Palladium-catalyzed, copper-mediated construction of benzene rings from the reactions of indoles with in situ generated enones†
Tenglong
Guo
a,
Quanbin
Jiang
a,
Fei
Huang
a,
Jiping
Chen
a and
Zhengkun
Yu
*ab
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, Liaoning 116023, P. R. China. E-mail: zkyu@dicp.ac.cn; Fax: +86-411-8437-9227
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Road, Shanghai 200032, P. R. China
First published on 13th June 2014
Abstract
Construction of the benzene ring in carbazoles was efficiently realized through a domino dehydrochlorination/alkenylation/cycloaddition–oxidation sequence by means of palladium(II)-catalyzed, copper(II)-mediated reactions of N-protected 2,3-unsubstituted indoles with 3-chloropropiophenones in the presence of a base. 3-Alkenylated indole was confirmed to be formed as the reaction intermediate which then underwent Diels–Alder cycloaddition to the initially in situ generated enone from a 3-chloropropiophenones substrate, and the subsequent dehydrogenative aromatization yielded the carbazole product. The strategy to employ in situ generated enones as the reactive species avoided the use of a large excess of labile substrates and lessened the side reactions.
Construction of benzene rings is usually the key step to establish important aromatic systems.1 Functionalized aromatics are traditionally prepared through aromatic electrophilic substitution reactions or by transition-metal catalyzed multistep transformations generally involving alkynes.2 Polycyclic (hetero)-arenes have demonstrated valuable utility in synthetic chemistry, and their concise synthesis is strongly desired from the reactions of readily available building blocks, among which formal oxidative cross-coupling of three alkenes seems to be a promising route to a benzene ring.3
Carbazoles are an important class of aromatic heterocyclic nuclei as structural motifs in many synthetic compounds and naturally occurring alkaloids.4 The cross-coupling reactions of C–H/C–X bonds (X = halo, N, O, C, etc.) have been employed for their synthesis.5–9 Oxidative intramolecular C–H/C–H cross-coupling of prefunctionalized diarylamines was realized for the same purpose.10 2-Aryl and 2-heteroarylindoles were reported to react with alkynes to prepare carbazoles.11,12 Using a trimetallic system, i.e., Pd(OAc)2/Cu(OAc)2/Ag(OCOCF3), N-methylindoles reacted with 10 equiv. of methyl or phenyl vinyl ketone to form diacetyl- and dibenzoylcarbazoles, respectively.13 Although indoles can react with α,β-unsaturated carbonyls,14 synthesis of carbazoles from their reactions with electron-deficient alkenes is still challenging because aryl vinyl ketones (enones) are usually not commercially available due to their susceptibility to heat, light and oxygen during preparation and storage. Palladium-catalyzed dehydrogenation of alkyl ketones was thus documented to prepare enones, but the synthesis generally requires high catalyst loading and relatively harsh conditions.15 As an alternative route, in situ generation of enones has recently attracted attention using saturated alkyl ketones and β-keto esters as the alkene sources.15c,16
