
Selective synthesis of secondary benzylic (Z)-allylboronates by Fe-catalyzed 1,4-hydroboration of 1-aryl-substituted 1,3-dienes†‡
Yanchun
Cao
,
Yanlu
Zhang
,
Lei
Zhang
,
Dan
Zhang
,
Xuebing
Leng
and
Zheng
Huang
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, 345 Lingling Road, Shanghai, 200032, China. E-mail: huangzh@sioc.ac.cn; Fax: +86 5492 5533; Tel: +86 5492 5522
First published on 23rd September 2014
Abstract
We have prepared and characterized a series of new iminopyridine iron complexes with a bulky diphenylphosphinomethyl-ketimine substituent. Using one of these iron complexes as the precatalyst, the hydroboration of 1-substituted 1,3-dienes containing aromatic groups with pinacolborane occurs regio- and stereoselectively to form secondary (Z)-allylboronates. In addition, we report the first examples of Suzuki–Miyaura cross-coupling of secondary allylboronates with aryl bromides. The reactions catalyzed by Pd(dba)2/Ad2PnBu yield the coupling products with excellent regioselectivity (γ/α > 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and E-selectivity of the olefin geometry (E/Z > 99:1).
:1) and E-selectivity of the olefin geometry (E/Z > 99:1).
Introduction
Allylboronates are versatile intermediates in organic synthesis. These compounds are widely used in carbonyl allylation reactions, in which the olefinic geometry of the reagents can be stereochemically transferred.1 In addition, the primary and secondary allylboron compounds can now be used as coupling partners in Pd-catalyzed allylic substitution reactions, producing allylated arenes that are an important class of substituted aromatic compounds.2 These applications have created an increasing demand for rapid construction of allylboron compounds with diverse structures.Classic methods for the synthesis of allylboronates involve the stoichiometric reactions of organolithium or organomagnesium reagents with suitable boron reagents.1b,3 Recently, transition metal-catalyzed borylations have received increasing attention for the preparation of allylboronates due to their wide functional group compatibility. For example, Miyaura,4 Aggarwal and Szabó,5 and Morken6 have independently reported Pd- or Ni-catalyzed allylic substitutions with bis(pinacolato)diboron (B2(Pin)2) to form primary allylboronates.7 Recently, Ito and Sawamura have developed a Cu-catalyzed γ-selective and stereospecific substitution of allylic carbonates with B2(Pin)2 to form secondary (E)-allylboronates.8
Because of its high atom economy and mild reaction conditions, catalytic 1,4-hydroboration of conjugated dienes is an attractive method to prepare allylborons.9 Pioneering work by Suzuki and Miyaura showed that Pd(PPh3)4 and Rh4(CO)12 catalyze 1,4-hydroboration of 2-substituted or 2,3-disubstituted dienes with catechoborane to form primary (Z)-allylboronates (Scheme 1a).9a Recently, Ritter reported iminopyridine-ligated iron catalysts for regio- and stereoselective hydroboration of 2-substituted dienes with pinacolborane (HBPin) to form primary (E)-allylboronates, but for 1-substituted dienes is restricted to 1,3-cyclohexadiene (Scheme 1b).9b Compared to the reactions of 2-substituted dienes, transition metal-catalyzed hydroboration of 1-substituted dienes is likely more complex because of the propensity for 1,2-addition.9d,e Nevertheless, Morken developed a Ni-catalyzed 1,4-hydroboration of 1-substituted dienes to form primary (Z)-allylboronates (Scheme 1c).9c To our knowledge, however, there is no example of 1,4-hydroboration of 1-substituted dienes to produce secondary allylboronates.
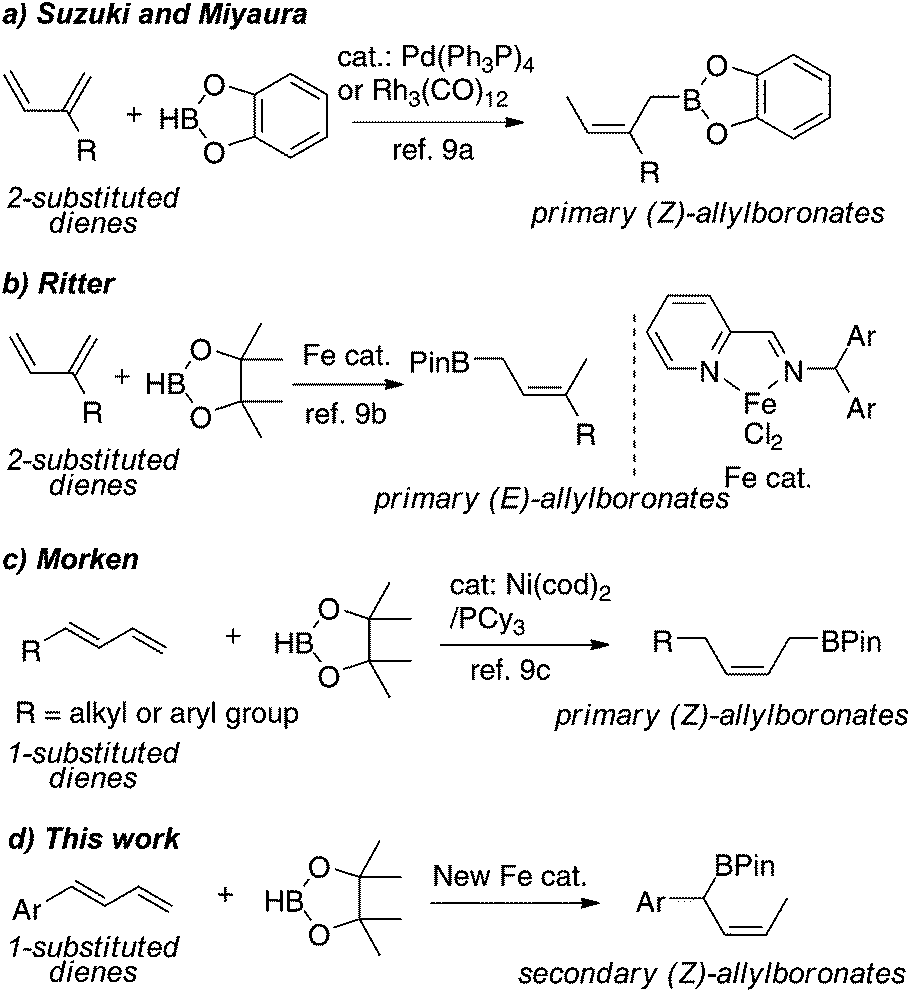 | ||
| Scheme 1 Transition metal-catalyzed 1,4-hydroboration of 1,3-dienes. | ||
Our group has been interested in developing well-defined non-precious metal complexes for catalytic alkene hydrofunctionalizations.10 Herein we report novel iminopyridine iron complexes for catalytic 1,4-hydroboration of 1,3-dienes. Significantly, the borylation of 1-substituted dienes containing aromatic substituents occurs regioselectively at the more sterically hindered carbon attached to the aryl group, furnishing the secondary benzylic (Z)-allylboronates. We also describe a mild protocol for Suzuki–Miyaura cross-coupling of the secondary allylboronates with aryl bromides that give the allylated arene products in a regio- and stereoselective mode.
