
N-Allenylnitrone acts as 2-azadiene in the Cu-catalyzed cascade reaction of O-propargylic oximes with azodicarboxylates†
Itaru
Nakamura
*a,
Takeru
Jo
b,
Dong
Zhang
b and
Masahiro
Terada
ab
aResearch and Analytical Center for Giant Molecules, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. E-mail: itaru-n@m.tohoku.ac.jp
bDepartment of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan
First published on 7th July 2014
Abstract
The reaction of O-propargylic oximes with azodicarboxylates efficiently afforded 1,2,4-triazine oxides in good yields. The key intermediate, N-allenylnitrone, acted as 2-azadiene, undergoing stepwise [4 + 2] cycloaddition.
π-Acidic metal-catalyzed rearrangements have been efficiently utilized for cascade reactions because the transformations proceed under mild reaction conditions and tolerate a wide variety of functional groups to afford reactive intermediates that are inaccessible by conventional organic synthesis.1,2 The key intermediates often feature multiple reactivities as seen in the homoallyl cation–cyclopropylcarbenoid–cyclobutyl cation in the enyne cycloisomerization3 and allene–enol–vinylcarbenoid in the catalytic skeletal rearrangement of propargylic esters.4 Those multiple reactivities are typically controlled by the sophisticated design of the starting materials or the appropriate choice of reagents for the synthesis of diverse organic molecules. Thus, the discovery of new reaction modes of the reactive intermediates is a fundamental challenge in the development of catalytic cascade reactions. We have recently demonstrated that N-allenylnitrone intermediate A, which is generated from O-propargylic oxime 1via a metal-catalyzed 2,3-rearrangement, acts as a nitrogen analog of vinylallene and undergoes further transformations, such as thermal electrocyclization (type a)5 and aza-metallacyclic formation6 (type b). Moreover, we have disclosed that N-allenylnitrone intermediate A shows a second reactivity as a 1,3-dipolar reagent in the cascade reaction with dipolarophiles, such as electron-deficient olefins and isocyanates (type c).7 However, Denmark's pioneering report8a demonstrating the [4 + 2] cycloaddition of N-vinylnitrone species inspired us to come up with the idea that key intermediate A possesses a third reactivity as “2-azadiene” (type d). Specifically, we envisioned that the reaction of N-allenylnitrone A with the diazene (RN
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR) would proceed via the [4 + 2] cycloaddition9 preferentially, rather than the [3 + 2] dipolar cycloaddition,10 because the latter reaction would proceed through an unfavorable transition state due to electrostatic repulsion between lone pairs on the nitrone O atom and the diazene N atom, forming a weak N–O bond.11 Moreover, since the expected [4 + 2] cycloadducts, 1,2,4-triazines, have been frequently utilized in pharmaceutical and agrochemical fields,12 the envisioned transformation appears promising from the synthetic viewpoint. Herein, we report that the copper-catalyzed intermolecular reaction of O-propargylic oximes 1 with azodicarboxylates 2 afforded the corresponding 1,2,3,6-tetrahydro-1,2,4-triazine oxides 3 in good to high yields (Scheme 1d).
NR) would proceed via the [4 + 2] cycloaddition9 preferentially, rather than the [3 + 2] dipolar cycloaddition,10 because the latter reaction would proceed through an unfavorable transition state due to electrostatic repulsion between lone pairs on the nitrone O atom and the diazene N atom, forming a weak N–O bond.11 Moreover, since the expected [4 + 2] cycloadducts, 1,2,4-triazines, have been frequently utilized in pharmaceutical and agrochemical fields,12 the envisioned transformation appears promising from the synthetic viewpoint. Herein, we report that the copper-catalyzed intermolecular reaction of O-propargylic oximes 1 with azodicarboxylates 2 afforded the corresponding 1,2,3,6-tetrahydro-1,2,4-triazine oxides 3 in good to high yields (Scheme 1d).