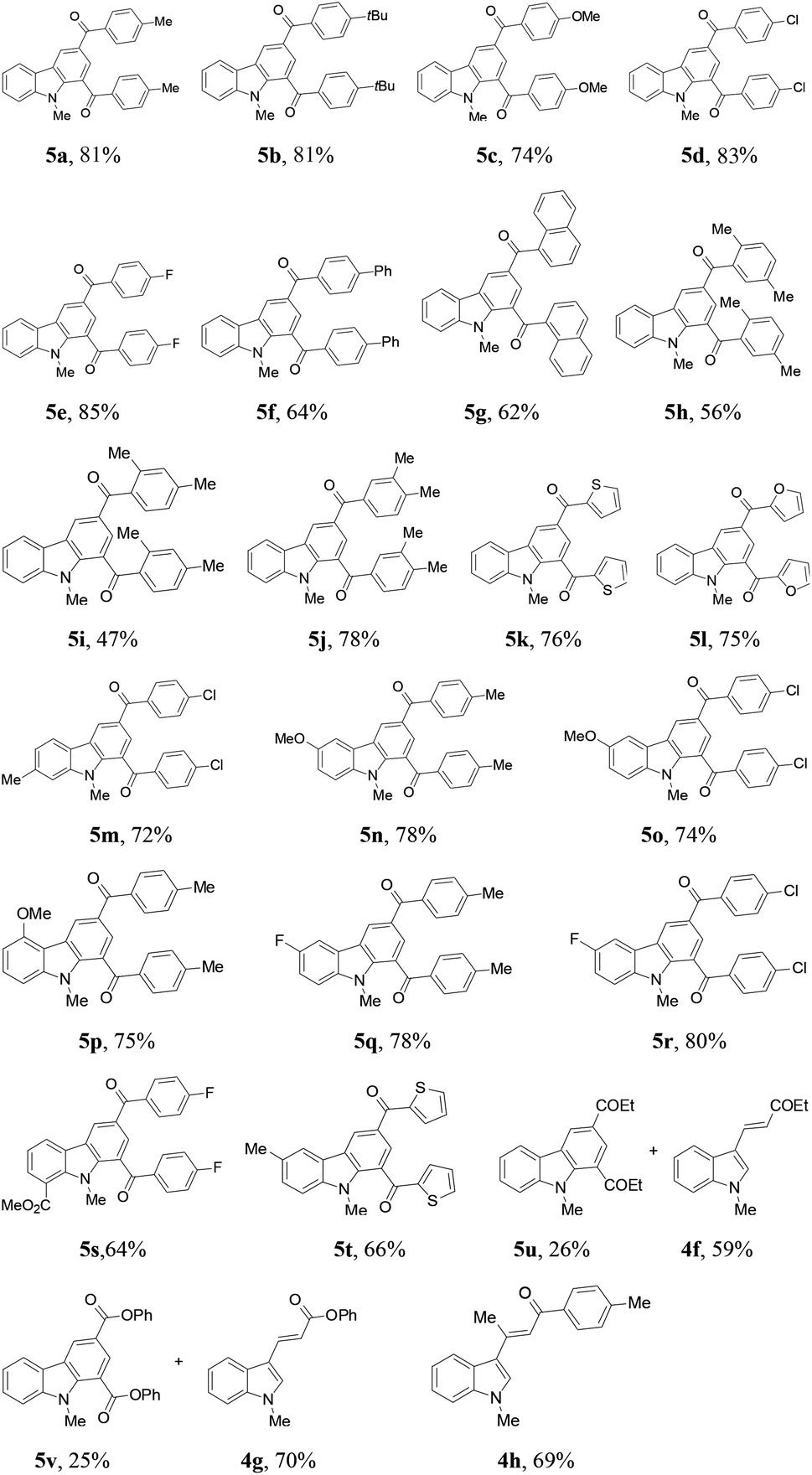
Recently, we found that β-chloroalkyl aryl ketones and their ester and amide derivatives can be utilized as precursors to α,β-unsaturated carbonyls in Rh(I)-catalyzed conjugate addition by arylboronic acids.17 Under an oxygen atmosphere, PdCl2-catalyzed trimerization of phenyl vinyl ketone afforded 1,3,5-tribenzoylbenzene in 25% yield.3b We thus envisioned that 3-chloropropiophenone might undergo the same reaction via the in situ generated enone intermediate. To our delight, such a reaction occurred to give the target trimerization product in 20% yield (Scheme 1a). Retrosynthetic analysis suggests that construction of a benzene ring is plausible from the reaction of a 3-chloroalkyl carbonyl (as the enone precursor) with indole (as an alkene building block) because palladium-catalyzed indole alkenylation with electron-deficient acrylates and alkenes has been well-known.18 Herein, we report the synthesis of functionalized carbazoles by the reactions of 3-chloroalkyl ketones with indoles through a domino sequence19 (Scheme 1b).
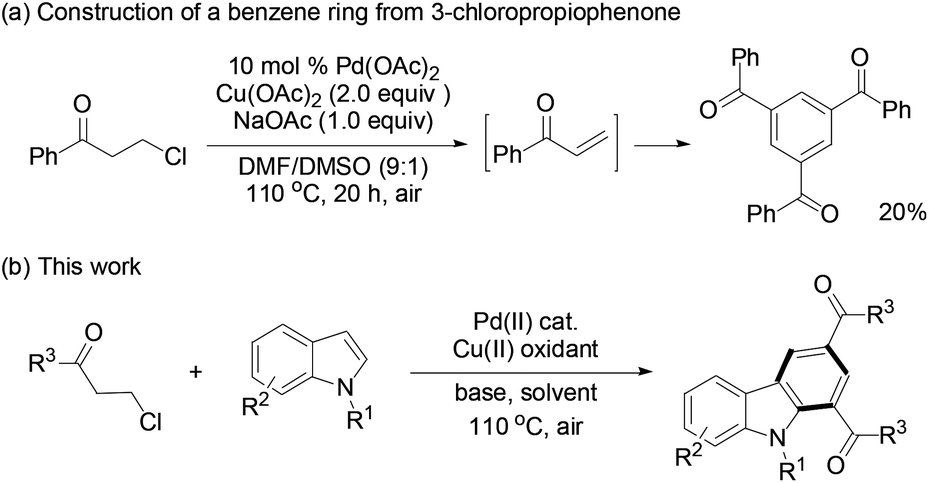 | ||
| Scheme 1 Synthesis of carbazoles via in situ generated enones. | ||
Initially, N-methylindole (1a) was reacted with 3-chloropropiophenone (2a) in dioxane at 100 °C under a nitrogen atmosphere using 10 mol% Pd(OAc)2 as the catalyst, Cu(OAc)2 as the oxidant, and Na2CO3 as the base, giving the desired product 3a in 10% yield (Table 1, entry 1). Changing the solvent to DMSO or DMF improved the reaction efficiency, and a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DMF and DMSO promoted the reaction to form 3a in 74% yield (Table 1, entries 2–4). Cu(OAc)2 acted as the most effective oxidant among those screened, i.e., Cu(OAc)2, Cu(OAc)2·H2O, AgOAc, AgCO3, benzoquinone (BQ), and tBuOOtBu, while the reaction hardly occurred without an added oxidant or by using CuOAc as the sole oxidant (Table 1, entries 5 and 6). Elevating the temperature to 110 °C improved the reaction (Table 1, entry 7). A catalyst was necessary for the reaction, and Pd(OAc)2 was shown to be the most efficient one among the screened Pd(II) and Pd(0) sources (see the ESI†). A base was required for the reaction, and NaOAc behaved better than Na2CO3. An air atmosphere slightly improved the yield of 3a (82%), but oxygen as the sole oxidant did not facilitate the reaction (Table 1, entry 12). Using less Cu(OAc)2 reduced the yield of 3a (Table 1, entry 13). Although both CuOAc and oxygen cannot be used as the sole oxidants, their combination worked well (Table 1, entry 14). A reduced loading of the catalyst (5 mol%) led to a lower yield of 3a (70%) (Table 1, entry 15). It should be noted that only a trace amount of 4a was detected during the reaction.
