
Controllable mono-/di-alkenylation of aryl alkyl thioethers tuned by oxidants via Pd-catalysis†
Xi-Sha
Zhang
a,
Yun-Fei
Zhang
a,
Kang
Chen
a and
Zhang-Jie
Shi
*ab
aBeijing National Laboratory of Molecular Sciences (BNLMS) and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Green Chemistry Center, Peking University, Beijing, 100871, China. E-mail: zshi@pku.edu.cn; Web: http://www.shigroup.cn/ Fax: (+86)-10-6276-0890
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, China
First published on 2nd September 2014
Abstract
Oxidant-controlled selective mono-/di-alkenylation of aryl C–H bonds via Pd-catalysis is reported. The substrate scopes for both the mono- and di-alkenylation were good. Thioether was used as the directing group and the product can be transformed to a useful sulfoxide-olefin ligand by simple oxidation in quantitative yield.
Transition-metal-catalyzed oxidative alkenylation (Fujiwara reaction) has attracted much attention in recent years1 due to its high atom-economy, high efficiency, low waste production and potential applications in complex molecule synthesis.2 To approach the high regio- and chemo-selectivity, a directing strategy3 has been well applied in such alkenylations. While two C–H bonds beside the directing group exist in the molecule, it is always difficult to control the selectivity between mono- and di-alkenylation and a mixture of them was usually obtained (Scheme 1, (1)). In the past, some successful strategies have been well established to approach high selectivity between mono- and di-alkenylation, such as by the amount of alkenylating reagents, oxidants, reaction time and temperature,4,5 by solvent effects6,7 and by others,8–11 although in almost all cases multi-factors