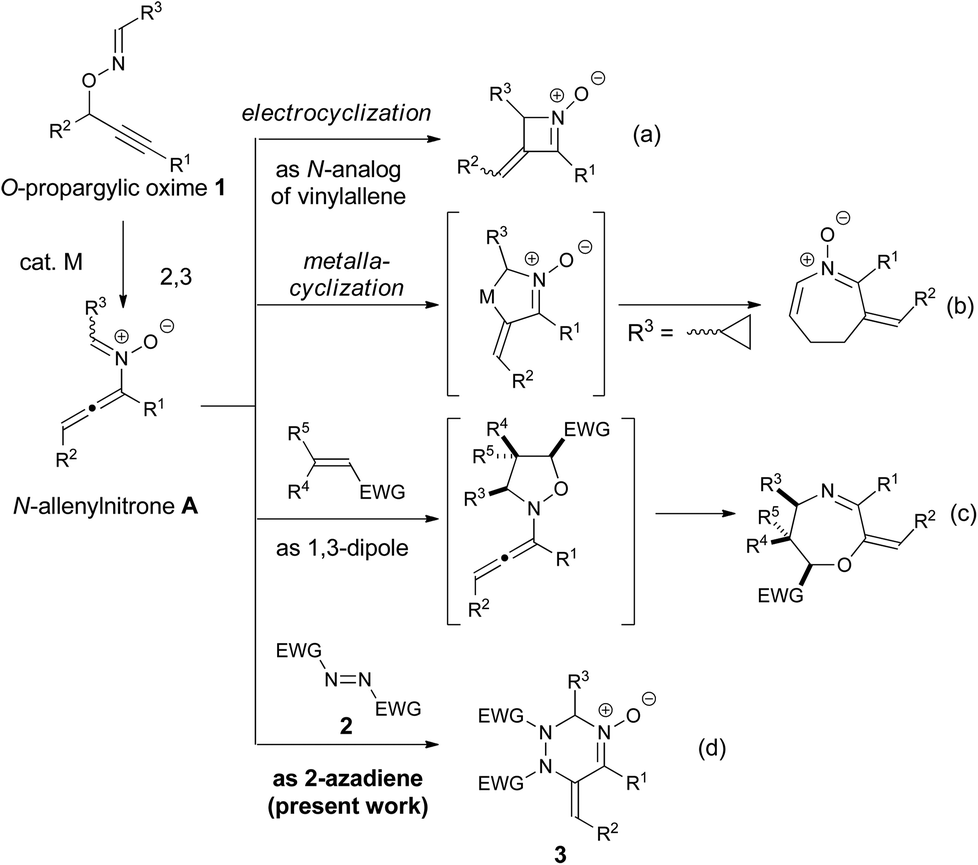 | ||
| Scheme 1 Reactivity of N-allenylnitrone A generated from O-propargylic oxime 1; N-analog of vinylallene (type a and b), 1,3-dipolar reagent (type c), and 2-azadiene (type d, present work). | ||
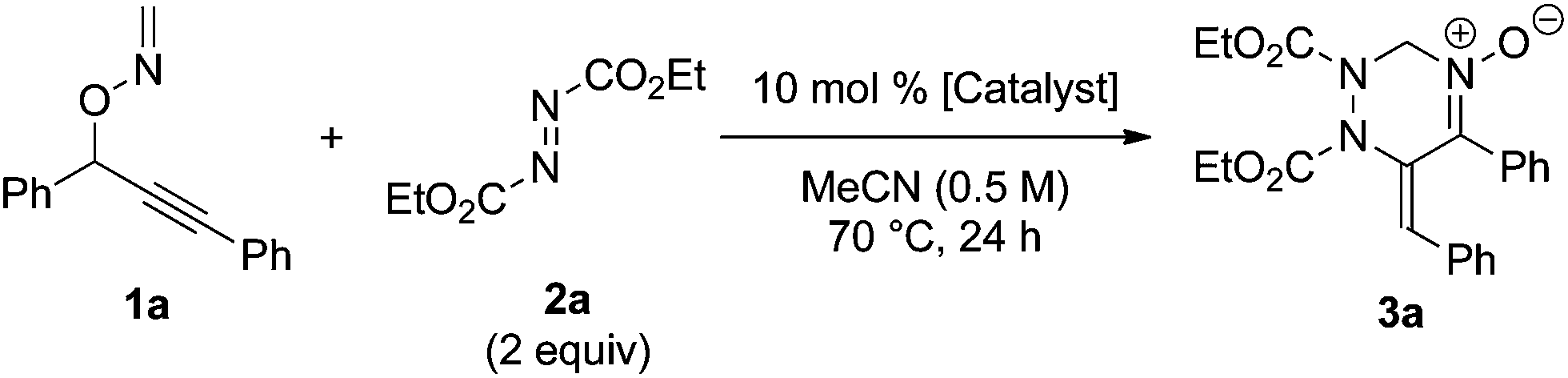
Initially, the reaction of 1a with diethyl azodicarboxylate 2a (2 equivalents) was carried out in the presence of a catalytic amount of CuCl (10 mol%) in acetonitrile (0.5 M) at 70 °C to afford tetrahydrotriazine 3a in 63% yield (Table 1, entry 1). The use of CuCl, [CuCl(cod)]2, and CuCl2 as the catalyst was effective, whereas CuBr and CuOTf exhibited low catalytic activities (entries 2–5). AgOTf and RhCl(PPh3)3 hardly promoted the reaction, whereas the use of PtCl2 and AuCl led to complete decomposition of the starting material (entries 6–9). The reaction in the absence of a metal catalyst did not afford the desired product and 85% of the starting material 1a was recovered (entry 10). In terms of solvent effects, the use of acetonitrile was the most effective in the present reaction, whereas the use of other solvents, such as 1,4-dioxane and toluene, resulted in low chemical yields (see ESI†).