:1 mixture of DMF and DMSO promoted the reaction to form 3a in 74% yield (Table 1, entries 2–4). Cu(OAc)2 acted as the most effective oxidant among those screened, i.e., Cu(OAc)2, Cu(OAc)2·H2O, AgOAc, AgCO3, benzoquinone (BQ), and tBuOOtBu, while the reaction hardly occurred without an added oxidant or by using CuOAc as the sole oxidant (Table 1, entries 5 and 6). Elevating the temperature to 110 °C improved the reaction (Table 1, entry 7). A catalyst was necessary for the reaction, and Pd(OAc)2 was shown to be the most efficient one among the screened Pd(II) and Pd(0) sources (see the ESI†). A base was required for the reaction, and NaOAc behaved better than Na2CO3. An air atmosphere slightly improved the yield of 3a (82%), but oxygen as the sole oxidant did not facilitate the reaction (Table 1, entry 12). Using less Cu(OAc)2 reduced the yield of 3a (Table 1, entry 13). Although both CuOAc and oxygen cannot be used as the sole oxidants, their combination worked well (Table 1, entry 14). A reduced loading of the catalyst (5 mol%) led to a lower yield of 3a (70%) (Table 1, entry 15). It should be noted that only a trace amount of 4a was detected during the reaction.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Oxidant | Base | Temp (°C) | Yieldb of 3a (%) |
|
a Reaction conditions: 1a (0.2 mmol), 2a (0.8 mmol), catalyst (0.02 mmol), base (0.8 mmol), oxidant (1.2 mmol), solvent (2.5 mL, DMF–DMSO, v/v = 9:1), 0.1 MPa N2, 20 h.
b Isolated yield.
c In dioxane.
d In DMSO.
e In DMF.
f In air.
g Cu(OAc)2 (1.0 mmol) was used.
h Using 5 mol% catalyst.
|
|||||
| 1c | Pd(OAc)2 | Cu(OAc)2 | Na2CO3 | 100 | 10 |
| 2d | Pd(OAc)2 | Cu(OAc)2 | Na2CO3 | 100 | 43 |