were changed to tune the selectivity. Herein, we report a controllable selective mono- or di-alkenylation of aryl C–H bonds by changing only one factor for the first time, with thioether as a new directing group. The oxidants with different anions were found to be crucial to approach high selectivity. Although oxidants have been found to tune the regio-selectivities of C–H activation,12 no report on the oxidant controlled mono-/di-functionalization selectivity exists.
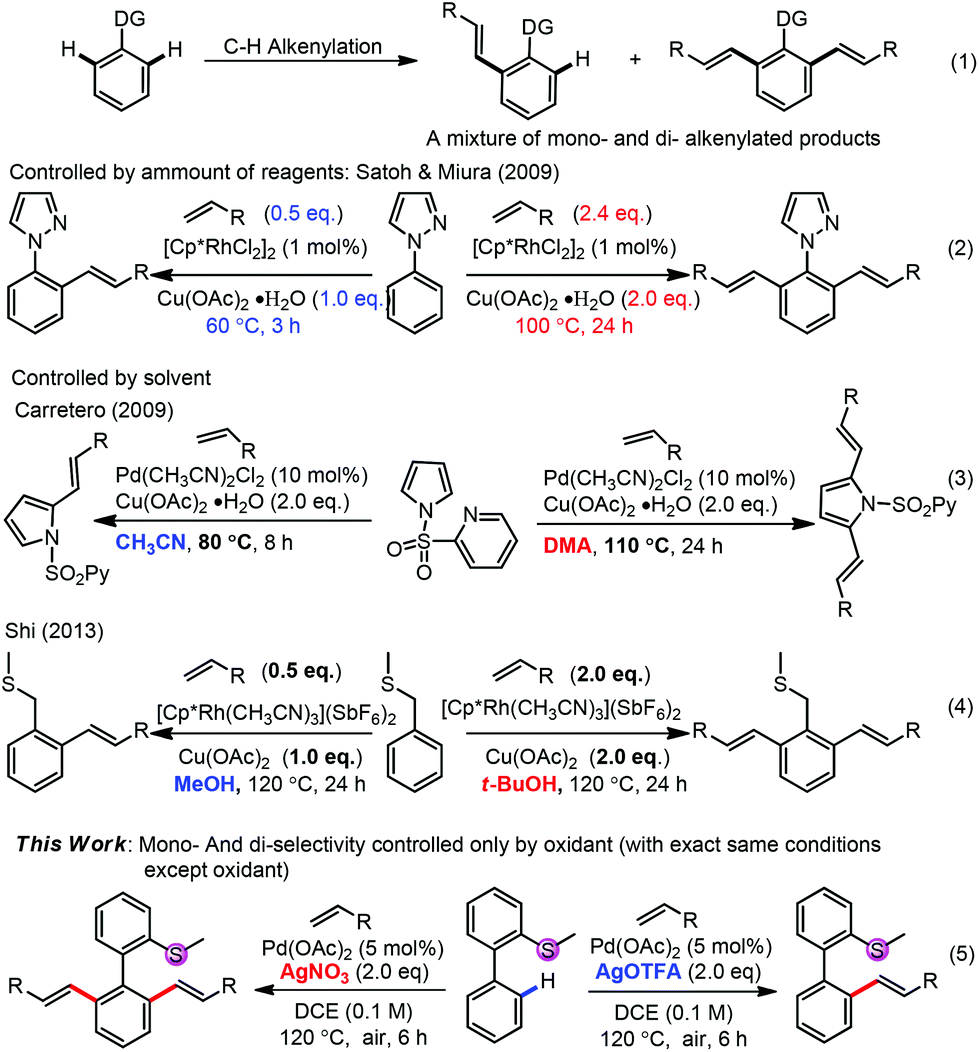 | ||
| Scheme 1 Selectivities of mono-functionalization and di-functionalization in the transition-metal-catalyzed C–H bond activation. | ||
Although S-contained groups were considered not to be good directing groups in C–H activation due to their easy oxidation and potential ability to poison the late transition-metal catalysts, recent advances unveiled the success to adapt such groups as directing groups in C–H activation.13,14 We recently reported the alkenylation of benzyl methyl sulfide with thioether as a directing group, in which a five-membered rhodacycle was considered as a key intermediate.6b The extension of such studies showed that homobenzyl methyl sulfide showed very low reactivity, probably arising from the less rigid 6-membered metallocycle intermediate.13d We envisioned that biphenyl-2-yl(methyl)sulfane (1a) could be the appropriate substrate by enhancing the rigidity of the key intermediate to promote the efficiency. Structurally, the desired product is indeed important since the sulfide group can be transformed into different functionalities, for example, the sulfoxide-olefin ligands.15,16