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yieldb (%) | Recoveryc (%) |
| a The reaction of 1a (0.4 mmol) with 2a (0.8 mmol) was conducted in the presence of 10 mol% of a catalyst in acetonitrile (0.8 mL) at 70 °C for 24 hours. b Isolated yield. c 1H NMR yield using CH2Br2 as the internal standard. d 5 mol% of [CuCl(cod)]2 was used. | |||
| 1 | CuCl | 63 | <1 |
| 2 | [CuCl(cod)]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
63 | <1 |
| 3 | CuBr | 26 | <1 |
| 4 | CuOTf | 44 | <1 |
| 5 | CuCl2 | 56 | <1 |
| 6 | AgOTf | 22 | <1 |
| 7 | RhCl(PPh3)3 | 15 | 37 |
| 8 | PtCl2 | <1 | <1 |
| 9 | AuCl | <1 | <1 |
| 10 | None | <1 | 85 |
Next, the reactivity of diazenes 2 was evaluated in the reaction of 1a at 50 °C using CuCl as the catalyst, as summarized in Table 2. The use of a bulky ester improved the chemical yield (entries 1–3). In particular, the reaction with di-tert-butyl azodicarboxylate (DBAD) 2c afforded the desired product 3c in good yield (entry 3). It should be noted that the use of 1 equivalent of 2c resulted in a decrease of the chemical yield (entry 4). Indeed, 1a was gradually degraded by the copper catalyst at 50 °C within one day in the absence of azodicarboxylate 2c. Dibenzyl ester 2d, trichloroethyl ester 2e, and 1,1′-azodicarbonyldipiperidine (ADDP) 2f were not effective diazenes, affording the corresponding products in poor yields (entries 5–7). Then, various O-propargylic formaldoximes 1 were employed as the substrate for the copper-catalyzed reaction with 2 equivalents of DBAD 2c (entries 8–16). The reaction with substrates 1b and 1c, which possess an aryl substituent at the alkyne terminus (R1), produced the corresponding products 3g and 3h, respectively, in good yields (entries 8 and 9). Alkyl substitution at R1 was also efficient (entries 10–12). In particular, the reaction of 1f having a cyclohexyl group afforded the desired product 3k in an excellent yield (entry 12). The reaction of 1h having an electron-deficient aryl group at the propargylic position (R2) proceeded much faster than that of 1g having an electron-rich p-anisyl group (entries 13 and 14). Substrate 1j bearing an alkyl group at R2 was also efficiently converted into product 3o in good yield (entry 16). It is noteworthy that except for 3k and 3o, products 3 showed only the E configuration at the exo-olefin moiety. The structures of 3 were characterized by various spectroscopic methods, such as NMR (1H and 13C), IR, and HRMS (see ESI†). Moreover, the structure of 3c was unambiguously determined by X-ray crystallographic analysis, as shown in Fig. 1.13
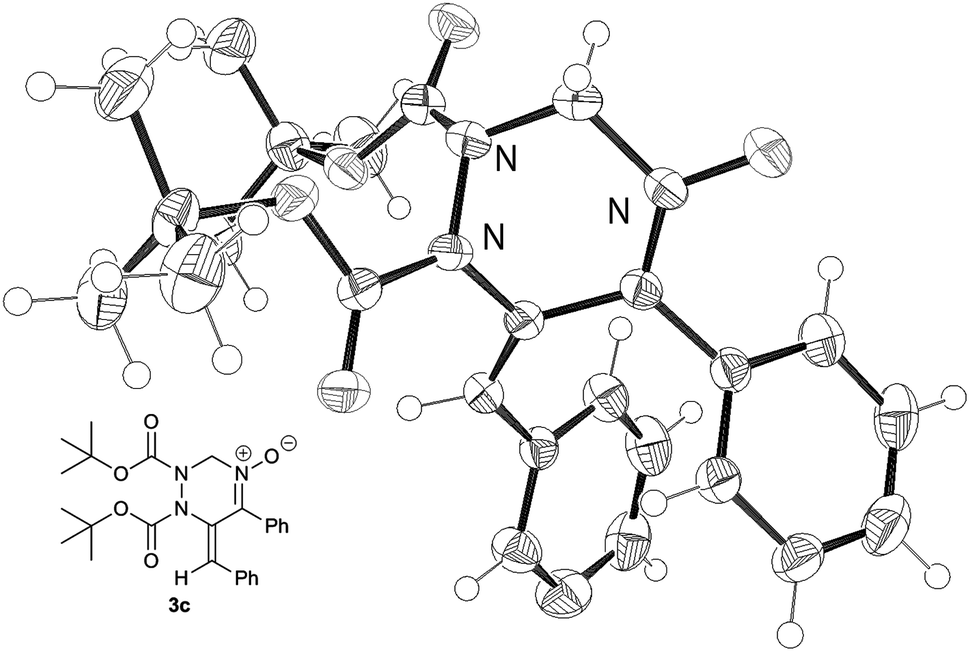 | ||
| Fig. 1 ORTEP drawing of 3c (shown with 50% probability ellipsoid). | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| 1 | R1 | R2 | 2 | Time (h) | 3 | Yieldb (%) | |
|
a The reaction of 1 (0.4 mmol) with 2 (0.8 mmol) was conducted in the presence of 10 mol% of CuCl in acetonitrile (1.6 mL) at 50 °C.
b Isolated yield.
c 1 equivalent of 2c was used.
d An 85:15 mixture of E/Z stereoisomers was obtained.
e A 66:34 mixture of E/Z stereoisomers was obtained.