| 3e | Pd(OAc)2 | Cu(OAc)2 | Na2CO3 | 100 | 51 |
| 4 | Pd(OAc)2 | Cu(OAc)2 | Na2CO3 | 100 | 74 |
| 5 | Pd(OAc)2 | Na2CO3 | 100 | <1 | |
| 6 | Pd(OAc)2 | CuOAc | Na2CO3 | 100 | <1 |
| 7 | Pd(OAc)2 | Cu(OAc)2 | Na2CO3 | 110 | 77 |
| 8 | Pd(OAc)2 | Cu(OAc)2 | Na2CO3 | 120 | 73 |
| 9 | Cu(OAc)2 | Na2CO3 | 110 | 0 | |
| 10 | Pd(OAc)2 | Cu(OAc)2 | NaOAc | 110 | 81 |
| 11f | Pd(OAc)2 | Cu(OAc)2 | NaOAc | 110 | 82 |
| 12f | Pd(OAc)2 | Air | NaOAc | 110 | <1 |
| 13f,g | Pd(OAc)2 | Cu(OAc)2 | NaOAc | 110 | 63 |
| 14f | Pd(OAc)2 | CuOAc | NaOAc | 110 | 51 |
| 15f,h | Pd(OAc)2 | Cu(OAc)2 | NaOAc | 110 | 70 |
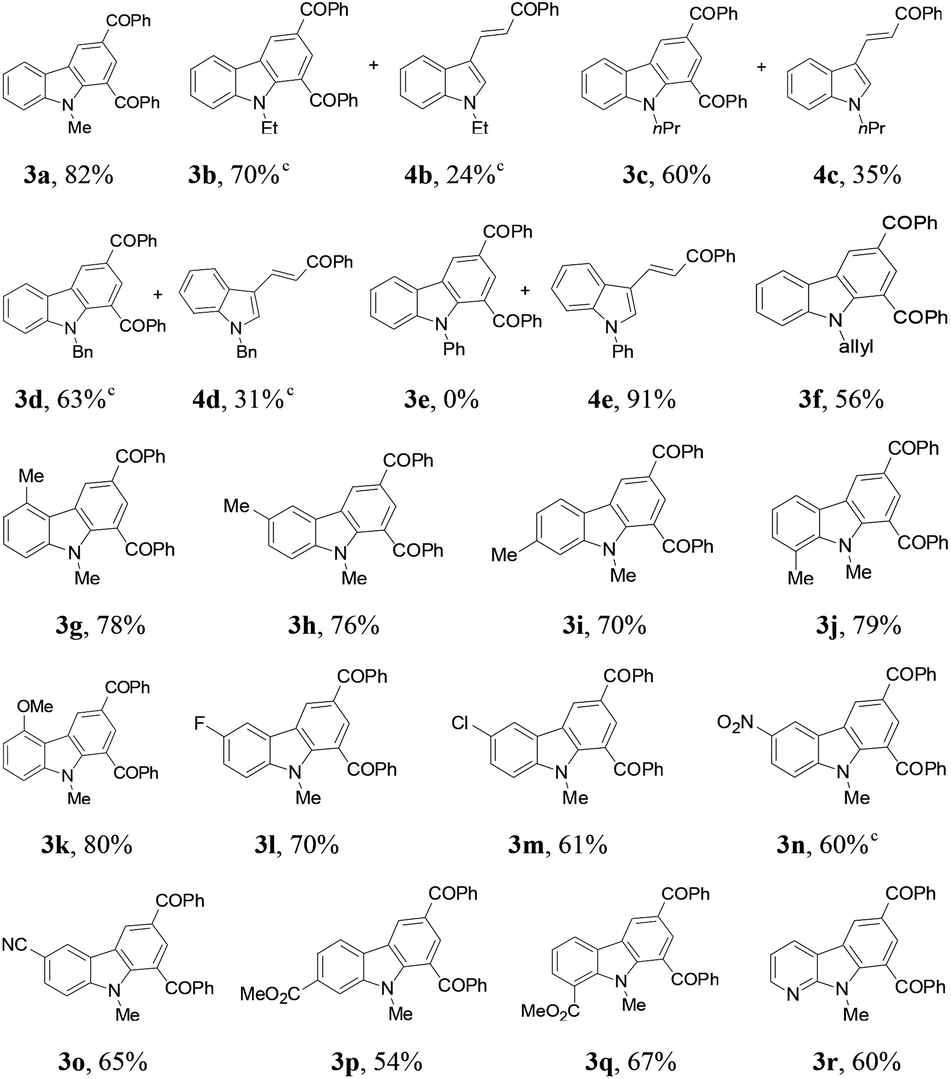
Next, the indole scope was explored to probe the protocol generality (Table 2). As the steric hindrance of the N-R moiety in indoles was increased from methyl to ethyl, n-propyl, benzyl, and phenyl, yields of the desired products were gradually decreased from 82% (R = Me for 3a) to 0% (R = Ph for 3e). 3-Alkenylated indoles 4b–4d were formed as the minor products (24–35%), while in the case of N-phenylindole, 4e was formed as the only product (91%). The reaction of N-allylindole with 2a afforded 3f (56%) with its allyl functional group unchanged. The electron-donating substituents on the aryl rings of the indole substrates did not obviously affect the reaction efficiency as 3g–3k were obtained in 70–80% yields, whereas the electron-withdrawing fluoro, chloro, nitro, cyano, and CO2Me groups lessened the formation of the desired products 3l–3q (54–70%). N-Methyl-7-azaindole also underwent the reaction with 2a to form 3r (60%). The molecular structures of compounds 3 were further confirmed by X-ray single crystal structural determination of 3i (Fig. 1).