Results and discussion
Preparation of iminopyridine iron dihalide complexes
During the course of the preparation of PNN pincer ligands,10c we found that the reaction of iminopyridine 1 with LDA and diphenylchlorophosphine gave compound 3, instead of the PNN pincer ligand 2 (Scheme 2). The result indicates that phosphination occurs more readily at the (imino)methyl group than the 2-methyl group on the pyridine ring under the reaction conditions. Treatment of 1 and 3 with FeCl2 generated four-coordinate Fe(II) dichloride complexes 4 and 5, respectively. Both the single crystal structures of 4 and 5 display a distorted tetrahedral geometry. The structure of 5 shows two nitrogen atoms coordinate to the iron center, while the phosphorus atom remains intact. The dihedral angle between the arylimino plane and the iminopyridine plane is −73.80° (see Fig. 1). As a comparison, the arylimino plane in 4 is almost orthogonal to the iminopyridine backbone with a dihedral angle of −93.18° (see Fig. 1), similar to those observed in related bis(imino)pyridine iron complexes.11 Apparently, the steric repulsion between the diphenylphosphinomethyl group and the arylimino group in 5 causes the decreased dihedral angle (by ∼16° from 90° in 5). We envisioned that the diphenylphosphinomethyl group in 5 could suppress the rotation or fluctuation of the CAr–N bond in solution.12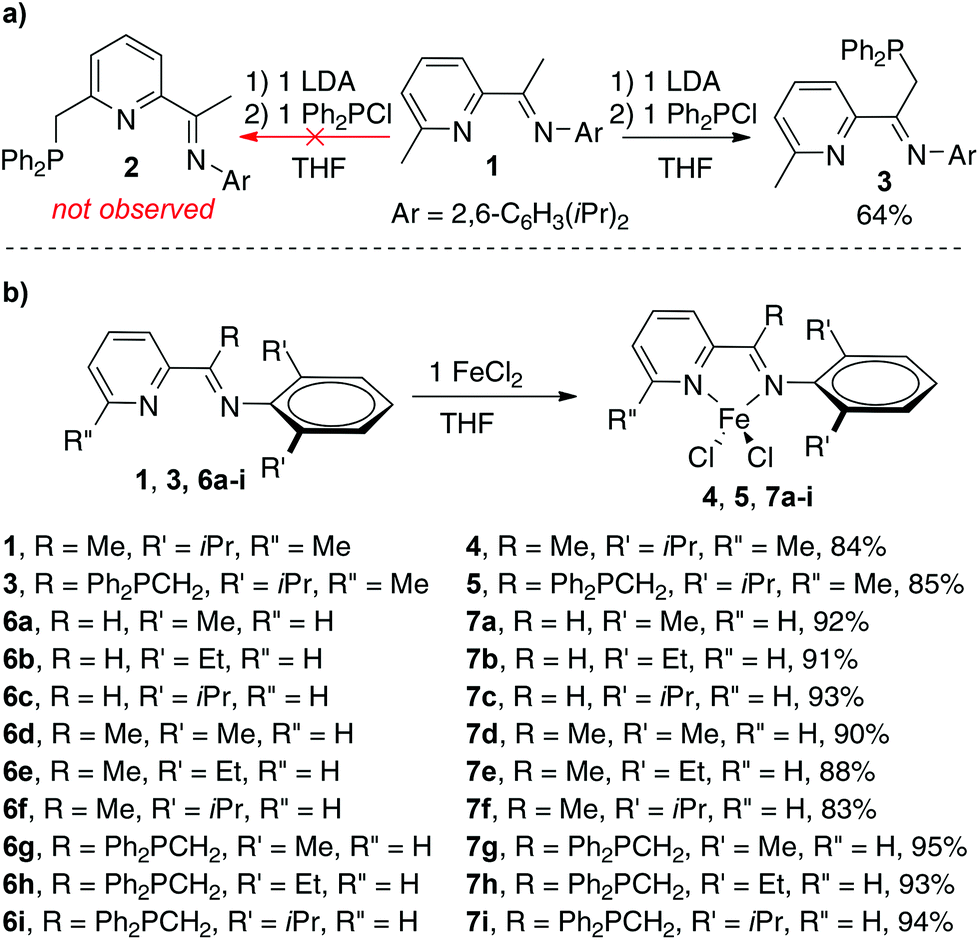 | ||
| Scheme 2 (a) Preparation of ligand 3; (b) preparation of iminopyridine iron complexes 4, 5, and 7a–i. | ||
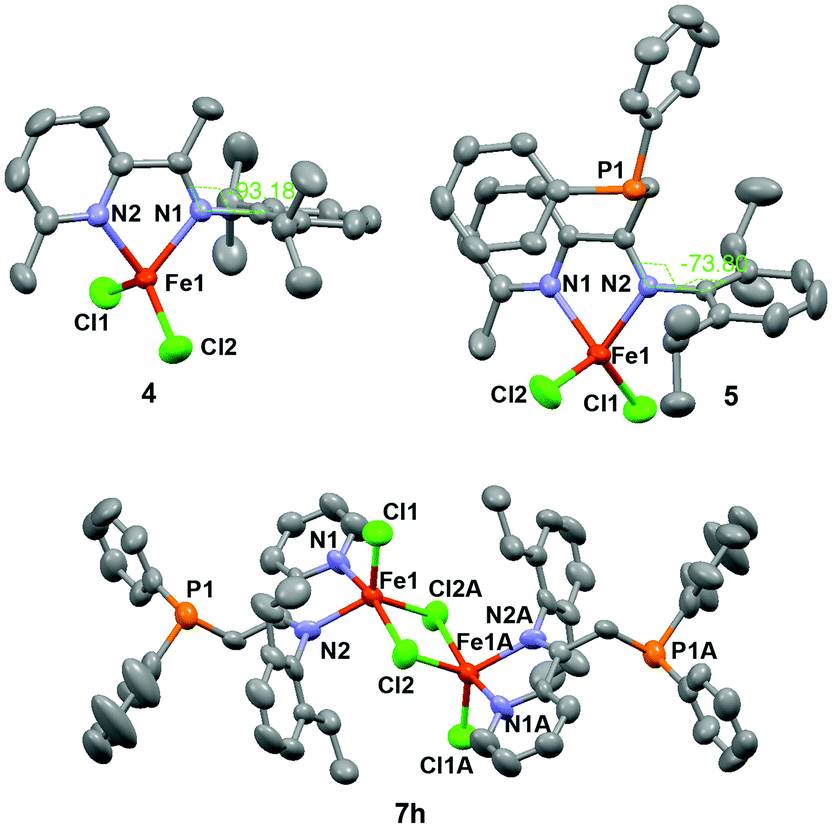 | ||
| Fig. 1 Single crystal structures of iron complexes 4, 5 and 7h. | ||
Inspired by Ritter's seminal work on (imino)pyridine iron-catalyzed hydroboration of 2-substituted 1,3-dienes (vide supra),9b and driven by the possible advantage of using more steric bulky iron complexes for selective hydroboration of terminally substituted 1,3-dienes, we prepared additional nine iron complexes (7a–h) with various ketimine substituents and N-aryl substituents (Scheme 2). Complex 7c has been previously reported9b and the others are new iron complexes. In contrast to the mononuclear structure of complexes 4 and 5, X-ray diffraction analysis of the solid-state structure of 7h revealed a dinuclear framework, in which two five-coordinated Fe(II) centers are connected by two Cl atoms (Fig. 1).13