|
|||||||
| 1 | 1a | Ph | Ph | 2a | 30 | 3a | 62 |
| 2 | 1a | Ph | Ph | 2b | 24 | 3b | 75 |
| 3 | 1a | Ph | Ph | 2c | 10 | 3c | 78 |
| 4 | 1a | Ph | Ph | 2c | 10 | 3c | 69 |
| 5 | 1a | Ph | Ph | 2d | >100 | 3d | 36 |
| 6 | 1a | Ph | Ph | 2e | 72 | 3e | 17 |
| 7 | 1a | Ph | Ph | 2f | 26 | 3f | 25 |
| 8 | 1b | p-MeOC6H4 | Ph | 2c | 8 | 3g | 78 |
| 9 | 1c | p-F3CC6H4 | Ph | 2c | 12 | 3h | 60 |
| 10 | 1d | nPr | Ph | 2c | 2 | 3i | 74 |
| 11 | 1e | H2CCH(CH2)2 |
Ph | 2c | 6 | 3j | 64 |
| 12 | 1f | Cy | Ph | 2c | 6 | 3k | 94d |
| 13 | 1g | Ph | p-MeOC6H4 | 2c | 36 | 3l | 77 |
| 14 | 1h | Ph | p-F3CC6H4 | 2c | 5 | 3m | 65 |
| 15 | 1i | Ph | 2-Naphthyl | 2c | 12 | 3n | 74 |
| 16 | 1j | Ph | nPr | 2c | 30 | 3o | 65e |
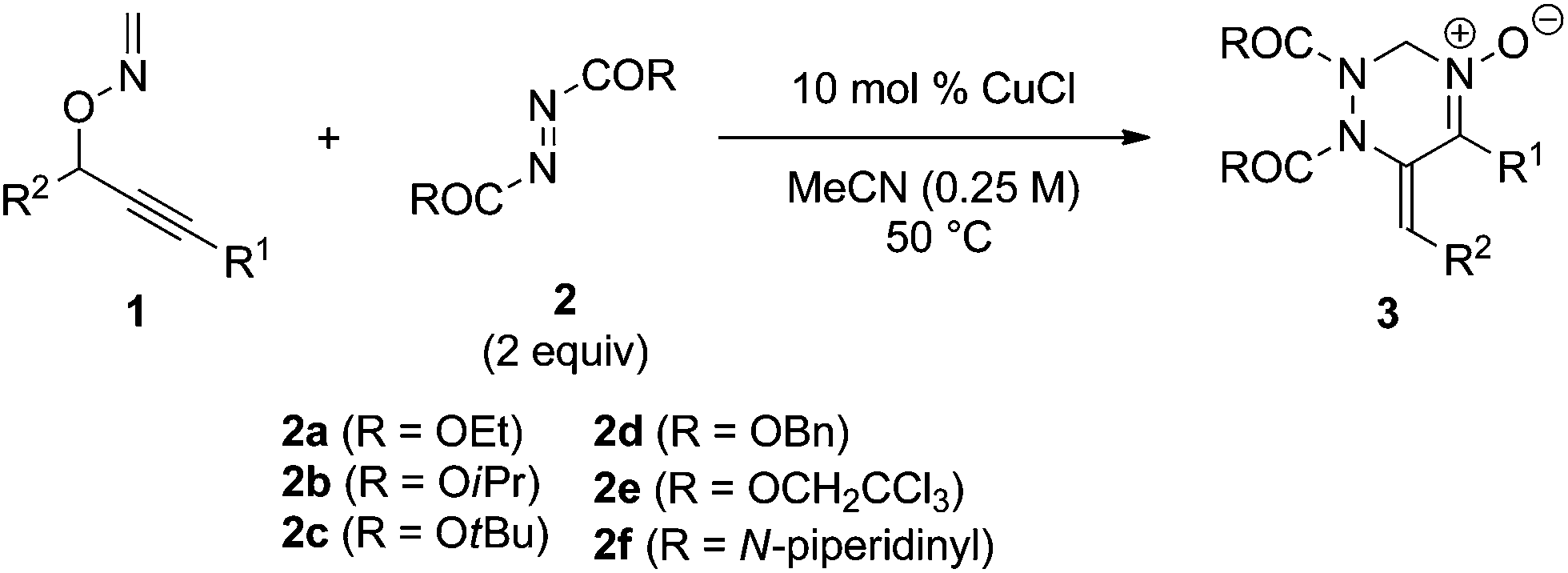
The reaction of acetaldoximes (E)-1k afforded the corresponding cycloadduct 3p in good yield (Table 3, entry 1), although prolonged reaction time was required in comparison with the formaldoxime 1a (Table 2, entry 3). It is noteworthy that both E and Z stereoisomers of butyraldoximes 1l and 1m were converted into identical products 3q and 3r, respectively (entries 2–5). The reaction of (Z)-1m in the absence of a copper catalyst at elevated temperature (70 °C) afforded only four-membered cyclic nitrone 4m and the desired cycloadduct 3r was not formed (entry 6).5d The reaction of cyclohexane-carbaldoxime required elevated reaction temperature (70 °C) to obtain 3s in good yield (entry 7). In contrast, the reaction of benzaldoxime (E)-1o did not afford the desired product; the reaction at 50 °C resulted in the quantitative recovery of 1o, whereas that at 100 °C solely afforded undesired four-membered cyclic nitrone 4o (entries 8 and 9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 | R2 | R3 | Time (h) | 3 (%) | 4 (%) |
| a The reaction of 1 (0.4 mmol) with 2c (0.8 mmol) was conducted in the presence of 10 mol% of CuCl in acetonitrile (1.6 mL) at 50 °C. b Isolated yield in parenthesis. c Chiral substrates (S, >99% ee) were used. d 2% ee. e At 70 °C. f The reaction was carried out in the absence of a copper catalyst. g At 100 °C. | ||||||
| 1 | (E)-1k | Ph | Me | 7 days | 3p (78) | <1 |