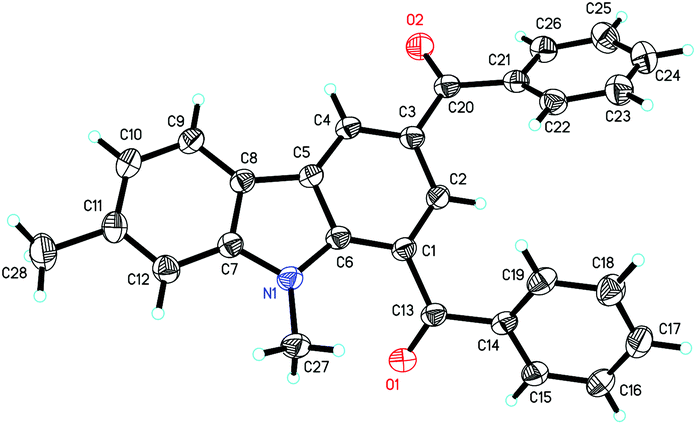 | ||
| Fig. 1 Molecular structure of 3i. | ||
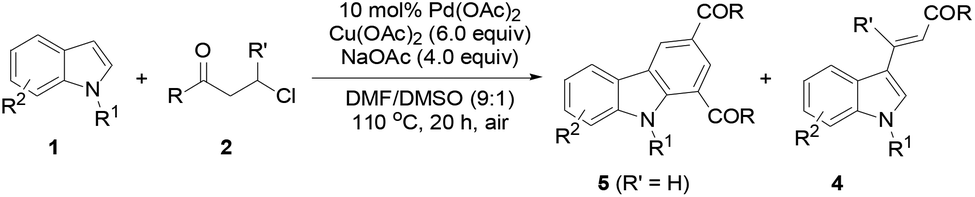
The protocol generality was then investigated by reacting indoles with various 3-chloroalkyl ketones (Table 3). Treatment of 1a with substituted 3-chloroalkyl ketones (2) formed the desired products 5a–5e in 74–85% yields. Increasing steric hindrance of the aryl moiety in 2 deteriorated the production of 5f–5i (47–64%), and the reduced steric hindrance from 3,4-dimethyl groups only had a slight influence on the yield of 5j (78%). Heteroaryl 3-chloropropiophenones underwent the same reactions to generate 1,3-diheteroaroylcarbazoles 5k (76%) and 5l (75%), respectively. Substituted N-methylindoles also efficiently reacted with 3-chloroalkyl aryl ketones to afford products 5m–5r (72%–80%). A 7-CO2Me in 1 or thienyl in 2 led to reduced yields for 5s (64%) and 5t (66%), respectively. However, the reaction of 5-chloropentan-3-one with 1a only gave 5u in 26% yield with 3-alkenylated indole 4f (59%) as the major product. Similar results were obtained in the case of 3-chloropropionic acid phenyl ester, forming 5v (25%) and 4g (70%). The β-methyl group in 2 completely inhibited the target reaction, resulting in 4h (69%) as the only product.
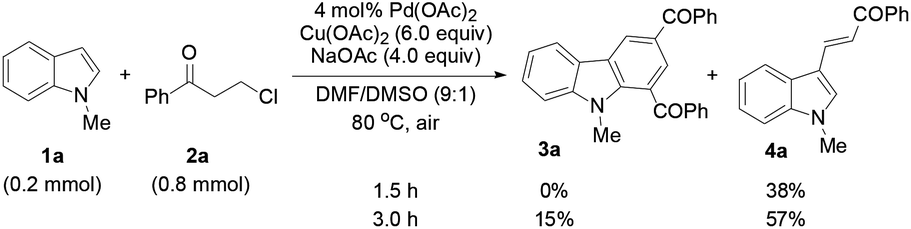
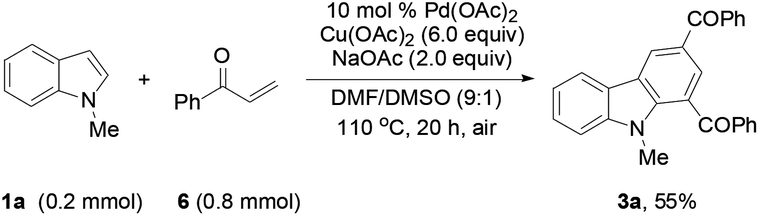
To determine whether the 3-alkenylated indole of type 4 was an intermediate, the reaction of 1a with 2a was conducted at 80 °C using 4 mol% catalyst (eqn (1)). It was found that the reaction initially formed 4a, which was then gradually converted to the desired product 3a. Analysis of the reaction mixture at 3 h by GC revealed the presence of unreacted enone 6.17 Reacting 1a with 6 under the typical conditions afforded 3a in 55% yield (eqn (2)), which is comparable with the 53% yield of the same reaction with 10 equiv. of 6 by means of the trimetallic system.13 These results have revealed that the present bimetallic system employing in situ generated enone 6 is efficient for carbazole synthesis.
 | (1) |
 | (2) |
To probe further into the reaction pathway of the 3-alkenylated indole intermediate with the in situ generated enone species, the controlled reaction of 4a with 2a was investigated (Table 4). Deviation of the standard conditions by omitting one or two reaction parameters changed the formation of 3a. Under the standard conditions as shown in eqn (3), 3a was formed in 75% yield (Table 4, entry 1). Without Pd(OAc)2 as the catalyst, the reaction was not affected much to give 3a (78% yield in air, 83% yield under atmospheric nitrogen) (Table 4, entries 2 and 3). Without Cu(OAc)2, or in the absence of both Pd(OAc)2 and Cu(OAc)2, 3a was still obtained in 48–51% yields, whereas the same reaction became complicated under a nitrogen atmosphere (Table 4, entries 4–6), suggesting that an added oxidant or air should be used as the oxidant for the reaction to occur properly. These results have revealed that a palladium catalyst is not necessary for the reaction as shown in eqn (3), and the reaction can occur without Cu(OAc)2 in air, but an added oxidant such as Cu(OAc)2 promotes the reaction better than air.