Iron-catalyzed hydroboration of 1-substituted dienes
The iron complexes 4, 5, and 7a–i were then tested as precatalysts for the hydroboration of terminally substituted 1,3-dienes, 1-phenylbutadiene (8a), with pinacolborane (HBPin). The active catalysts were generated in situ upon the addition of NaBEt3H to the precursors (two equiv. relative to Fe).14 As summarized in Table 1, the regioselectivity for the addition varies significantly depending on the substituents in the iron complexes. Precatalysts 4 and 5 with a methyl substituent at the 2-position of the pyridine ring gave the 1,2-hydroboration product 9a′′ as the major hydroboration product (entries 1 and 2). In contrast, precatalysts (7a–i) without the methyl substituent favour the 1,4-hydroboration products (entries 3–11). The most sterically crowded precatalyst 7i containing a diphenylphosphinomethyl-ketimine substituent and bulky iPr substituent at the 2,6-positions of the N-aryl group gave the primary (Z)-allylboronate 9a′ as the major product (entry 11). In contrast, all other eight precatalysts (7a–h) gave the secondary (Z)-allylboronate 9a as the major product. Among these, the reactions with 7d, 7e, 7g, and 7h are most selective for the formation of 9a; the branched product constitutes ∼95% of the total hydroboration products. In most reactions, the combined yields of the hydroboration products are much lower than the conversion of the diene (multiple unidentified species were observed by GC/MS in these reactions). To our delight, the precatalyst 7h with the diphenylphosphinomethyl group and the ortho-aryl Et substituent provided 9a in high yield with high selectivity (entry 10). A comparison with the results obtained using precatalysts 7b and 7e indicates that the bulky diphenylphosphinomethyl group in 7h has a beneficial effect on the hydroboration of 1-substituted dienes (entry 10 vs. entries 4 and 7).|
|
||||
|---|---|---|---|---|
| Entry | Precat. | Conv.b (%) | Regioselectivity (9a:9a′:9a′′)b |
Yieldb (%) |
| a Reaction conditions: 8a (0.25 mmol), HBPin (0.26 mmol), precatalyst (1 mol%) and NaBEt3H (2.5 mol%) in Et2O (1 mL) at 25 °C. b The conversion of 8a, the ratio of the hydroboration products, and the total yields of the hydroboration products were determined by GC. Average of two runs. | ||||
| 1 | 4 | 90 | 19:15:66 |
24 |
| 2 | 5 | 49 | 28:5:67 |
12 |
| 3 | 7a | >99 | 84:6:10 |