Although initial screenings of rhodium catalysts based on our previous work showed low efficiency (see ESI†), palladium catalysts showed very good activity. With AgOAc as the oxidant, the mono-alkenylated product could be obtained as the major product in 78% NMR yield (Table 1, entry 1). Interestingly and importantly, further screening showed that the selectivity of the mono-alkenylation and di-alkenylation could be controlled by the oxidant. When AgOTFA was used as an oxidant, the mono-alkenylated product was obtained in 92% NMR yield, while only changing the oxidant to AgNO3 gave the di-alkenylated product in 93% NMR yield (Table 1, entries 6 and 7). Further investigation revealed that the catalyst loading could be reduced to 5 mol% and the yield was not affected when the reaction was conducted on a 0.2 mmol scale (Table 1, entries 12 and 13).
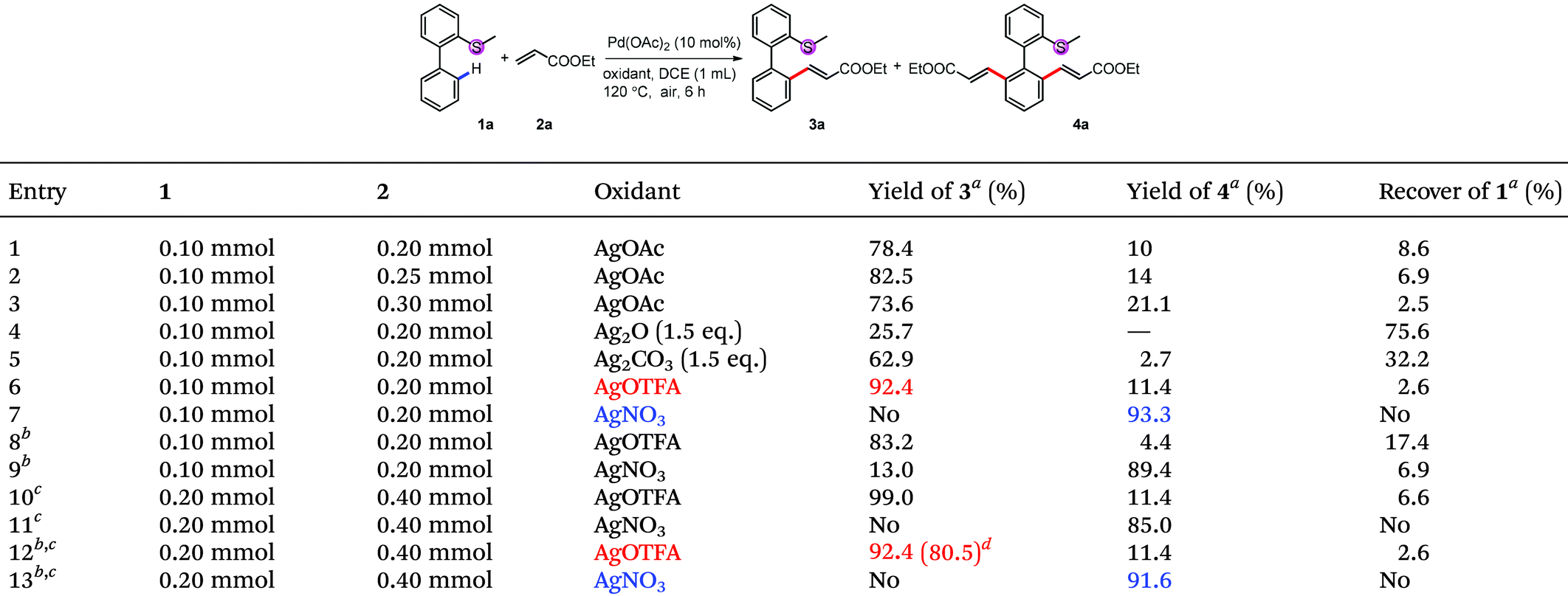
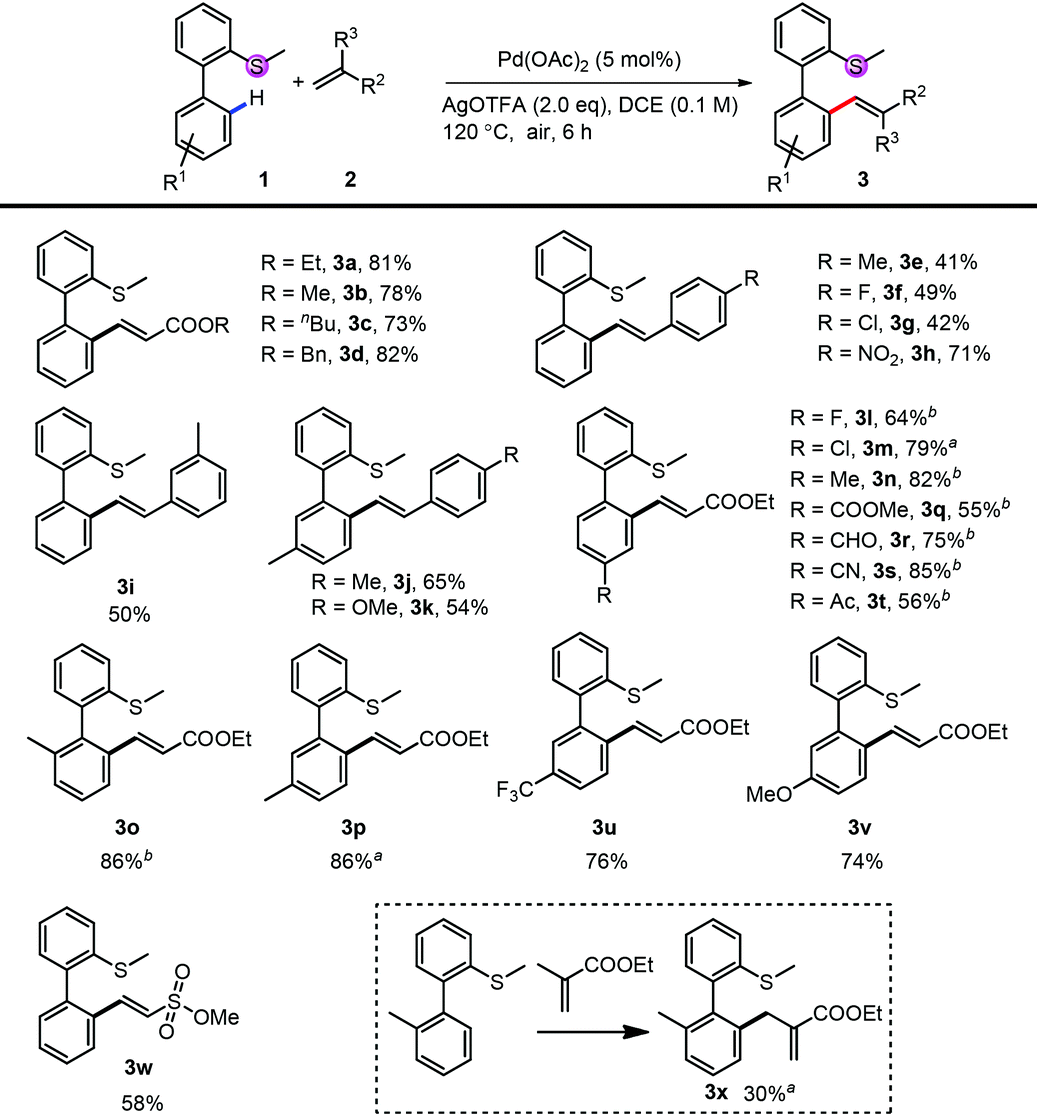
We first investigated the substrate scope for the mono-alkenylation (Table 2). For the thioethers, both electron rich (3n, 3o, 3p and 3v) and electron poor (3l, 3m, 3q–3t, 3u) ones are tolerated very well. The group compatibility of this monoalkenylation (3l–3m, 3q–3v) showed its wide substrate scope. Notably, methyl ethenesulfonate is also a suitable alkenylating reagent in this reaction (3w), expanding the application of this method. It should be noted that the di-substituted alkene was also an applicable coupling partner, and the isomerized product was obtained in relative lower efficiency (3x). Apart from a variety of acrylates (3a–3d), styrene derivatives also reacted smoothly in moderate to good yields (3e–3k). Although the efficiency of electron-rich styrene was low, the yield can be improved by blocking the other ortho-position with the methyl group (3j and 3k).