| 2 | (E)-1l | Ph | nPr | 7 days | 3q (77) | <1 |
| 3 | (Z)-1l | Ph | nPr | 48 | 3q (56) | <1 |
| 4 | (E)-1mc | p-ClC6H4 | nPr | 6 days | 3r (72)d | <1 |
| 5 | (Z)-1m | p-ClC6H4 | nPr | 16e | 3r (41) | <1 |
| 6 | (Z)-1m | p-ClC6H4 | nPr | 6 dayse,f | <1 | 4m (48) |
| 7 | (E)-1n | Ph | Cy | 100e | 3s (63) | Trace |
| 8 | (E)-1o | Ph | Ph | 100 | Trace | <1 |
| 9 | (E)-1o | Ph | Ph | 14g | <1 | 4o (66) |
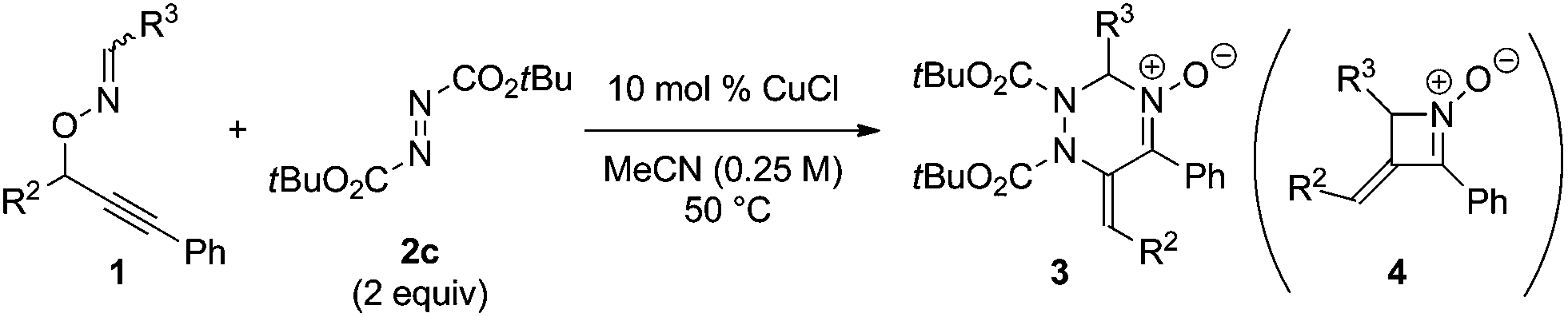
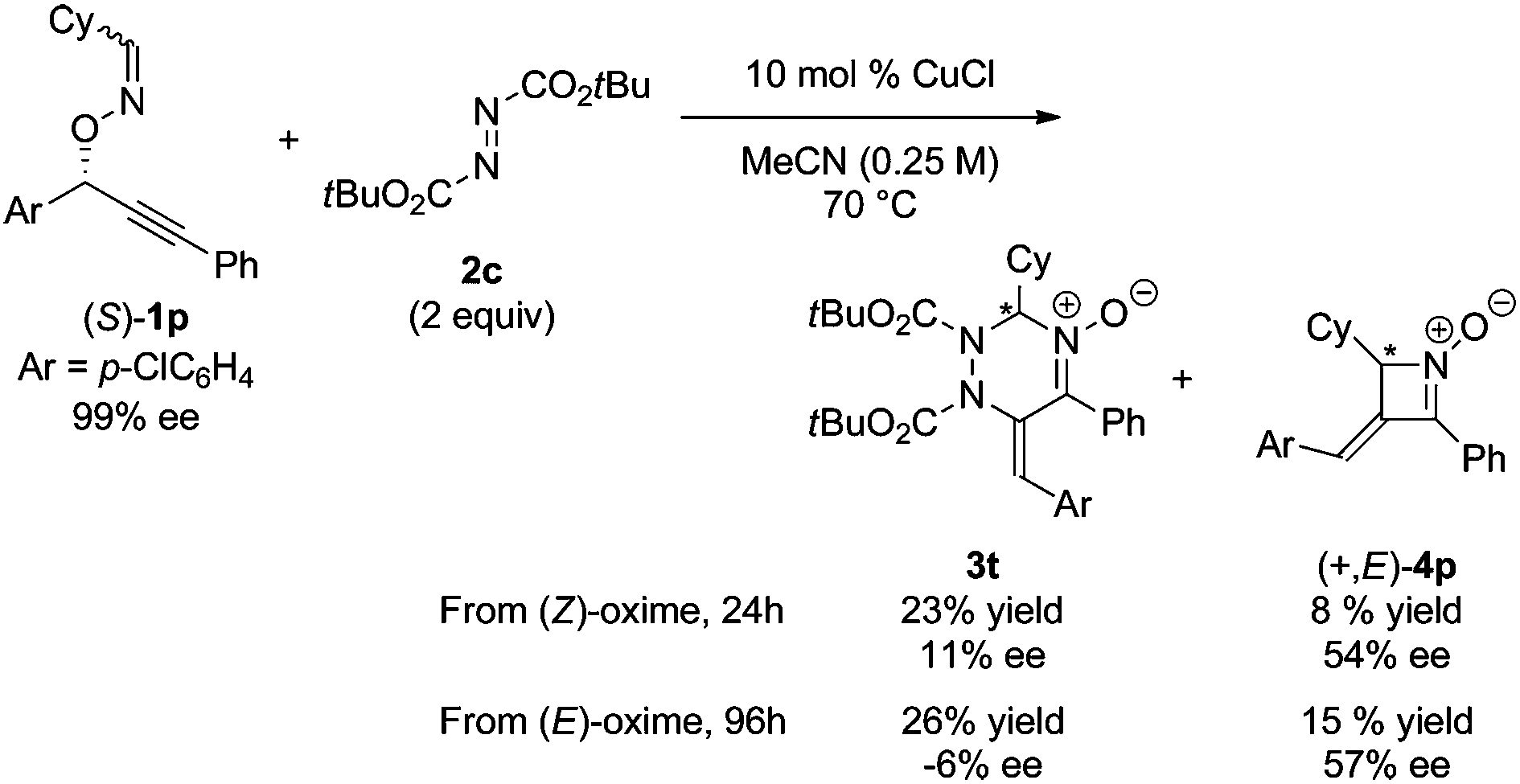
In order to gain an insight into the reaction mechanism, the copper-catalyzed reaction of enantiomerically pure substrate (S)-1p with DBAD 2c was carried out to afford the corresponding product 3t with excellent E stereoselectivity at the exo-olefin moiety and low enantioselectivity at the sp3 carbon in the triazine ring, regardless of the configuration at the oxime moiety of (S)-1p (eqn (1)).14,15 It should be noted that the reaction of both stereoisomers (S,E)- and (S,Z)-1p afforded the same enantiomer of four-membered cyclic nitrone byproduct (+,E)-4p with a sufficient level of chirality transfer. Moreover, the starting material (S)-1p was not racemized under the reaction conditions.16
 | (1) |
A plausible mechanism for the copper-catalyzed reaction between 1 and 2 is illustrated in Scheme 2. First, the π-acidic copper catalyst coordinates to the triple bond of 1, forming π-complex 5. Nucleophilic attack of the oxime nitrogen atom on the electrophilically activated alkyne moiety proceeds in a 5-endo manner, yielding cyclized vinylcopper intermediate 6. Cleavage of the C–O bond and elimination of the copper catalyst generate N-allenylnitrone 7. The nitrone carbon of the key intermediate 7 nucleophilically attacks the azodicarboxylate activated by the copper catalyst, forming zwitterionic species 8.17 Finally, intramolecular addition of the copper amidate to the oxoammonium-conjugated CC bond from the less hindered allene face produces 3 having an exo-E-olefin.18 The reaction of (Z)-oxime in the absence of a copper catalyst afforded only four-membered cyclic nitrone 4 derived from 2,3-rearrangement followed by thermal 4π-electrocyclization (Table 3, entry 6). According to our knowledge that the 