14 |
| 4 | 7b | 73 | 79:11:10 |
8 |
| 5 | 7c | 88 | 88:10:2 |
27 |
| 6 | 7d | 76 | 95:4:1 |
26 |
| 7 | 7e | 91 | 95:5:— |
45 |
| 8 | 7f | >99 | 69:31:— |
82 |
| 9 | 7g | 65 | 94:5:1 |
41 |
| 10 | 7h | >99 | 95:5:— |
91 |
| 11 | 7i | 70 | 42:57:1 |
47 |
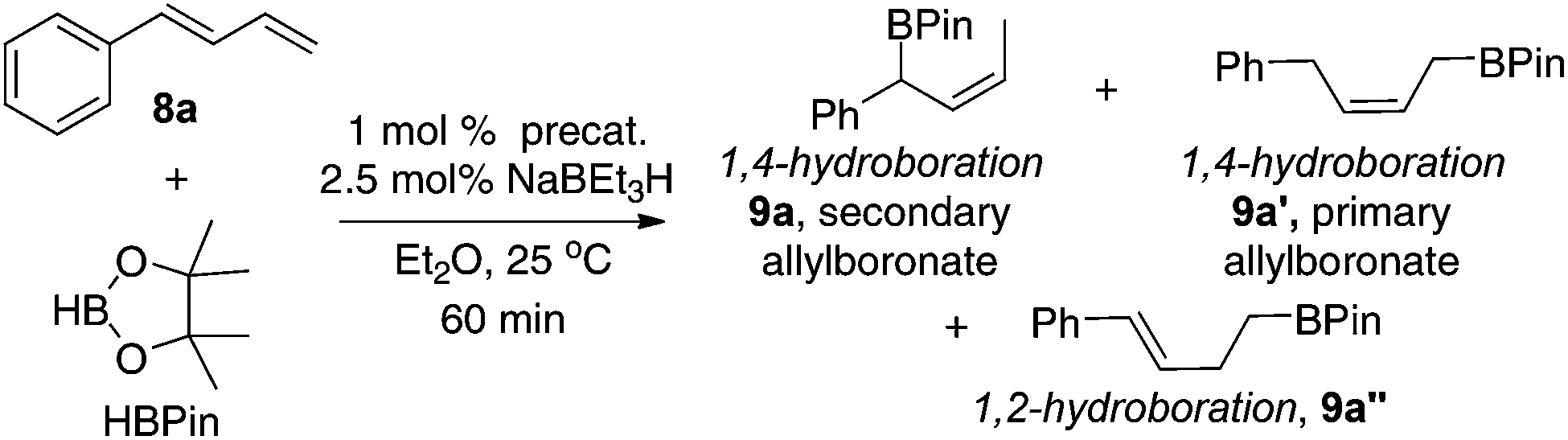
The regioselective formation of secondary (Z)-allylboronates via Fe-catalyzed hydroboration of 1-substituted dienes is of interest because it can afford stereodefined secondary allylboron compounds in a convenient fashion. The method is complement to the Ni-catalyzed hydroboration, in which substrates with both aromatic or alkyl substituents were hydroborated to give the primary (Z)-allylboronates (vide supra).9c In addition, the Fe-catalyzed 1,4-hydroboration of dienes provides an easy access to secondary (Z)-disubstituted allylic alcohols that are important synthetic intermediates.15 For example, a one-pot, two-step hydroboration–oxidation reaction converted 1-phenylbutadiene 8a into the (Z)-allylic alcohol 10 in 80% isolated yield (eqn (1)).
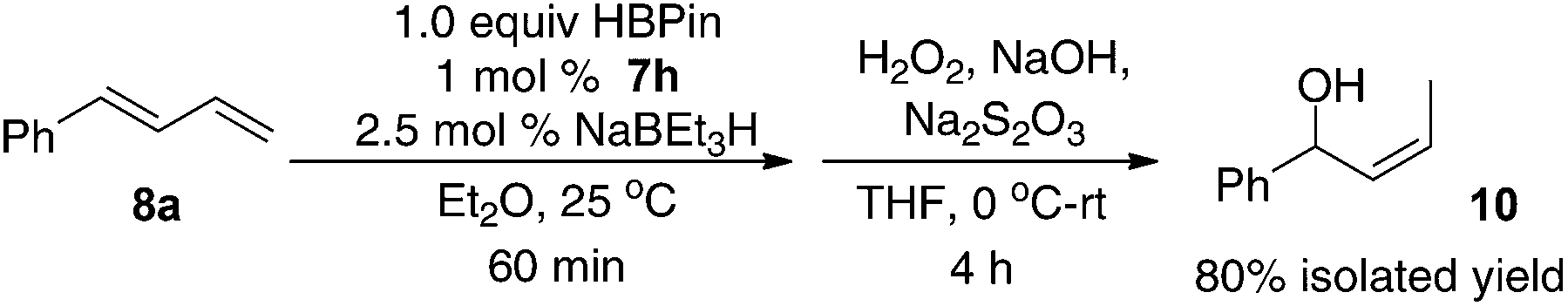 | (1) |
Next, the scope of the Fe-catalyzed hydroboration of 1-substituted dienes was expanded as listed in Scheme 3. The reactions of substrates containing aromatic groups with HBPin occurred smoothly to provide the secondary (Z)-allylboronate esters with high level of regio- and stereocontrol. Substrates with both electron-donating and -withdrawing groups at the para and meta positions of the aryl rings gave the branched products in high yields (9a–i). The hydroborations of dienes bearing naphthayl (8j) and 1,3-benzodioxole (8k) groups also afforded the secondary (Z)-allylboronate esters with high regio- and stereoselectivity.
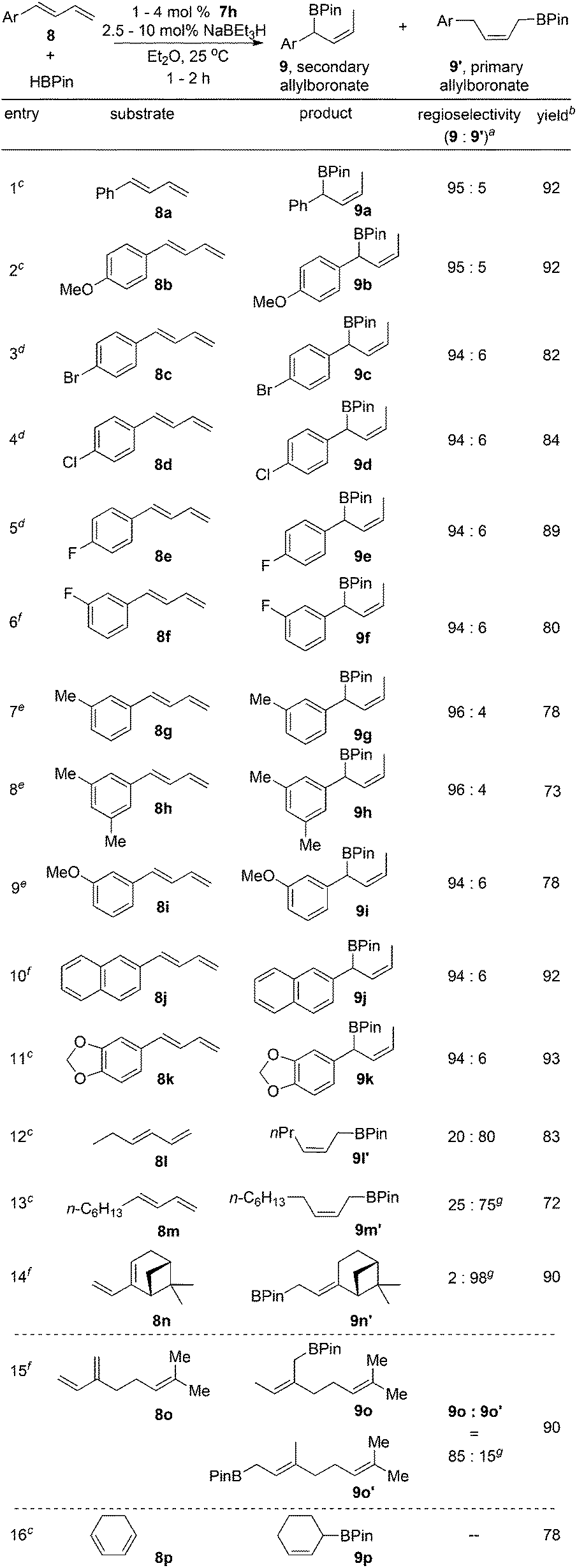 | ||
| Scheme 3
7h-catalyzed hydroboration of 1,3-dienes with HBPin. Reaction conditions: 8 (0.25 mmol) and HBPin (0.26 mmol) in Et2O (1 mL) at 25 °C. aThe ratio of the hydroboration products was determined by 1H NMR. bIsolated yields. cWith 1 mol% 7h and 2.5 mol% NaBEt3H, 1 h. dWith 4 mol% 7h and 10 mol% NaBEt3H, 2 h. eWith 2 mol% 7h and 5 mol% NaBEt3H, 1 h. fWith 2 mol% 7h and 5 mol% NaBEt3H, 2 h. gThe ratio of the hydroboration products were determined by GC. | ||
