
Kinetic resolution of citronellal by chiral aluminum catalysts: L-menthol synthesis from citral†
Hisanori
Itoh
ab,
Hironori
Maeda
a,
Shinya
Yamada
a,
Yoji
Hori
*a,
Takashi
Mino
*b and
Masami
Sakamoto
b
aTaksasago International Corporation, 4-11, Nishi-Yawata 1-Chome, Hiratsuka City, Kanagawa 254-0073, Japan. E-mail: hisanori_itoh@takasago.com; yoji_hori@takasago.com; Fax: +81-463-25-2093; Tel: +81-463-25-2000
bDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, Yayoi-cho, Inage-ku, Chiba 263-8522, Japan. E-mail: tmino@faculty.chiba-u.jp; Tel: +81-43-290-3385
First published on 11th September 2014
Abstract
A highly reactive catalytic ring-closing ene reaction is discussed. This reaction is catalyzed via novel optically active aluminum BINOL and TADDOL complexes. The kinetic resolution of the racemic analogs of citronellal was affected by these Al catalysts. The BINOL-Al catalyst afforded 68% ee of a diastereomer of isopulegol and 62% ee of citronellal at 47% conversion. The reaction mechanism proposed assumes that the optically active catalyst possesses a metal center between two parallel aromatic rings. We postulate that the edge of the aromatic rings can recognize the methyl group at the 3-position of citronellal, as the rings are oriented in a pseudoparallel orientation. We utilized the kinetic resolution for the synthesis of L-menthol from citral.
Introduction
A number of asymmetric intermolecular or intramolecular ene reactions have been reported for the synthesis of optically active products.1 The ene and Prins reactions between carbonyl and olefinic groups have been utilized and reported previously.1–3Another common application for the ring-closing ene reaction is the synthesis of (5R)-isopulegols (2) from (R)-citronellal ((R)-1) (Scheme 1). Citronellal and isopulegol are not only important aroma chemicals but are also valuable intermediates for the synthesis of L-menthol (3). A variety of methods have been reported for the cyclization of citronellal to isopulegol.4–6 