanz-Cervera, A. Simón-Fuentes, A. C. Cuñat, S. Villanova and M. Murguía, J. Org. Chem., 2008, 73, 3523 CrossRef CAS PubMed.
- V. K. Rao, R. Tiwari, B. S. Chhikara, A. N. Shirazi, K. Parang and A. Kumar, RSC Adv., 2013, 3, 15396 RSC.
- D. Vuluga, J. Legros, B. Crousse and D. Bonnet-Delpon, Green Chem., 2009, 11, 156 RSC.
- V. K. Aggarwal, J. de Vicente and R. V. Bonnert, J. Org. Chem., 2003, 68, 5381 CrossRef CAS PubMed.
- L.-L. Wu, Y.-C. Ge, T. He, L. Zhang, X.-L. Fu, H.-Y. Fu, H. Chen and R.-X. Li, Synthesis, 2012, 1577 CAS.
- Y. Kong, M. Tang and Y. Wang, Org. Lett., 2013, 16, 576 CrossRef PubMed.
- P. Liu, Q.-Q. Xu, C. Dong, X. Lei and G.-q. Lin, Synlett, 2012, 2087 CAS.
- A. Kumar, M. K. Muthyala, S. Choudhary, R. K. Tiwari and K. Parang, J. Org. Chem., 2012, 77, 9391 CrossRef CAS PubMed.
- A. Corradi, C. Leonelli, A. Rizzuti, R. Rosa, P. Veronesi, R. Grandi, S. Baldassari and C. Villa, Molecules, 2007, 12, 1482 CrossRef CAS.
- G. Zhang, H. Ni, W. Chen, J. Shao, H. Liu, B. Chen and Y. Yu, Org. Lett., 2013, 15, 5967 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data of ionic liquid-supported sulfonyl hydrazones 9, copies of 1H NMR and 13C NMR of ionic liquid-supported sulfonyl hydrazones 9, and pyrazoles 11 & 13. See DOI: 10.1039/c4qo00092g |
| This journal is © the Partner Organisations 2014 |