We attributed the unusual regioselectivity for secondary allylboronates to the presence of the aromatic substituents in the dienes. Consistent with our hypothesis, the reactions of 1-substituted dienes 8l–n containing alkyl groups at terminal positions with HBPin provided the primary allylboronates as the major products. The hydroboration of 2-substituted diene, myrcene (8o), produced the branched allylboronate 9o as the major product (9o:9o′ = 85:15). Similar regioselectivity was observed for the reaction using 7c as the precatalyst in previous work.9b The reaction with 1,3-cyclohexadiene gave the hydroboration product 9p in good yield.
The Fe-catalyzed hydroboration of 1,4-disubstituted internal dienes also produces the benzylic allylic boronates with good selectivity (Scheme 4). The disubstituted internal dienes, which were obtained as an E,E- and E,Z-mixture by Wittig reactions, react more slowly with HBPin than the 1-substituted dienes. However, the hydroboration of 11a–d catalyzed with 7e as the precatalyst (5 mol%) and magnesium metal as the activator (12.5 mol%) afforded the product 12 in good yields (entries 1–4).16 As shown in Scheme 4, the boron atom was added primarily to the more hindered aryl-substituted carbons. The 1,1,4-trisubstituted internal diene 11e, however, is unreactive under the reaction conditions, presumably due to the steric hindrance effect of the diene (entry 5).
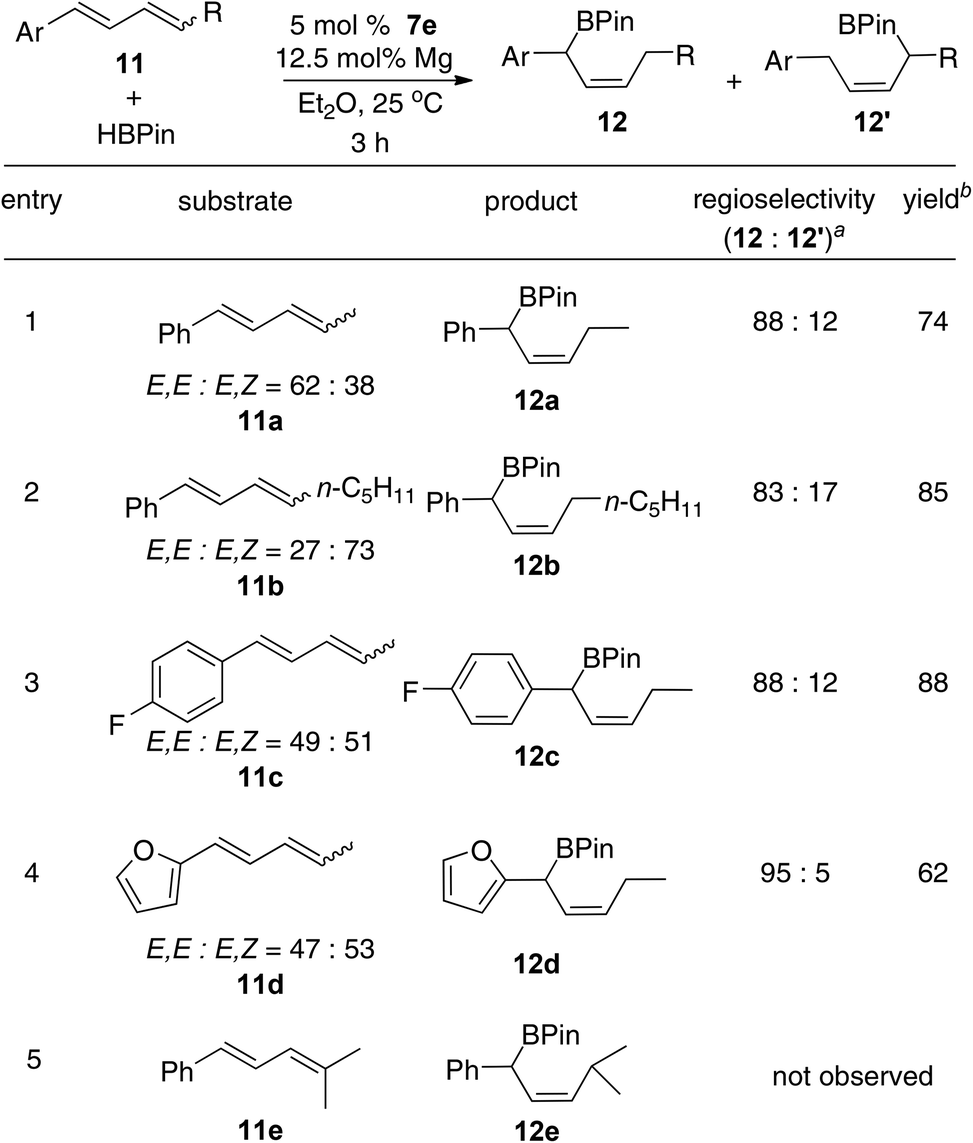 | ||
| Scheme 4
7e-catalyzed hydroboration of internal dienes with HBPin. Reaction conditions: 11 (0.25 mmol), HBPin (0.5 mmol), 7h (5 mol%), and Mg (12.5 mol%) in Et2O (1 mL) at 25 °C. aThe ratio of the hydroboration products were determined by 1H NMR. bIsolated yields. | ||
On the basis of our preliminary data and precedent regarding transition metal-catalyzed hydroborations of 1,3-dienes,9b,c we propose the pathway of iron-catalyzed hydroborations of 1-aryl-substituted 1,3-dienes depicted in Scheme 5. Following the bonding of 1,3-diene to the iron center, oxidative addition of HBPin to the η4-diene species A forms a formal 18 electron iron boryl hydride compound B. Migratory insertion into either the Fe–H or Fe–B would give the allyl intermediate C or C′, respectively. Although we currently cannot distinguish between these two pathways, the former mode may be facilitated by the formation of the conjugation system. Subsequent η3–η1 rearrangement and reductive elimination would then generate the secondary allylboronate product.