Several chiral organometallic Zn and Ti complexes have been employed for the asymmetric cyclization of 1.7–10 The stoichiometric ring-closing ene reaction of citronellal using alkylaluminum chloride has also been reported previously,11 and various other aluminum complexes have also been applied to this end, with excellent diastereoselectivity.12–15
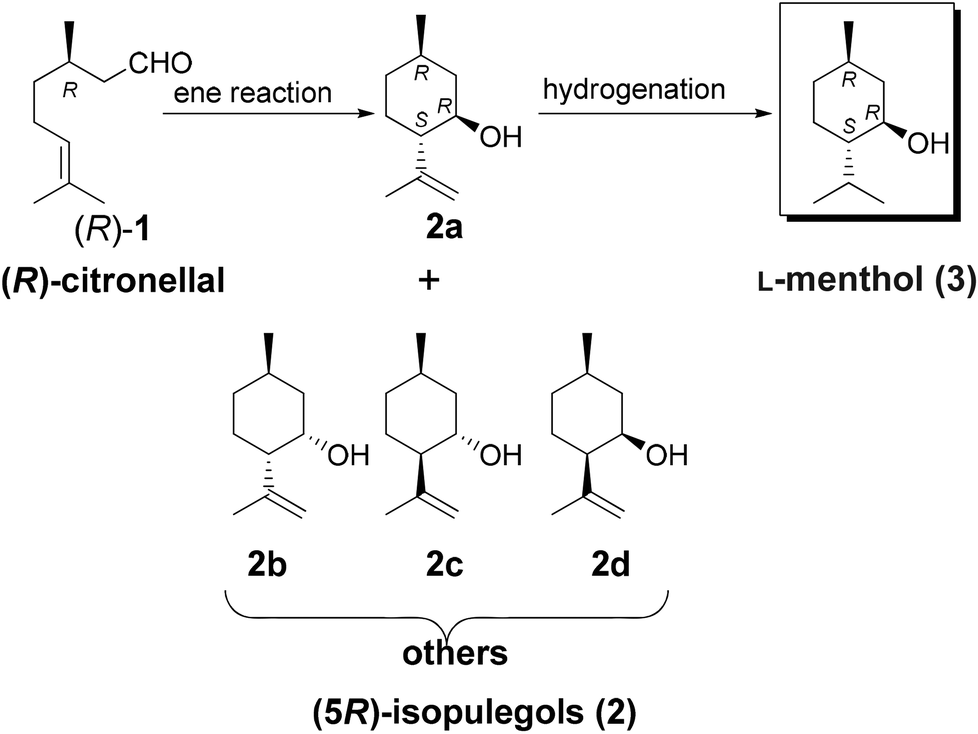 | ||
| Scheme 1 L-Menthol synthesis from (R)-citronellal. | ||
Kinetic resolution of racemic compounds, where one enantiomer reacts faster than the other, is an important method in asymmetric synthesis, and in recent years, a lot of research has been dedicated to the discovery and development of novel methods to achieve kinetic resolution. This is largely due to the cost associated with the purchase of optically pure starting materials and the expertise required for their asymmetric assembly. Kinetic resolution can take many forms such as enzymatic or biocatalytic,16 thermal (high or low temperature),17 and organic or organometallic.18,19 Kagan et al. found a variety of kinetic resolution methods and laid the foundation for the modern study of kinetic resolution.20
Kinetic resolution can be divided into roughly six categories, hydrolysis, reduction, oxidation, ring-opening, ring-closing, and other substituent changing reactions.20–22 However, to the best of our knowledge, neither the cyclization nor the kinetic resolution of citronellal derivatives with optically active aluminum complexes has been reported.
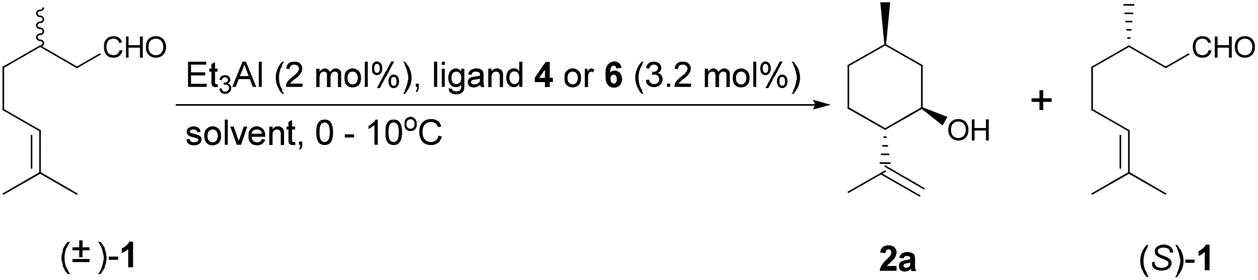
We describe herein the results of our investigation into the highly diastereoselective kinetic resolution of citronellal analogs catalyzed by chiral aluminum complexes such as BINOL-Al. These catalysts afforded isopulegol analogs in good diastereo- and enantioselectivities. It is expected that this process can be utilized for the synthesis of 3 from racemic starting materials (Scheme 2). A mechanistic analysis of the kinetic resolution is also discussed.