2,3-rearrangement of (Z)-oxime proceeds thermally, this result strongly implies that the copper catalyst promotes not only the 2,3-rearrangement but also the [4 + 2] cycloaddition process from 7 to 3. The low enantioselectivity of the reaction of (S)-1p (eqn (1)) is presumably because azodicarboxylate 2 approaches allenylnitrone 7 from both Si and Re faces of the (Z)-nitrone moieties, which are far from the chiral allene group.7a Alternatively, loss of chirality may occur when the concerted [4 + 2] cycloaddition takes place from the opposite side of the chiral allene substituent (R2) of (Z)- and (E)-allenylnitrones 7 almost equally.19 However, the simultaneous formation of two C–N bonds is unlikely because the same enantiomer of four-membered cyclic nitrone (+,E)-4p was obtained as the byproduct of both (Z)- and (E)-1p with sufficient enantioselectivities, indicating that the configuration of allenylnitrone intermediate 7 is biased toward the thermodynamically more stable (Z)-isomer, (Z)-7.
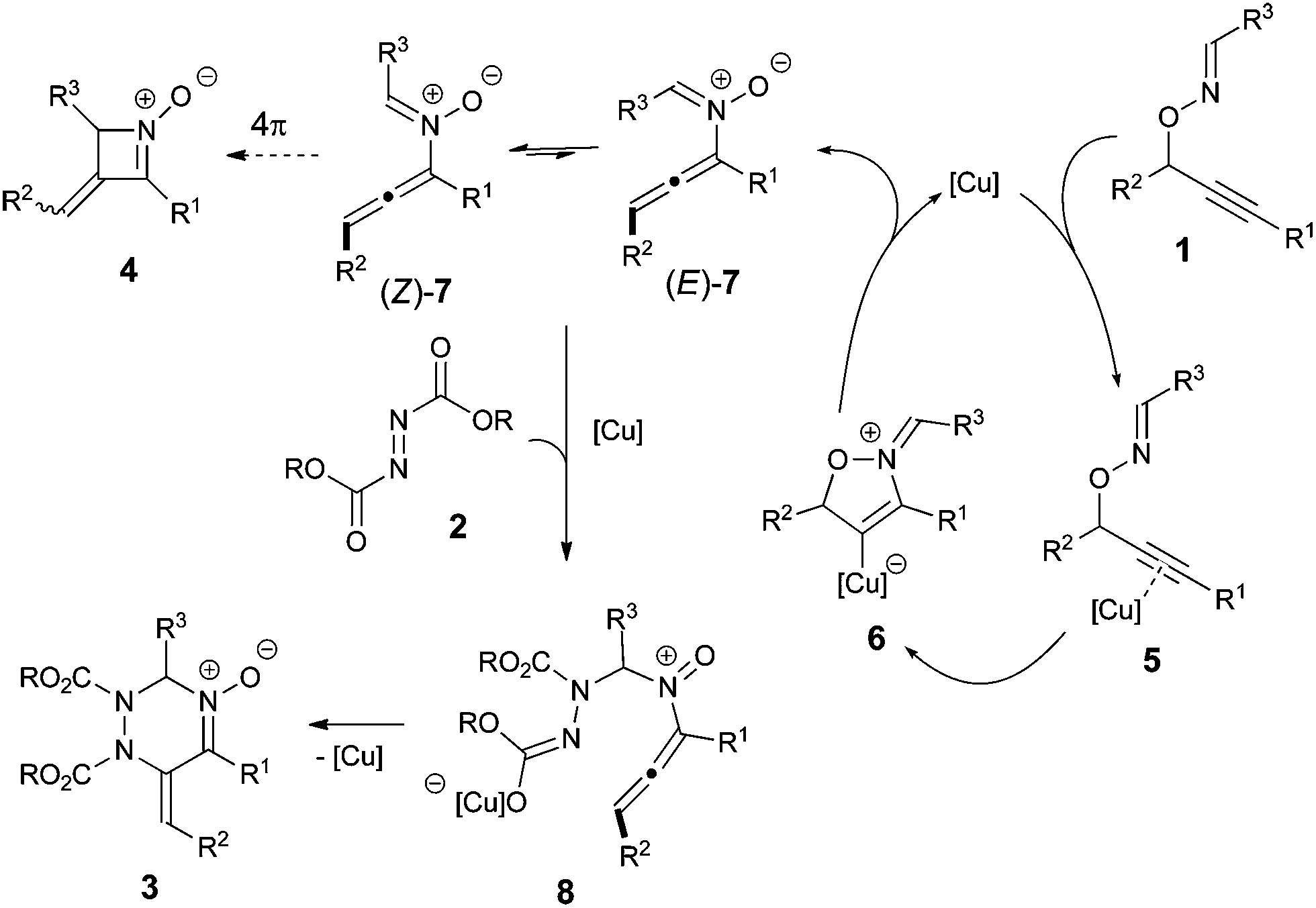 | ||
| Scheme 2 A plausible mechanism. | ||
Conclusions
In conclusion, we have developed an entirely new approach to the tetrahydrotriazine framework, which involves the catalytic cascade reaction from O-propargylic oximes. The key intermediate, N-allenylnitrone, can be efficiently generated in situ by catalytic 2,3-rearrangement, in contrast to N-vinylnitrone, which requires several synthetic steps including the use of toxic reagents, such as aluminum amalgam and organoselenium.8a Therefore, the present method is useful to synthesize multisubstituted triazine derivatives in an efficient manner.Notes and references
- (a) S. F. Kirsch, Synthesis, 2008, 3183 CrossRef CAS PubMed; (b) H. C. Shen, Tetrahedron, 2008, 64, 7847 CrossRef CAS PubMed; (c) B. Crone and S. F. Kirsch, Chem. – Eur. J., 2008, 14, 3514 CrossRef CAS PubMed.