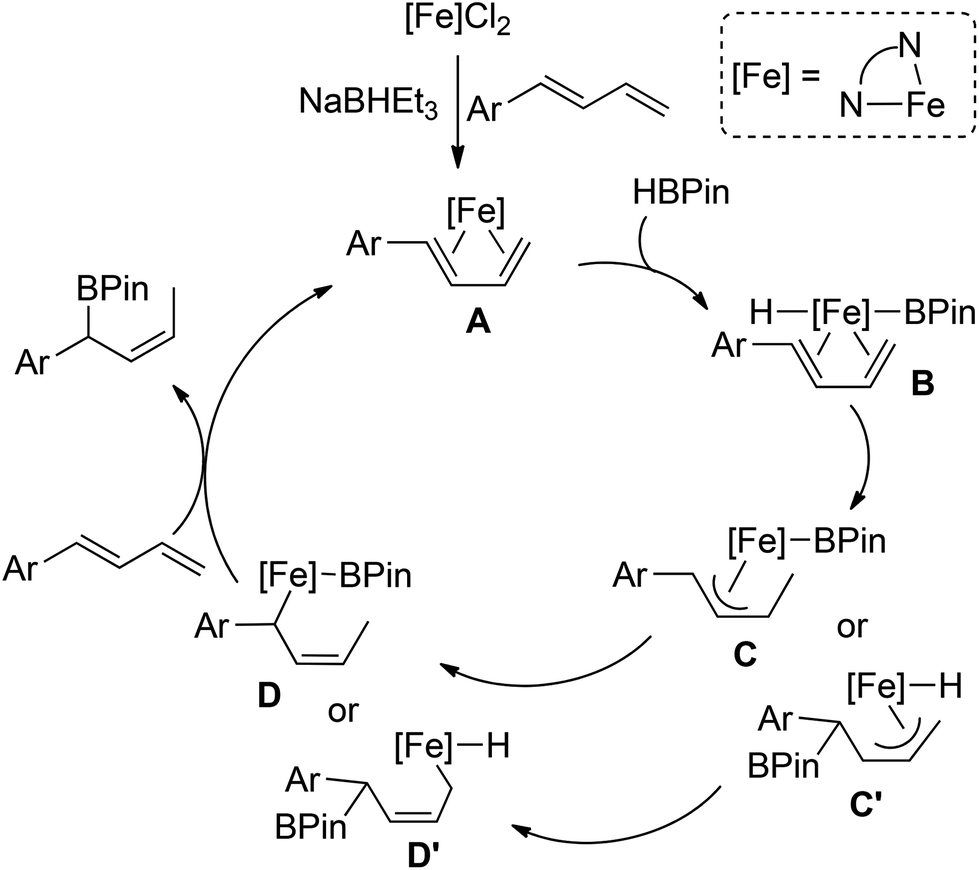 | ||
| Scheme 5 Proposed mechanism for the iron-catalyzed hydroboration of 1-aryl-substituted 1,3-dienes. | ||
Suzuki–Miyaura cross-coupling of secondary allylboronate esters with aryl bromides
Having established an efficient approach to access secondary benzylic allylboronates, it was of interest to explore their applications in Suzuki–Miyaura cross-coupling reactions. Recently, Crudden reported the first examples of Pd-catalyzed couplings of secondary allylboronates with aryl iodides.2h The reactions of secondary benzylic allylboronates catalysed by Pd(PPh3)4/PPh3 in the presence of Ag2O and K2CO3 provided γ/α selectivity of ∼90:10. The use of Ag2O was believed to be critical for the transformation because it may facilitate the transmetallation step.2h Using the standard reaction conditions, more recently Crudden and Aggarwad developed an enantiospecific and regioselective coupling of enantiomerically enriched secondary allylboronates with aryl iodides.2j To our surprise, the reaction employing aryl bromide as the coupling reagent has not yet been reported.
Using the Ag2O/K2CO3-mediated reaction conditions as reported in previous work,2b,c the Pd-catalyzed couplings between 4-methylphenyl bromide 13b and allylboronate 9a (1.5 equiv.) with various monophosphine and biphosphine ligands gave the cross-coupling products in low to moderate yields (see Table S2 in ESI‡). An extensive screening of the ligands and additives (see ESI‡ for details) revealed that the catalyst generated from Ad2PnBu in the presence of KF provided a high yield of the coupling product 14b; excellent regioselectivity (γ/α > 99:1) and E selectivity with respect to the geometry of the olefin (E/Z > 99:1) were observed using 2 mol% of Pd. Under the optimized conditions, aryl bromides 13 and secondary benzylic allylboronates 9 (1.5 equiv.) bearing both electron-donating and -withdrawing groups could be effectively coupled in DME at 25 °C to produce the allylated arenes 14 in high isolated yields (Scheme 6).