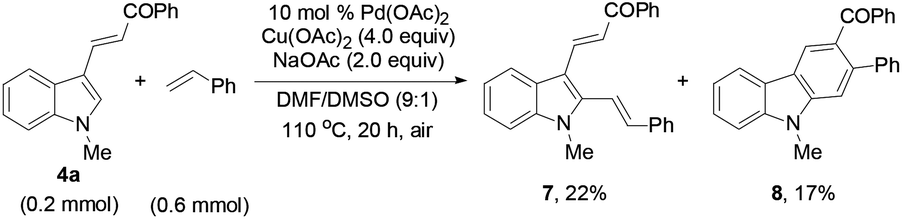
Reacting 4a with styrene under the standard conditions (eqn (4)) afforded 2,3-dialkenylated indole 7 (22%) and carbazole 8 (17%) through multiple C–H activation of the indole substrate. The X-ray single crystal structure of 8 was also confirmed (Fig. 2). No product of type 7 was obtained from the reaction of 4a with 2a, excluding a C–H activation pathway. These results led us to deduce that Diels–Alder cycloaddition and subsequent oxidation were involved in forming the benzene ring in the overall reaction.
 | (4) |
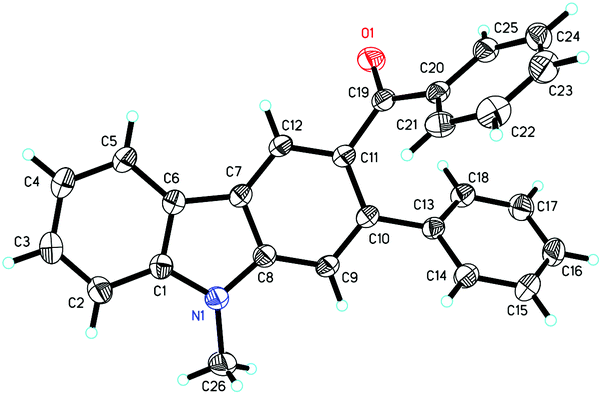 | ||
| Fig. 2 Molecular structure of 8. | ||
A proposed mechanism is depicted by the reaction of 1a with 2a (Scheme 2). The indole substrate initially undergoes palladation at its 3-position to form a palladated species A and HOAc. A reacts with the in situ generated enone 6 from 2a to yield an alkene insertion species B, which undergoes reductive elimination to produce 3-alkenylated indole 4a and a Pd(0) species. A Diels–Alder cycloaddition of 6 to 4a forms tetrahydrocarbazole C, which is subsequently oxidized to form the desired product 3a. Air facilitates regeneration of the Cu(II) oxidant and the Pd(II) catalyst.
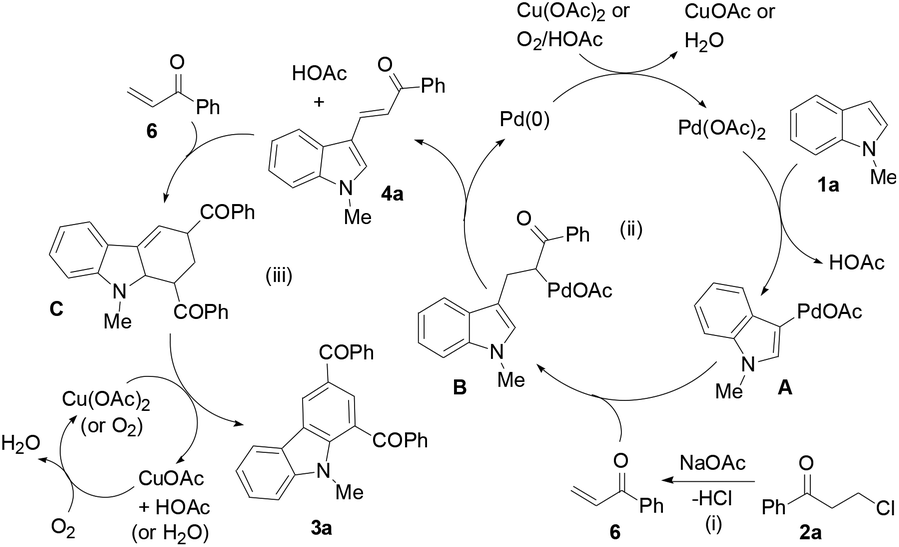 | ||
| Scheme 2 Proposed mechanism. | ||
Finally, the protocol was extended to the reactions of N-unprotected indoles. It was found that the N-unprotected analogue of 4a reacted with 2a in the presence of NaOAc to form the N-(3-oxo-3-phenylpropyl)-substituted product 9 (39%), affording no desired carbazole product (eqn (5)). For the versatile synthesis of carbazoles, the reactions of 4a with 3-chloroalkyl aryl ketones were performed, producing carbazoles 10a (74%) and 10b (78%) bearing two different substituents (eqn (6)). A one-pot, two-step procedure was also established to synthesize the mixed aroyl-substituted carbazoles 10a–10d (55–63%) (Scheme 3), providing a potentially applicable route to functionalized carbazoles.