- Recent reviews of π-acidic metal catalysis: (a) C. Fehr, Synlett, 2012, 990 CrossRef CAS PubMed; (b) J. Xiao and X. Li, Angew. Chem., Int. Ed., 2011, 50, 7226 CrossRef CAS PubMed; (c) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (d) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (e) T. de Haro and C. Nevado, Synthesis, 2011, 2530 CAS; (f) H. Huang, Y. Zhou and H. Liu, Beilstein J. Org. Chem., 2011, 7, 897 CrossRef CAS PubMed; (g) S. Hummel and S. F. Kirsch, Beilstein J. Org. Chem., 2011, 7, 847 CrossRef CAS PubMed; (h) A. Pradal, P. Y. Toullec and V. Michelet, Synthesis, 2011, 1501 CAS; (i) N. D. Shapiro and F. D. Toste, Synlett, 2010, 675 CAS; (j) S. Wang, G. Zhang and L. Zhang, Synlett, 2010, 692 CAS; (k) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (l) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (m) Y. Yamamoto, J. Org. Chem., 2007, 72, 7817 CrossRef CAS PubMed; (n) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed.
- Selected examples: (a) C. Nieto-Oberhuber, M. P. Munoz, E. Bunuel, C. Nevado, D. J. Cárdenas and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 2402 CrossRef CAS PubMed; (b) H. Teller and A. Fürstner, Chem. – Eur. J., 2011, 17, 7764 CrossRef CAS PubMed; (c) P. R. McGonigal, C. de León, Y. Wang, A. Homs, C. R. Solorio-Alvarado and A. M. Echavarren, Angew. Chem., Int. Ed., 2012, 51, 13093 CrossRef CAS PubMed. Reviews: (d) C. Aubert, O. Buisine and M. Malacria, Chem. Rev., 2002, 102, 813 CrossRef CAS PubMed; (e) S. T. Diver and A. J. Giessert, Chem. Rev., 2004, 104, 1317 CrossRef CAS PubMed; (f) L. Añorbe, G. Domínguez and J. Pérez-Castells, Chem. – Eur. J., 2004, 10, 4938 CrossRef PubMed; (g) C. Bruneau, Angew. Chem., Int. Ed., 2005, 44, 2328 CrossRef CAS PubMed; (h) L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271 CrossRef CAS; (i) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268 CrossRef CAS PubMed; (j) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (k) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300 RSC.
- For example: (a) A. S. K. Hashmi, W. Yang, Y. Yu, M. M. Hansmann, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2013, 52, 1329 CrossRef CAS PubMed. Reviews: (b) X.-Z. Shu, D. Shu, C. M. Schienebeck and W. Tang, Chem. Soc. Rev., 2012, 41, 7698 RSC; (c) N. Marion and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2750 CAS; (d) A. Correa, N. Marion, L. Fensterbank, M. Malacria, S. P. Nolan and L. Cavallo, Angew. Chem., Int. Ed., 2008, 47, 718 CrossRef CAS PubMed.