| Conditions: 1 (0.1 mmol or 0.2 mmol), 2 (2.0 eq.), Pd(OAc)2 (5 mol%) and AgOTFA (2.0 eq.) were mixed in DCE (0.1 M) at 120 °C under air for 6 h.a 10 mol% of Pd(OAc)2 was used.b After the reaction, another 2 (2.0 eq.), Pd(OAc)2 (5 mol%) and AgOTFA (1.0 eq.) were added to react for further 10 h for full conversion. |
|---|
|
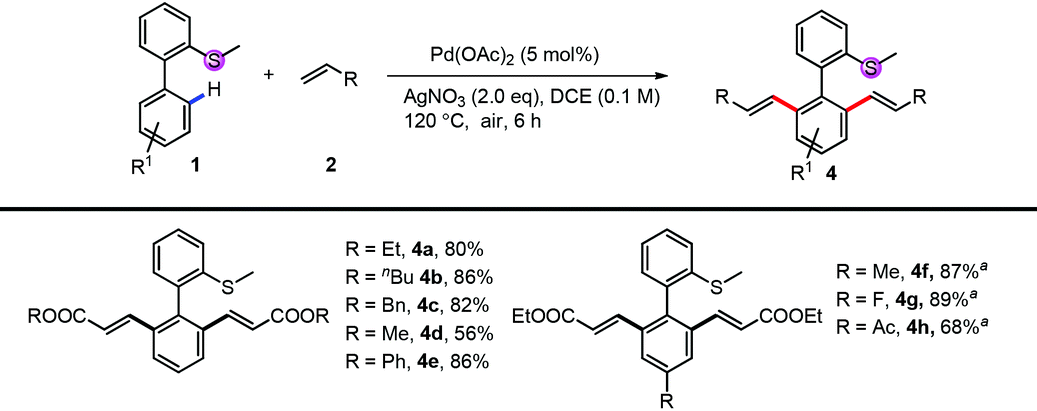
Under di-alkenylation conditions, with only the change of the oxidant to AgNO3, a variety of alkenes and bi-phenyl arenes were tolerated very well, providing good to excellent yields (Table 3). A variety of acrylates, both alkyl (4a–4d) and aryl acrylates (4e), reacted smoothly to give the desired products in high yields. For the thioethers, both electron-rich and electron-deficient substituents can be tolerated very well (4f, 4g). Functional groups like acyl were untouched (4h).
| Conditions: 1 (0.1 mmol or 0.2 mmol), 2 (2.0 eq.), Pd(OAc)2 (5 mol%) and AgNO3 (2.0 eq.) were mixed in DCE (0.1 M) at 120 °C under air for 6 h.a 10 mol% of Pd(OAc)2 was used. |
|---|
|
To explore the potential applications, both reactions (mono-alkenylation and di-alkenylation) were scaled up to the 1 mmol scale and the yields were not affected intensively, although for the di-alkenylation a longer reaction time was needed (Scheme 2, eqn (1) and (2)). In addition, the mono-alkenylated product can be transformed to the sulfoxide-olefin ligand in quantitative yield by simple oxidation (dr = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40) (Scheme 2, eqn (3)).
:40) (Scheme 2, eqn (3)).