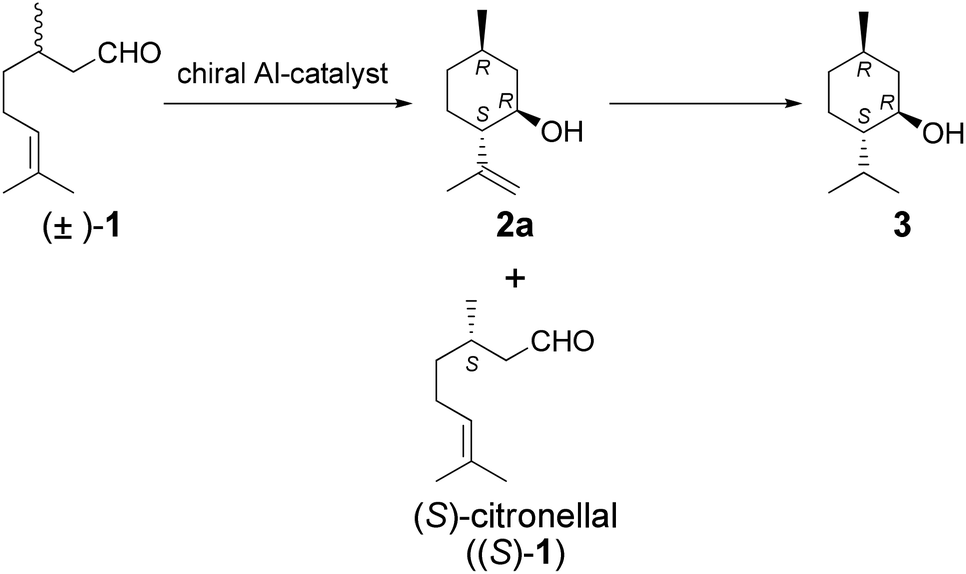 | ||
| Scheme 2 Kinetic resolution of racemic citronellal by the chiral Al-catalyst. | ||
Results and discussion
Ring-closing ene reaction of (R)-citronellal using BINOL-Al
As a first step, we carried out the reaction of (R)-citronellal ((R)-1) with BINOL (4a)-Al catalysts under various conditions. The results are summarized in Table 1. The conversions were calculated as the total consumption of substrate 1. Diastereoselectivity was calculated as the ratio of (5R)-n-isopulegol (2a) to the total (5R)-isopulegols (2). As can be seen from Table 1 (entries 1 and 2), the reactivities of (R)-4a-Al and (S)-4a-Al were significantly different. This result indicated that the BINOL-Al catalysts recognized the steric structure of 1, resulting in the different reactivity. The chiral recognition of the catalyst strongly affected the cyclization; the mismatched combination of (S)-4a-Al and (R)-1) afforded 2a in moderate reactivity (entry 2, Table 1). The racemic BINOL ((±)-4a)-Al catalyst afforded a 80% selectivity of dimer ester 5 by the Tishchenko reaction23,24 and 6% selectivity of 2 (entry 3, Table 1). We have further investigated and optimized the details of this reaction.|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | BINOL (4a) | Conv.a,b (%) | Selectivity of (5R)-isopulegols (2)c (%) | Ratio of 2a in 2c (%) |
| a Determined from the GC areas and corrected by the ratio of the GC areas and the molecular volumes of (R)-citronellal ((R)-1), (5R)-isopulegols (2), and dimer 5. b The conversions were calculated as the total consumption of (R)-1. c Determined from the GC areas. d Reacted for 19 h. e 5 mol% of (R)-4a was used. f 25 mol% of (R)-4a was used. g To a solution of LiAlH4 (19 mg, 0.50 mmol, 5 mol%) in THF (5 mL) a solution of (R)-4a (300 mg, 1.05 mmol, 10 mol%) in THF (5 mL) was added at 0 °C. After stirring for 30 min at 0 °C, (R)-1 (1.54 g, 10.0 mmol) was added dropwise at a temperature below 5 °C. The mixture was stirred for 1 h. The reaction products were analyzed using GC.25 | |||||
| 1 | Toluene | (R) | 97 | 93 | 98 |
| 2 | Toluene | (S) | 33 | 100 | 69 |
| 3d | Toluene | (±) | 90 | 6 + 80(5) | 79 |
| 4 | Toluene | (R)e | 98 | 3 + 84(5) | n.d. |
| 5 | Toluene | (R)f | 86 | 100 | 94 |
| 6g | Toluene | (R) | Trace | — | — |
| 7 | CH2Cl2 | (R) | 98 | 100 | 97 |
| 8 | Heptane | (R) | 83 | 85 | 88 |
| 9 | THF | (R) | 1 | 100 | n.d. |
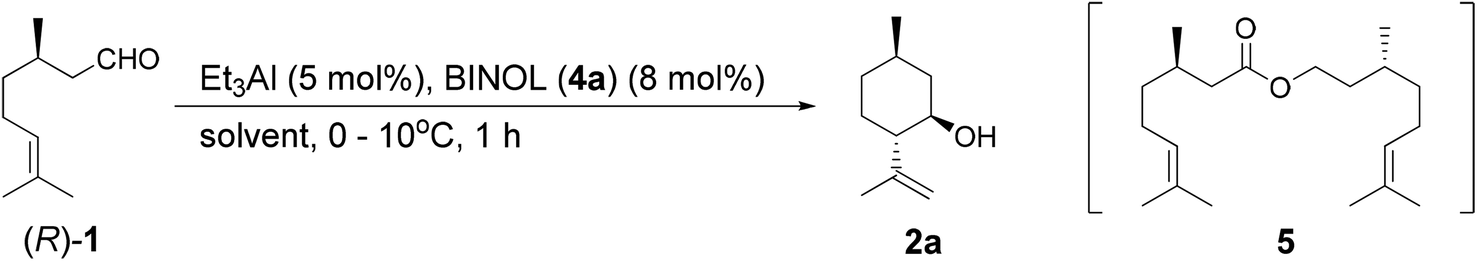
Screening of ligand equivalents