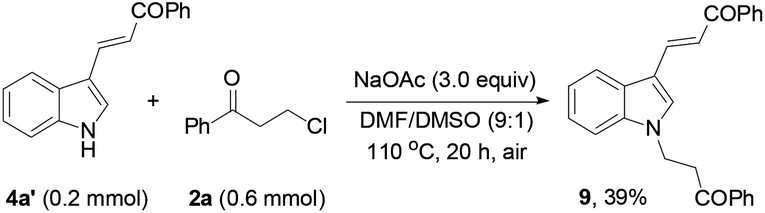 | (5) |
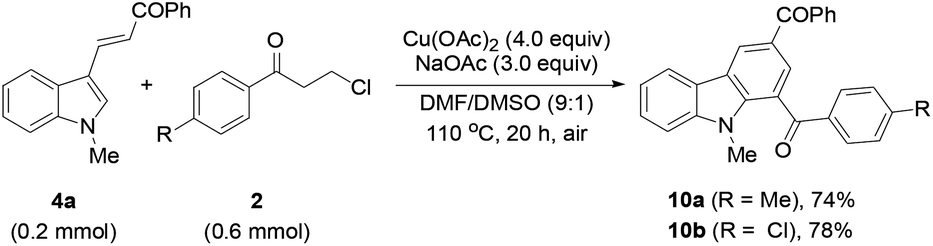 | (6) |
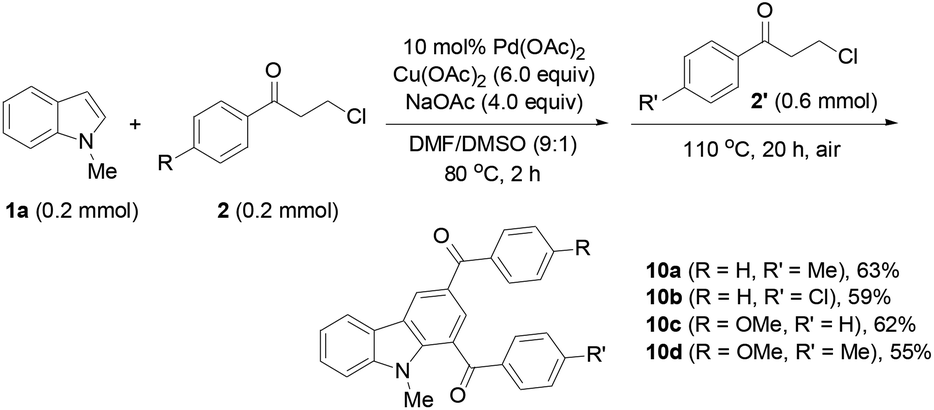 | ||
| Scheme 3 One-pot synthesis of functionalized carbazoles. | ||
In summary, palladium(II)-catalyzed, copper(II)-mediated synthesis of carbazoles has been efficiently realized through a domino dehydrochlorination/alkenylation/cycloaddition–oxidation sequence by means of the reactions of N-protected indoles with 3-chloropropiophenones in the presence of a base. The strategy employed in situ generated α,β-unsaturated carbonyls to avoid using a large excess of labile substrates and to lessen side reactions. The present method provides a concise route to functionalized carbazoles.
Experimental section
A typical procedure for the synthesis of carbazoles: synthesis of 3a
A mixture of N-methylindole (1a) (26 mg, 0.2 mmol), 3-chloropropiophenone (2a) (133 mg, 0.8 mmol), Pd(OAc)2 (4.5 mg, 0.02 mmol), Cu(OAc)2 (218 mg, 1.2 mmol), and NaOAc (66 mg, 0.8 mmol) in 2.5 mL DMF–DMSO (v/v = 9:1) was stirred at 110 °C under an air atmosphere for 20 h. After cooling to ambient temperature, 10 mL CH2Cl2 was added and the resulting mixture was filtered through a short pad of silica gel, followed by rinsing with 20 mL CH2Cl2. The combined filtrate was washed with brine (15 mL) and separated. The organic phase was dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (eluent: petroleum ether (60–90 °C)–EtOAc–CH2Cl2 = 30:1:2, v/v/v) to afford 3a as a white solid (64 mg, 82%).
Acknowledgements
We are grateful to the National Natural Science Foundation of China (21272232) for financial support of this research.Notes and references
- R. E. Maleczka Jr., Science, 2009, 323, 1572–1573 CrossRef PubMed.
- P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307–2327 CrossRef CAS.
- (a) P. Hu, S. J. Huang, J. Xu, Z.-J. Shi and W. P. Su, Angew. Chem., Int. Ed., 2011, 50, 9926–9930 CrossRef CAS PubMed; (b) H.-F. Jiang, Y.-X. Shen and Z.-Y. Wang, Tetrahedron Lett., 2007, 48, 7542–7545 CrossRef CAS PubMed; (c) C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2010, 132, 17710–17712 CrossRef CAS PubMed.
- A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193–3328 CrossRef CAS PubMed.