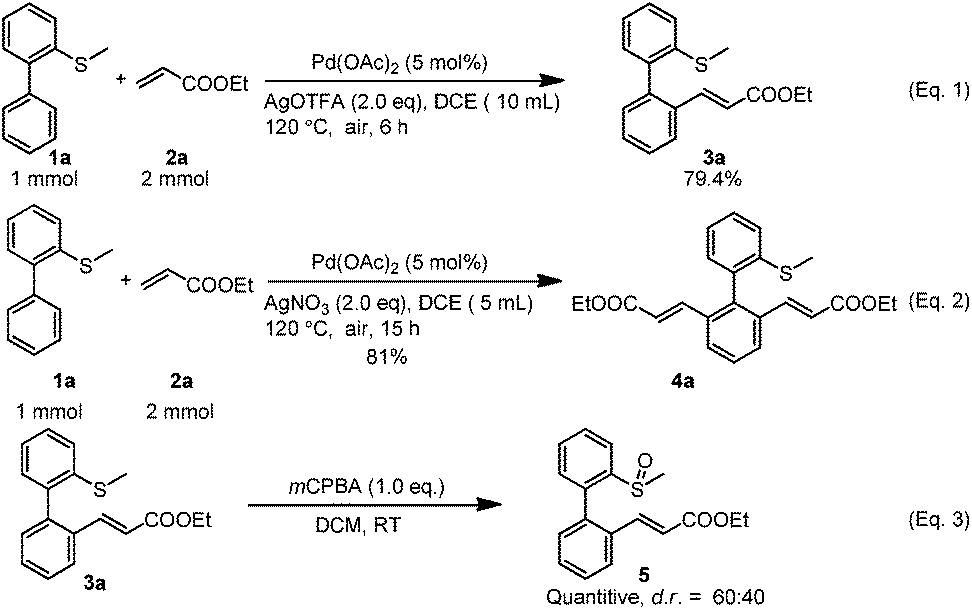 | ||
| Scheme 2 Large scale reaction and transformation of the product to synthesize the ligand. | ||
In order to gain some insights into the mechanism, we conducted several control experiments. When the S-atom was changed to the O-atom, the ether directed alkenylation also occurred, albeit in a lower yield and low selectivity, showing the importance of the S-atom (Scheme 3, eqn (4)).13a,17 The competing experiments showed that the electron rich arenes reacted much faster (Scheme 4, eqn (5) and eqn (6)). The deuterium labeling experiment showed that the C–H bond activation was not reversible and gave a KIE value of 5.6, indicating that the C–H activation was possibly involved in the turnover limiting step (Scheme 4, eqn (7) and eqn (8)). As a result, the C–H bond activation was proposed to undergo an electrophilic metalation pathway.
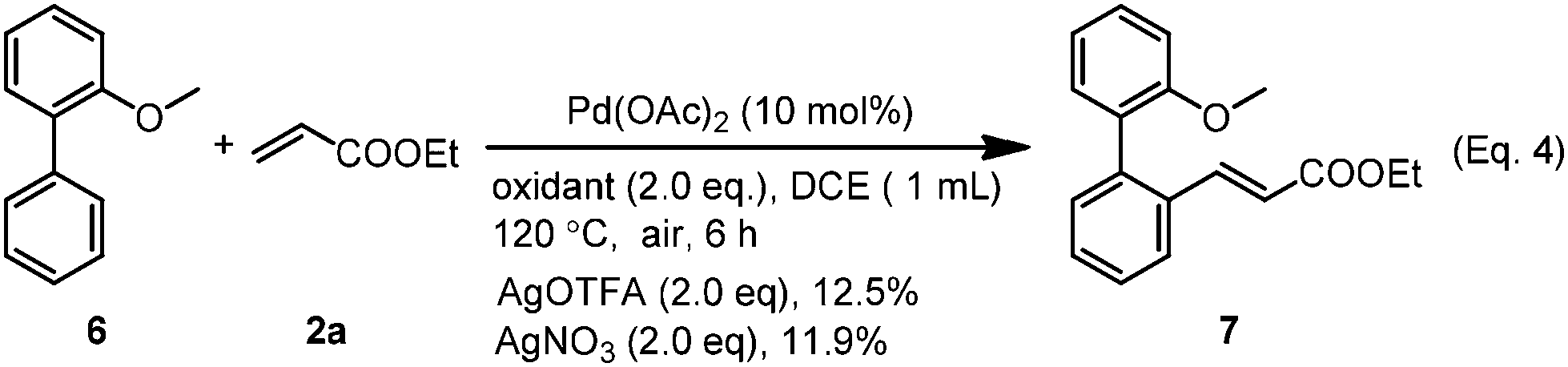 | ||
| Scheme 3 Ether as the directing group. | ||
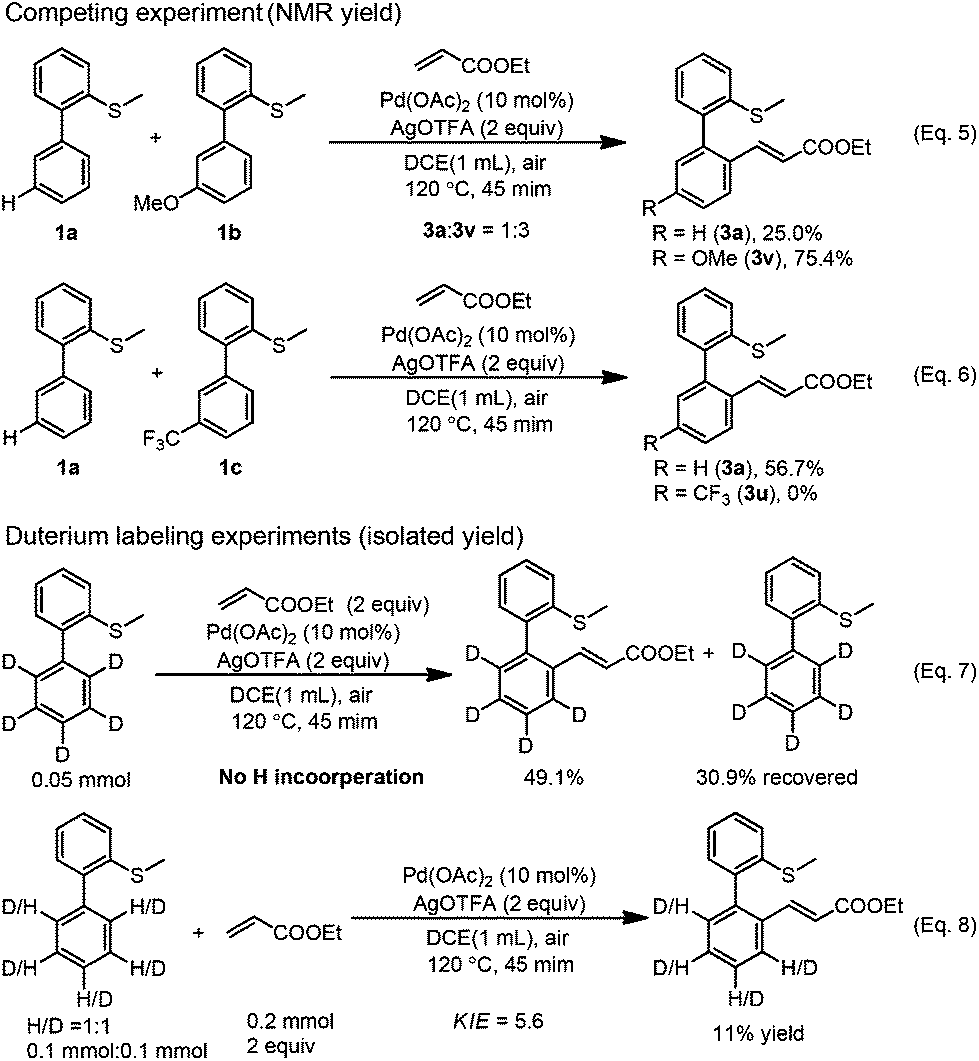 | ||
| Scheme 4 Mechanistic studies. | ||
On the other hand, the oxidant controlled selective C–H bond mono- and di-functionalization is interesting and the reason is not clear at this stage. To clarify the reason for the oxidant controlled selectivity, we investigated the additive effect (Scheme 5) by adding some inorganic salts. We found that when NaNO3 was added to the mono-alkenylation system, the ratio of mono-alkenylated product reduced. And when NaOAc was added to the di-alkenylation system, the ratio of the di-alkenylated product reduced. However, when NaOTFA was added to the di-alkenylation system, no obvious change in mono-/di-selectivity was observed. Based on these results, we conclude that it is possibly the coordinating ability of the counter anion of the oxidant that affected the selectivity.