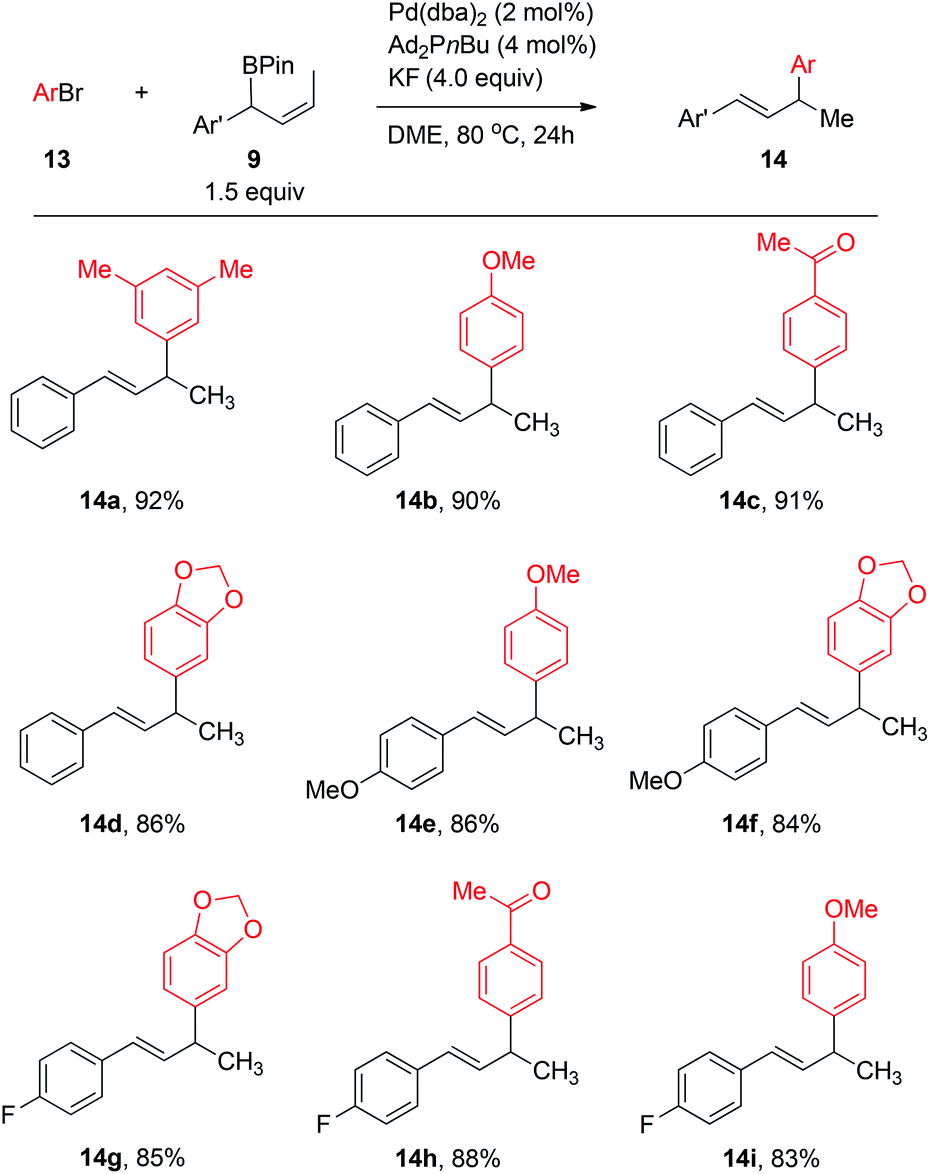 | ||
| Scheme 6 Pd-catalyzed Suzuki–Miyaura cross-coupling of benzylic (Z)-allylboronates with aryl bromides. Reaction conditions: aryl bromides 13 (0.203 mmol), allylboronate esters 9 (0.304 mmol), Pd(dba)2 (2 mol%), Ad2PnBu (4 mol%) and KF (4.0 equiv.) in DME (1 mL) at 80 °C. Yields shown are of isolated products. | ||
Conclusions
In summary, we have prepared novel iminopyridine iron complexes containing a diphenylphosphinomethyl-ketimine substituent. The iron complex can be applied to an unprecedented regio- and stereoselective catalytic hydroboration of 1-substituted 1,3-dienes with pinacolborane that allow for the convenient synthesis of secondary (Z)-allylboronates. The products would be difficult or tedious to access by other means. In addition, we have disclosed a selective protocol for the Suzuki–Miyaura cross-coupling of secondary allylboronates with aryl bromides. Further mechanistic studies to determine the origin of the regioselectivity for the formation of secondary allylboronates are undergoing in our laboratory.We gratefully acknowledge the financial support from the National Natural Science Foundation of China (no. 21272255 and 21121062).
Notes and references
- For examples, see: (a) V. Rauniyar, H. Zhai and D. G. Hall, J. Am. Chem. Soc., 2008, 130, 8481 CrossRef CAS PubMed; (b) M. Althaus, A. Mahmood, J. R. Suárez, S. P. Thomas and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 4025 CrossRef CAS PubMed; (c) M. J. Hesse, S. Essafi, C. G. Watson, J. N. Harvey, D. Hirst, C. L. Willis and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 6145 CrossRef CAS PubMed.
- For Pd-catalyzed cross-couplings with allyboron derivatives: (a) Y. Yamamoto, S. Takada and N. Miyaura, Chem. Lett., 2006, 35, 704 CrossRef CAS; (b) Y. Yamamoto, S. Takada and N. Miyaura, Chem. Lett., 2006, 35, 1368 CrossRef CAS; (c) Y. Yamamoto, S. Takada and N. Miyaura, Organometallics, 2009, 28, 152 CrossRef CAS; (d) S. Sebelius, V. J. Olsson and K. J. Szabó, J. Am. Chem. Soc., 2005, 127, 10478 CrossRef CAS PubMed; (e) V. J. Olsson, S. Sebelius, N. Selander and K. J. Szabó, J. Am. Chem. Soc., 2006, 128, 4588 CrossRef CAS PubMed; (f) S. Sebelius, V. J. Olsson, O. A. Wallner and K. J. Szabó, J. Am. Chem. Soc., 2006, 128, 8150 CrossRef CAS PubMed; (g) D. C. Gerbino, S. D. Mandolesi, H.-G. Schmalz and J. C. Podestá, Eur. J. Org. Chem., 2009, 3964 CrossRef CAS; (h) B. W. Glasspoole, K. Ghozati, J. W. Moir and C. M. Crudden, Chem. Commun., 2012, 48, 1230 RSC; (i) J. L. Farmer, H. N. Hunter and M. G. Organ, J. Am. Chem. Soc., 2012, 134, 17470 CrossRef CAS PubMed; (j) L. Chausset-Boissarie, K. Ghozati, E. LaBine, J. L. Y. Chen, V. K. Aggarwal and C. M. Crudden, Chem. – Eur. J., 2013, 19, 17698 CrossRef CAS PubMed; (k) Y. Yang and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 10642 CrossRef CAS PubMed.