The ligand equivalent effects are shown in Table 1, entries 1, 4, and 5. The catalyst with one equivalent of (R)-4a to aluminum afforded 2 in only 3% selectivity and ester 5 in 84% selectivity (entry 4, Table 1). In contrast, the ring-closing ene reaction proceeded exclusively when the ratio of BINOL equivalents to aluminum was greater than 1.6 (entries 1 and 5, Table 1). BINOL-Al in an equimolar ratio has previously been used as a catalyst in the Tishchenko reaction.23 However, when the BINOL/Al ratio was 1.5, the complex changed its character to become a cyclization catalyst. Therefore, we only added a small excess of BINOL ligands in order to prevent the Tishchenko reaction. The details of the catalyst characteristics are described below in the section Reaction mechanism. Moreover, these results suggest that the structure of the catalyst used in the ring-closing ene reaction is quite different from that of the Al-Li-BINOL complex reported by Shibasaki (Table 1, entry 6).25Solvent effects
The results of solvent screening for the citronellal cyclization are shown in entries 1 and 7–9 of Table 1. As can be seen, the reaction did not proceed in THF (entry 9, Table 1), indicating that BINOL-Al catalysts could not be used in polar solvents. We believe that the lone-pair of polar solvents coordinates to aluminum and decreases the Lewis acidity of the catalyst. Therefore, we concluded toluene and dichloromethane to be suitable reaction solvents.Kinetic resolution
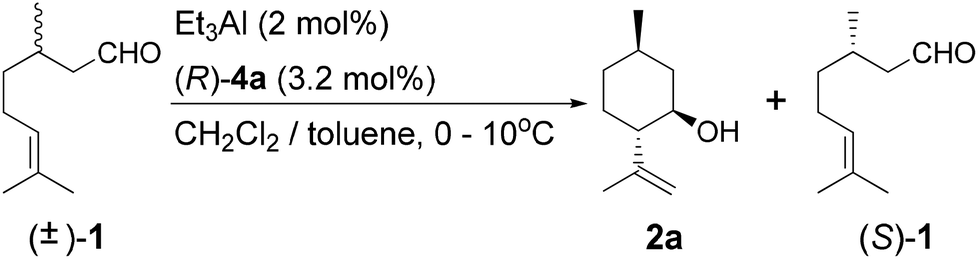
From the results of the cyclization of citronellal described above (Table 1), we hypothesized that kinetic resolution occurred in the presence of the BINOL-Al catalysts. The optical resolution of racemic citronellal ((±)-1) was carried out using Al catalysts. We also explain the details of the kinetic resolution.The value of krel, the ratio of the rate constants for conversion of the fast-reacting and slow-reacting enantiomers, controls the product distribution. For the purposes of calculating krel, catalytic reactions are often assumed to follow zero-order rate laws. However, kinetic resolutions can be explained by both zero-order and first-order reactions.21,26,27 Therefore, we decided to characterize the kinetic resolution of citronellal using both zero-order and first-order rate laws.16,19
The kinetic resolution of (±)-1 by (R)-4a-Al was carried out. The variation in the ee of (S)-1 with conversion is plotted in Fig. 1 and the results are summarized in Table 2. From Table 2, it appears that the resolution was complete with around 50% conversion in 90 min with 2 mol% catalyst loading (entry 4). The ee value of 2a gradually decreased as the reaction progressed (entries 1–5). It seems that the resolution of 2a is consistent with both zero-order and first-order reactions. However, the plot of the ee of (S)-1 with respect to conversion seems to follow first-order reaction kinetics, implying that the results of the kinetic resolution experiments are more consistent with a first-order reaction than it is with a zero-order reaction in the substrate and the product.16 Therefore, we evaluated the kinetic resolution as a first-order reaction in the substrate and the product. The krel value was found to gradually decrease from 26 to 5.5 as the reaction proceeded (entries 1–5).