- (a) I. Nakamura, D. Zhang and M. Terada, J. Am. Chem. Soc., 2010, 132, 7884 ( J. Am. Chem. Soc. , 2011 , 133 , 6862 ) CrossRef CAS PubMed (Additions and Corrections); (b) I. Nakamura, T. Araki, D. Zhang, Y. Kudo, E. Kwon and M. Terada, Org. Lett., 2011, 13, 3616 CrossRef CAS PubMed; (c) I. Nakamura, D. Zhang and M. Terada, Tetrahedron Lett., 2011, 52, 6470 CrossRef CAS PubMed; (d) I. Nakamura, Y. Kudo, T. Araki, D. Zhang, E. Kwon and M. Terada, Synthesis, 2012, 1542 CrossRef CAS; (e) I. Nakamura, Y. Ishida and M. Terada, Org. Lett., 2014, 16, 2562 CrossRef CAS PubMed.
- I. Nakamura, M. Okamoto, Y. Sato and M. Terada, Angew. Chem., Int. Ed., 2012, 51, 10816 CrossRef CAS PubMed.
- (a) I. Nakamura, Y. Kudo and M. Terada, Angew. Chem., Int. Ed., 2013, 52, 7536 CrossRef CAS PubMed; (b) I. Nakamura, T. Onuma, D. Zhang and M. Terada, Tetrahedron Lett., 2014, 55, 1178 CrossRef CAS PubMed.
- (a) S. E. Denmark and J. O. Montgomery, J. Org. Chem., 2006, 71, 6211 CrossRef CAS PubMed; (b) D.-L. Mo, D. A. Wink and L. L. Anderson, Org. Lett., 2012, 14, 5180 CrossRef CAS PubMed.
- (a) W. Ried, U. Reiher and J. W. Bats, Helv. Chim. Acta, 1987, 70, 1255 CrossRef CAS; (b) T. Sheradsky and N. Itzhak, J. Chem. Soc., Perkin Trans. 1, 1987, 1979 RSC; (c) J. Barluenga, F. J. González, S. Fustero, S. García-Granda and E. Pérez-Carreňo, J. Org. Chem., 1991, 56, 4459 CrossRef CAS; (d) C. Balsamini, A. Bedini, R. Galarini, G. Spadoni, G. Tarzia and M. Hamdan, Tetrahedron, 1994, 50, 12375 CrossRef CAS; (e) J. F. Sanz-Cervera, R. M. Williams, J. A. Marco, J. M. López-Sánchez, F. González, M. E. Martínez and F. Sancenón, Tetrahedron, 2000, 56, 6345 CrossRef CAS; (f) F. Palacios, C. Alonso, G. Rubiales, C. Tobillas and J. M. Ezpeleta, Heterocycles, 2003, 61, 493 CrossRef CAS PubMed; (g) F. Palacios, C. Alonso, M. Rodríguez, E. M. de Marigorta and G. Rubiales, Eur. J. Org. Chem., 2005, 1795 CrossRef CAS.
- (a) A. M. Nour-el-Din and A.-F. E. Mourad, J. Prakt. Chem., 1983, 325, 908 CrossRef CAS; (b) L. Eberson and O. Persson, Acta Chem. Scand., 1998, 52, 1081 CrossRef CAS PubMed.
- (a) A. A. Tabolin and S. L. Ioffe, Chem. Rev., 2014, 114, 5426 CrossRef CAS PubMed; (b) S. W. Benson, J. Chem. Educ., 1965, 42, 502 CrossRef CAS.
- (a) P. Zhang, X. Li, Z. Li, X. Chen, Y. Tian, W. Chen, X. Liu, C. Pannecouque and E. De Clercq, Bioorg. Med. Chem. Lett., 2012, 22, 7155 CrossRef PubMed; (b) Y. Song, P. Zhan, Q. Zhang and X. Liu, Curr. Pharm. Des., 2013, 19, 1528 CAS; (c) M. Congreve, S. P. Andrews, A. S. Dore, K. Hollenstein, E. Hurrell, C. J. Langmead, J. S. Mason, I. W. Ng, I. B. Tehan, A. Zhukov, M. Weir and F. H. Marshall, J. Med. Chem., 2012, 55, 1898 CrossRef CAS PubMed.
- CCDC 961608 (3c) contains the supplementary crystallographic data for this paper.
- (a) H. J. Reich, E. K. Eisenhart, W. L. Whipple and M. J. Kelly, J. Am. Chem. Soc., 1988, 110, 6432 CrossRef CAS; (b) C. Spino, C. Thibault and S. Gingras, J. Org. Chem., 1998, 63, 5283 CrossRef CAS.
- 3t was not racemized under the reaction conditions.
- The recovered (E,S)-1p, which was treated under the reaction conditions at 70 °C for 8 hours, had an enantiomeric excess of 99%.
- L. Chang, Y. Kuang, B. Qin, X. Zhou, X. Liu, L. Lin and X. Feng, Org. Lett., 2010, 12, 2214 CrossRef CAS PubMed.
- (a) R. K. Dieter and K. Lu, Tetrahedron Lett., 1999, 40, 4011 CrossRef CAS; (b) M. A. Silvestri, D. C. Bromfield and S. D. Lepore, J. Org. Chem., 2005, 70, 8239 CrossRef CAS PubMed; (c) P. Elsner, L. Bernardi, G. D. Salla, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2008, 130, 4897 CrossRef CAS PubMed.
- M. Sasaki, Y. Kondo, M. Kawahata, K. Yamaguchi and K. Takeda, Angew. Chem., Int. Ed., 2011, 50, 6375 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of the products 3 and 4. CCDC 961608. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00156g |
| This journal is © the Partner Organisations 2014 |