- For examples, see: (a) J. W. Clary, T. J. Rettenmaier, R. Snelling, W. Bryks, J. Banwell, W. T. Wipke and B. Singaram, J. Org. Chem., 2011, 76, 9602 CrossRef CAS PubMed; (b) D. S. Matteson and D. Majumdar, Organometallics, 1983, 2, 1529 CrossRef CAS; (c) D. S. Matteson, Tetrahedron, 1998, 54, 10555 CrossRef CAS; (d) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS PubMed; (e) R. P. Sonawane, V. Jheengut, C. Rabalakos, R. Larouche-Gauthier, H. K. Scott and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 3760 CrossRef CAS PubMed; (f) B. A. Ondrusek, J. K. Park and D. T. McQuade, Synlett, 2014, 239 CAS.
- T. Ishiyama, T.-A. Ahiko and N. Miyaura, Tetrahedron Lett., 1996, 37, 6889 CrossRef CAS.
- G. Dutheuil, N. Selander, K. J. Szabó and V. K. Aggarwal, Synthesis, 2008, 2293 CAS.
- P. Zhang, I. A. Roundtree and J. P. Morken, Org. Lett., 2012, 14, 1416 CrossRef CAS PubMed.
- For other examples of catalytic borylation of allylic compouds, see: (a) G. W. Kabalka, B. Venkataiah and G. Dong, J. Org. Chem., 2004, 69, 5807 CrossRef CAS PubMed; (b) M. Murata, S. Watanabe and Y. Masuda, Tetrahedron Lett., 2000, 41, 5877 CrossRef CAS.
- (a) H. Ito, C. Kawakami and M. Sawamura, J. Am. Chem. Soc., 2005, 127, 16034 CrossRef CAS PubMed; (b) H. Ito, S. Ito, Y. Sasaki, K. Matsuura and M. Sawamura, J. Am. Chem. Soc., 2007, 129, 14856 CrossRef CAS PubMed; (c) H. Ito, T. Miya and M. Sawamura, Tetrahedron, 2012, 68, 3423 CrossRef CAS PubMed; (d) Y. Sasaki, C. Zhong, M. Sawamura and H. Ito, J. Am. Chem. Soc., 2010, 132, 1226 CrossRef CAS PubMed.
- (a) M. Satoh, Y. Nomoto, N. Miyaura and A. Suzuki, Tetrahedron Lett., 1989, 30, 3789 CrossRef CAS; (b) J. Y. Wu, B. t. Moreau and T. Ritter, J. Am. Chem. Soc., 2009, 131, 12915 CrossRef CAS PubMed; (c) R. J. Ely and J. P. Morken, J. Am. Chem. Soc., 2010, 132, 2534 CrossRef CAS PubMed; (d) M. Zaidlewicz and J. Meller, Tetrahedron Lett., 1997, 38, 7279 CrossRef CAS; (e) Y. Matsumoto and T. Hayashi, Tetrahedron Lett., 1991, 32, 3387 CrossRef CAS.
- (a) L. Zhang, Z. Zuo, X. Leng and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 2696 CrossRef CAS PubMed; (b) L. Zhang, D. J. Peng, X. B. Leng and Z. Huang, Angew. Chem., Int. Ed., 2013, 52, 3676 CrossRef CAS PubMed; (c) D. Peng, Y. Zhang, X. Du, L. Zhang, X. Leng, M. D. Walter and Z. Huang, J. Am. Chem. Soc., 2013, 135, 19154 CrossRef CAS PubMed.
- (a) B. L. Small, M. Brookhart and A. M. A. Bennett, J. Am. Chem. Soc., 1998, 120, 4049 CrossRef CAS; (b) G. J. P. Britovsek, M. Bruce, V. C. Gibson, B. S. Kimberley, P. J. Maddox, S. Mastroianni, S. J. McTavish, C. Redshaw, G. A. Solan, S. Strömberg, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 8728 CrossRef CAS.
- F.-S. Liu, H.-B. Hu, Y. Xu, L.-H. Guo, S.-B. Zai, K.-M. Song, H.-Y. Gao, L. Zhang, F.-M. Zhu and Q. Wu, Macromolecules, 2009, 42, 7789 CrossRef CAS.
- CCDC 1013290 (4), 1013291 (5) and 1013292 (7h) contain the supplementary crystallographic data for this paper.
- This activation strategy was first reported by Chiril et al. for in situ activation of (PDI)FeCl2. See: (a) M. W. Bouwkamp, A. C. Bowman, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2006, 128, 13340 CrossRef CAS PubMed; (b) K. Kamata, A. Suzuki, Y. Nakai and H. Nakazawa, Organometallics, 2012, 31, 3825 CrossRef CAS.
- For reviews, see: (a) W. Adam and T. Wirth, Acc. Chem. Res., 1999, 32, 703 CrossRef CAS; (b) M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689 CrossRef CAS PubMed; (c) H. Lebel, J. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed.
- With increased amount of iron precatalyst and activator in the reactions of internal dienes, the reaction using NaBEt3H as the activator would result in a decent yield of the hydrogenation product. Thus we used Mg, instead of NaBEt3H, as the activator to prevent the hydrogenation of dienes.
Footnotes |
| † This work is dedicated to Prof. Lixin Dai on the occasion of his 90th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and product characterization. CCDC 1013290–1013292. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00206g |
| This journal is © the Partner Organisations 2014 |