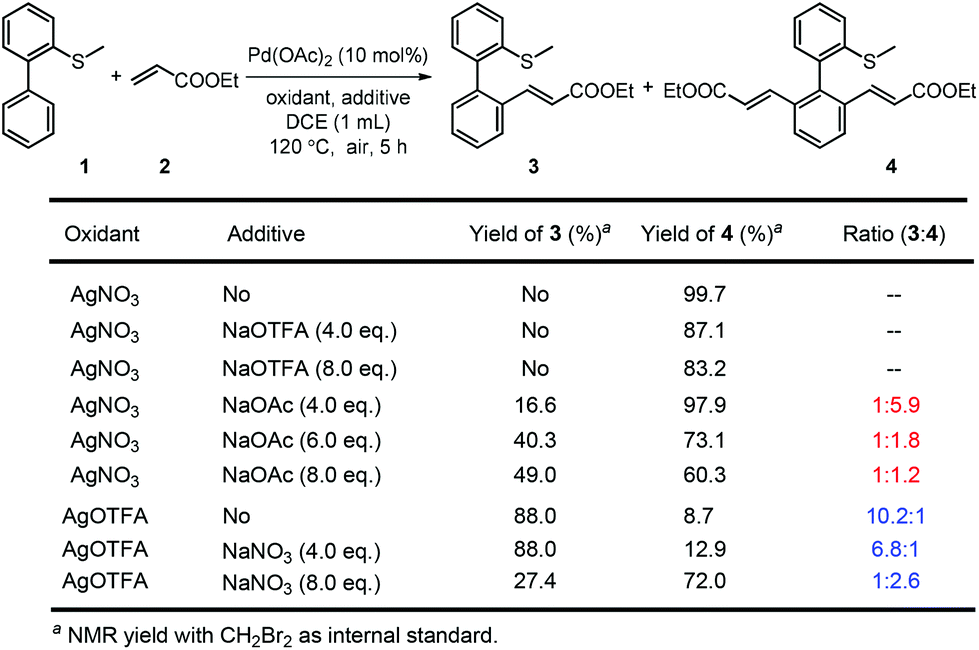 | ||
| Scheme 5 The effect of additives on the selectivity. | ||
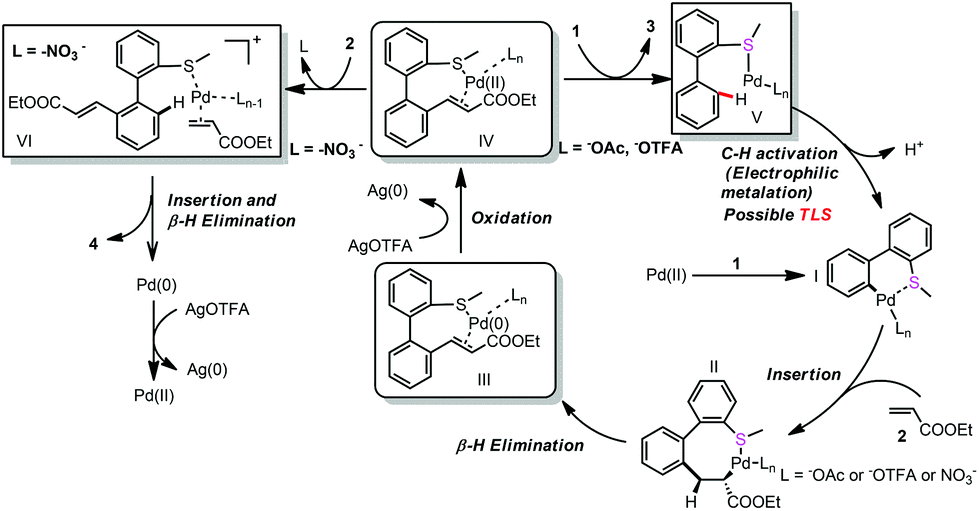
Based on the above mentioned experiments and mechanism studies, the detailed mechanism is depicted in Scheme 6. Pd-catalyzed first C–H bond activation formed intermediate I, alkene insertion and β-H elimination produced Pd(0) complex III with the mono-alkenylated product coordinating as a ligand, which was oxidized to Pd(II) complex IV. Weak coordinating NO3− can be exchanged by another molecule of alkene (2) and further promote the second C–H bond activation (Path a). However, the relatively strong coordinating −OAc could not be exchanged by another molecule of alkene, so the coordination of another molecule of thioether (1) released the mono-alkenylated product (3) (Path b).
 | ||
| Scheme 6 Proposed mechanism. | ||
In summary, with our thioether directing group, we realized the selective mono- and di-alkenylation of aryl C–H bonds in the bi-phenyl framework controlled by the oxidant, mainly by the coordinating ability of the counter anion of the oxidant. The substrate scope for both the mono-alkenylation and the di-alkenylation is very good. A widely used sulfoxide-olefin ligand was also obtained by simple transformation of the product. More detailed mechanism studies and other reactions based on the thioether directing group are underway.