 | ||
| Fig. 1 Kinetic resolution of racemic citronellal ((±)-1) using the (R)-BINOL((R)-4a)-Al catalyst: experimental values (dots); simulated data for the first-order reaction (lines). | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Time (min) | Conv.a,b (%) | Ratio of 2a in 2c (%) | ee of 2ac (%) | ee of (S)-1c (%) |
k
rel![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
| a Determined from the GC areas and corrected by the ratio of the GC areas and the molecular volumes of citronellal (1), (5R)-isopulegols (2), and ester 5. b The conversions were calculated as the total consumption of 1. c Determined from the GC areas. d Calculated from the first-order rate law using the theoretical conversion value.16 | ||||||
| 1 | 15 | 26 | 93 | 71 | 32 | 26 |
| 2 | 30 | 35 | 92 | 71 | 46 | 21 |
| 3 | 60 | 41 | 91 | 70 | 54 | 12 |
| 4 | 90 | 47 | 91 | 68 | 62 | 11 |
| 5 | 120 | 56 | 91 | 67 | 64 | 5.5 |
The conversion is over 50% only at 120 min. After 90 min, it is still 47%. The ee value of 2a gradually decreased as the reaction progressed. The ee of 2a was 71% at 26% conversion (entry 1, Table 2) and decreased linearly to 67% at 56% conversion (entry 5, Table 2). It seems that the resolution of 2a is consistent with both zero-order and first-order reactions. The plot of the ee of (S)-1 with respect to conversion seems to follow first-order reaction kinetics. The results of the kinetic resolution experiments are more consistent with a first-order reaction than it is with a zero-order reaction in the substrate and the product.16 Therefore, we evaluated the kinetic resolution as a first-order reaction in the substrate and the product. At the start of the reaction, the first-order krel value was 26 (entry 1, Table 2). As the reaction proceeded, the krel value gradually decreased from 26 to 11. The krel value decreased further to 5.5 when the reaction conversion was over 50% (entry 5, Table 2).
There was a difference between the estimated ee of 2a from the krel and the ee of 2a determined from the GC analysis. For example, the GC-analyzed ee of 2a was 68%, whereas the estimated ee from the krel value was 71% at 47% conversion (entry 4, Table 2). The GC-analyzed ee was 67% at 56% conversion (entry 5, Table 2), whereas the estimated ee decreased significantly to 50%. As the krel values were calculated assuming 2a to be the only product, we attribute the differences in the ee values to the formation of other products. The difference in the ee of 2a could also be because of the difference in the enantioselectivity of 2a and the other isomers of 2. There are 8 stereoisomers possible for 2, and the starting material does not convert cleanly to just one diastereomer. Another reason for the difference in the ee values is the side reaction to form 5. At 47% conversion the selectivity of 2 was 96%, but ester 5 was afforded by the Tishchenko reaction in 3.4% selectivity. This reaction consumed 3.4% of racemic citronellal ((±)-1) and contributed to the distortion between the analyzed and estimated values of ee of 2a.
A variety of ligands and substrates were utilized for the kinetic resolution and the results are shown in Table 3. Toluene was chosen as the solvent instead of dichloromethane from the viewpoint of environmental friendliness. The structures of the chiral ligands tested are shown in Fig. 2. The catalyst with (R)-4a ligands showed the best result. The kinetic resolution in toluene and CH2Cl2 with the catalyst with (R)-4a ligands afforded almost the same ratio of 2a in 2 and enantioselectivity of 2a as the reaction in a mixture of dichloromethane and toluene (entries 2 and 3, Table 3).