- (a) R. Y. Huang, P. T. Franke, N. Nicolaus and M. Lautens, Tetrahedron, 2013, 69, 4395–4402 CrossRef CAS PubMed; (b) T. Nanjo, C. Tsukano and Y. Takemoto, Org. Lett., 2012, 14, 4270–4273 CrossRef CAS PubMed; (c) S. Cacchi, G. Fabrizi, A. Goggiamani and A. Iazzetti, Org. Biomol. Chem., 2012, 10, 9142–9147 RSC; (d) D. Tsvelikhovsky and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14048–14051 CrossRef CAS PubMed; (e) L. Ackermann, A. Althammer and P. Mayer, Synthesis, 2009, 3493–3503 CrossRef CAS PubMed; (f) L. Ackermann and A. Althammer, Angew. Chem., Int. Ed., 2007, 46, 1627–1629 CrossRef CAS PubMed.
- (a) Y. Ou and N. Jiao, Chem. Commun., 2013, 49, 3473–3475 RSC; (b) Z. Qi and X. W. Li, Angew. Chem., Int. Ed., 2013, 52, 8995–9000 CrossRef CAS PubMed.
- (a) C. S. Nervig, P. J. Waller and D. Kalyani, Org. Lett., 2012, 14, 4838–4841 CrossRef CAS PubMed; (b) M. Abid, A. Spaeth and B. Török, Adv. Synth. Catal., 2006, 348, 2191–2196 CrossRef CAS.
- M. Yamashita, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 2337–2340 CrossRef CAS PubMed.
- (a) W. Q. Kong, C. L. Fu and S. M. Ma, Org. Biomol. Chem., 2012, 10, 2164–2173 RSC; (b) W. Q. Kong, C. L. Fu and S. M. Ma, Chem. Commun., 2009, 4572–4574 RSC.
- J. Zhao and R. C. Larock, J. Org. Chem., 2006, 71, 5340–5348 CrossRef CAS PubMed.
- Z. Z. Shi, B. Zhang, Y. X. Cui and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 4036–4041 CrossRef CAS PubMed.
- (a) T. Tsuchimoto, H. Matsubayashi, M. Kaneko, Y. Nagase, T. Miyamura and E. Shirakawa, J. Am. Chem. Soc., 2008, 130, 15823–15835 CrossRef CAS PubMed; (b) Z. Z. Shi, S. T. Ding, Y. X. Cui and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 7895–7898 CrossRef CAS PubMed.
- K. Ozaki, H. Zhang, H. Ito, A. W. Lei and K. Itami, Chem. Sci., 2013, 4, 3416–3420 RSC.
- (a) Q. Cai and S.-L. You, Org. Lett., 2012, 14, 3040–3043 CrossRef CAS PubMed; (b) S. R. Kandukuri, J. A. Schiffner and M. Oestreich, Angew. Chem., Int. Ed., 2012, 51, 7428–7431 CrossRef PubMed; (c) Q. Cai, Z.-A. Zhao and S.-L. You, Angew. Chem., Int. Ed., 2009, 48, 7428–7431 CrossRef CAS PubMed.
- For selected recent examples, see: (a) T. N. Diao, T. J. Wadzinski and S. S. Stahl, Chem. Sci., 2012, 3, 887–891 RSC; (b) W. M. Gao, Z. Q. He, Y. Qian, J. Zhao and Y. Huang, Chem. Sci., 2012, 3, 883–886 RSC; (c) Y. Moon, D. Kwon and S. Hong, Angew. Chem., Int. Ed., 2012, 51, 11333–11336 CrossRef CAS PubMed; (d) T. N. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566–14569 CrossRef CAS PubMed.
- (a) Y. P. Shang, X. M. Jie, J. Zhou, P. Hu, S. J. Huang and W. P. Su, Angew. Chem., Int. Ed., 2013, 52, 1299–1303 CrossRef CAS PubMed; (b) J. Zhou, G. Wu, M. Zhang, X. M. Jie and W. P. Su, Chem. – Eur. J., 2012, 18, 8032–8036 CrossRef CAS PubMed; (c) M. V. Leskinen, K.-T. Yip, A. Valkonen and P. M. Pihko, J. Am. Chem. Soc., 2012, 132, 5750–5753 CrossRef PubMed; (d) S. Ueno, R. Shimizu and R. Kuwano, Angew. Chem., Int. Ed., 2009, 48, 4543–4545 CrossRef CAS PubMed.
- Q. B. Jiang, T. L. Guo, Q. F. Wang, P. Wu and Z. K. Yu, Adv. Synth. Catal., 2013, 355, 1874–1880 CrossRef CAS.
- N. P. Grimster, C. Gauntlett, C. R. A. Godfrey and M. J. Gaunt, Angew. Chem., Int. Ed., 2005, 44, 3125–3129 CrossRef CAS PubMed.
- L. F. Tietze, Chem. Rev., 1996, 96, 115–136 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, compound characterization and NMR spectra. CCDC 973030–973031. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00122b |
| This journal is © the Partner Organisations 2014 |