Notes and references
- (a) V. T. Trepohl and M. Oestreich, Palladium-catalyzed arylation reactions of alkenes (Mizoroki-Heck reaction and related processes), Wiley-VCH Verlag GmbH & Co. KGaA, 2009, p. 221 Search PubMed; (b) B. Karimi, H. Behzadnia, D. Elhamifar, P. F. Akhavan, F. K. Esfahani and A. Zamani, Synthesis, 2010, 1399 CrossRef CAS PubMed; (c) R. Rossi, F. Bellina and M. Lessi, Synthesis, 2010, 4131 CrossRef CAS PubMed; (d) T. Satoh and M. Miura, Synthesis, 2010, 3395 CrossRef CAS PubMed; (e) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (f) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (g) J. Le Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef CAS PubMed; (h) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (i) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (j) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (k) S. I. Kozhushkov and L. Ackermann, Chem. Sci., 2013, 4, 886 RSC.
- (a) P. Thansandote and M. Lautens, Chem. – Eur. J., 2009, 15, 5874 CrossRef CAS PubMed; (b) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976 RSC; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (d) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed.
- (a) L. V. Desai, K. J. Stowers and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 13285 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (e) C. Wang and Y. Huang, Synlett, 2013, 145 CAS.
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS; (b) N. Umeda, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 7094 CrossRef CAS PubMed; (c) F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9982 CrossRef CAS PubMed.
- (a) A. V. Gulevich, F. S. Melkonyan, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2012, 134, 5528 CrossRef CAS PubMed; (b) T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Angew. Chem., Int. Ed. Engl., 1997, 36, 1740 CrossRef CAS; (c) Z. Ni, Q. Zhang, T. Xiong, Y. Zheng, Y. Li, H. Zhang, J. Zhang and Q. Liu, Angew. Chem., Int. Ed., 2012, 51, 1244 CrossRef CAS PubMed; (d) Z. Liang, R. Feng, H. Yin and Y. Zhang, Org. Lett., 2013, 15, 4544 CrossRef CAS PubMed.
- (a) A. Garcia-Rubia, R. G. Arrayas and J. C. Carretero, Angew. Chem., Int. Ed., 2009, 48, 6511 CrossRef CAS PubMed; (b) X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem. – Eur. J., 2013, 19, 11898 CrossRef CAS PubMed.
- (a) D. R. Armstrong, S. C. Ball, D. Barr, W. Clegg, D. J. Linton, L. C. Kerr, D. Moncrieff, P. R. Raithby, R. J. Singer, R. Snaith, D. Stalke, A. E. H. Wheatley and D. S. Wright, J. Chem. Soc., Dalton Trans., 2002, 2505 RSC; (b) K. S. L. Chan, M. Wasa, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 9081 CrossRef CAS PubMed.
- M. Aoyama, T. Fukuhara and S. Hara, J. Org. Chem., 2008, 73, 4186 CrossRef CAS PubMed.
- T.-S. Mei, R. Giri, N. Maugel and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 5215 CrossRef CAS PubMed.
- (a) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315 CrossRef CAS PubMed; (b) S. Harada, H. Yano and Y. Obora, ChemCatChem, 2013, 5, 121 CrossRef CAS; (c) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed.
- (a) X. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520 CrossRef CAS PubMed; (b) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350 CrossRef CAS PubMed; (c) M. Iwasaki, M. Iyanaga, Y. Tsuchiya, Y. Nishimura, W. Li, Z. Li and Y. Nishihara, Chem. – Eur. J., 2014, 20, 2459 CrossRef CAS PubMed.
- (a) T. A. Dwight, N. R. Rue, D. Charyk, R. Josselyn and B. DeBoef, Org. Lett., 2007, 9, 3137 CrossRef CAS PubMed; (b) D. R. Stuart, E. Villemure and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 12072 CrossRef CAS PubMed; (c) S. Potavathri, A. S. Dumas, T. A. Dwight, G. R. Naumiec, J. M. Hammann and B. DeBoef, Tetrahedron Lett., 2008, 49, 4050 CrossRef CAS PubMed; (d) Z. Liang, J. Zhao and Y. Zhang, J. Org. Chem., 2009, 75, 170 CrossRef PubMed; (e) Y. Li, W.-H. Wang, S.-D. Yang, B.-J. Li, C. Feng and Z.-J. Shi, Chem. Commun., 2010, 46, 4553 RSC.
- (a) B. Sezen, R. Franz and D. Sames, J. Am. Chem. Soc., 2002, 124, 13372 CrossRef CAS PubMed; (b) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed; (c) J. Yao, M. Yu and Y. Zhang, Adv. Synth. Catal., 2012, 354, 3205 CrossRef CAS; (d) M. Yu, Y. Xie, C. Xie and Y. Zhang, Org. Lett., 2012, 14, 2164 CrossRef CAS PubMed.
- (a) R. Samanta and A. P. Antonchick, Angew. Chem., Int. Ed., 2011, 50, 5217 CrossRef CAS PubMed; (b) T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139 CrossRef CAS; (c) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188 CrossRef CAS PubMed; (d) B. Wang, C. Shen, J. Yao, H. Yin and Y. Zhang, Org. Lett., 2014, 16, 46 CrossRef CAS PubMed.
- Y. Li and M.-H. Xu, Chem. Commun., 2014, 50, 3771 RSC.
- G. Chen, J. Gui, L. Li and J. Liao, Angew. Chem., Int. Ed., 2011, 50, 7681 CrossRef CAS PubMed.
- (a) A. Iglesias, R. Alvarez, A. R. de Lera and K. Muniz, Angew. Chem., Int. Ed., 2012, 51, 2225 CrossRef CAS PubMed; (b) J. Oyamada and Z. Hou, Angew. Chem., Int. Ed., 2012, 51, 12828 CrossRef CAS PubMed; (c) Z. Ren, F. Mo and G. Dong, J. Am. Chem. Soc., 2012, 134, 16991 CrossRef CAS PubMed; (d) G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00172a |
| This journal is © the Partner Organisations 2014 |