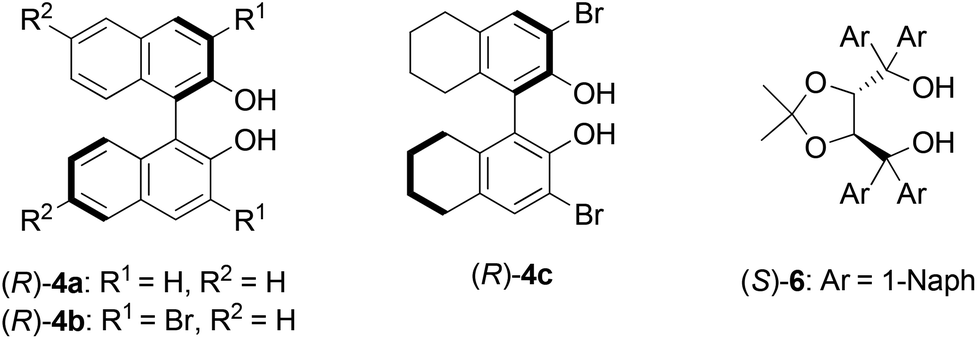 | ||
| Fig. 2 Chiral ligands. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Time (h) | Conv.a,b (%) | Ratio of 2a in 2c (%) | ee of 2ac (%) | ee of (S)-1c (%) |
k
reld |
| a Determined from the molecular ratio of substrates, products, and dimmers calculated by GC area. b The conversions were calculated as the total consumption of substrates. c Calculated from the GC area. d Calculated from the first-order rate law using the theoretical conversion value.16 e Trimethylaluminum was used as a reagent for the catalyst. f Triisobutylaluminum was used as a reagent for the catalyst. g 3 mol% of (S)-6 was used. | ||||||||
| 1 | (R)-4a | CH2Cl2–toluene | 1.5 | 47 | 91 | 68 | 62 | 11 |
| 2 | (R)-4a | Toluene | 3 | 50 | 88 | 72 | 61 | 7.3 |
| 3 | (R)-4a | CH2Cl2 | 4 | 42 | 87 | 69 | 50 | 8.9 |
| 4e | (R)-4a | CH2Cl2–toluene | 2 | 45 | 90 | 72 | 55 | 8.8 |
| 5f | (R)-4a | CH2Cl2–toluene | 1 | 41 | 89 | 76 | 35 | 4.2 |
| 6 | (R)-4b | CH2Cl2 | 7 | 31 | 65 | 43 | 23 | 3.9 |
| 7 | (R)-4c | CH2Cl2 | 0.5 | 57 | 87 | 17 | 5.4 | 1.1 |
| 8g | (S)-6 | Toluene | 5 | 40 | 93 | 20 | 17 | 1.9 |
A variety of alkyl aluminum substrates were investigated for the kinetic resolution. The results indicated that the alkyl groups on aluminum affected the performance of the catalysts with the best performance being observed for the catalyst with (R)-4a ligands (entries 4–7).
The kinetic resolution of (±)-1 was also carried out using 1-NaphTADDOL-Al catalysts. The results indicated that (S,S)-1-NaphTADDOL ((S)-6)-Al showed chiral recognition towards (±)-1 (entry 8, Table 3). However, the resolution ability was lower than that obtained with BINOL-Al.
Reaction mechanism
A tentative reaction mechanism is shown in Fig. 3. The reaction follows an intramolecular ene reaction mechanism catalyzed by an aluminum complex acting as a Lewis acid.11,12 The carbonyl group of (R)-1 is coordinated to the aluminum active site A. Then, a concerted reaction occurs to afford a 6-membered ring, which goes on to afford (5R)-isopulegols (2). When BINOL (4a) of the aluminum complex is substituted with 2 or any other alcohol, the reaction proceeds via another reaction path. The aluminum alkoxide complex (B) works as a catalyst for the Tishchenko reaction. The reaction of two molecules of (R)-1 with B affords dimer 5. The transformation from A to B increases the possibility of the Tishchenko reaction. The increase in the concentration of 2 results in this transformation. For an equimolar ratio of aluminum and BINOL (4a), the transformation will immediately occur by the substitution of an ethyl substituent on aluminum with 2. Hence, the Tishchenko reaction is initiated in the initial stage of the reaction and affords dimer 5. It is assumed that a bulky ligand such as 1-NaphTADDOL (6) can prevent this substitution. Therefore, 1-NaphTADDOL (6)-Al acted as a good cyclization catalyst with an equimolar ratio of ligand and aluminum.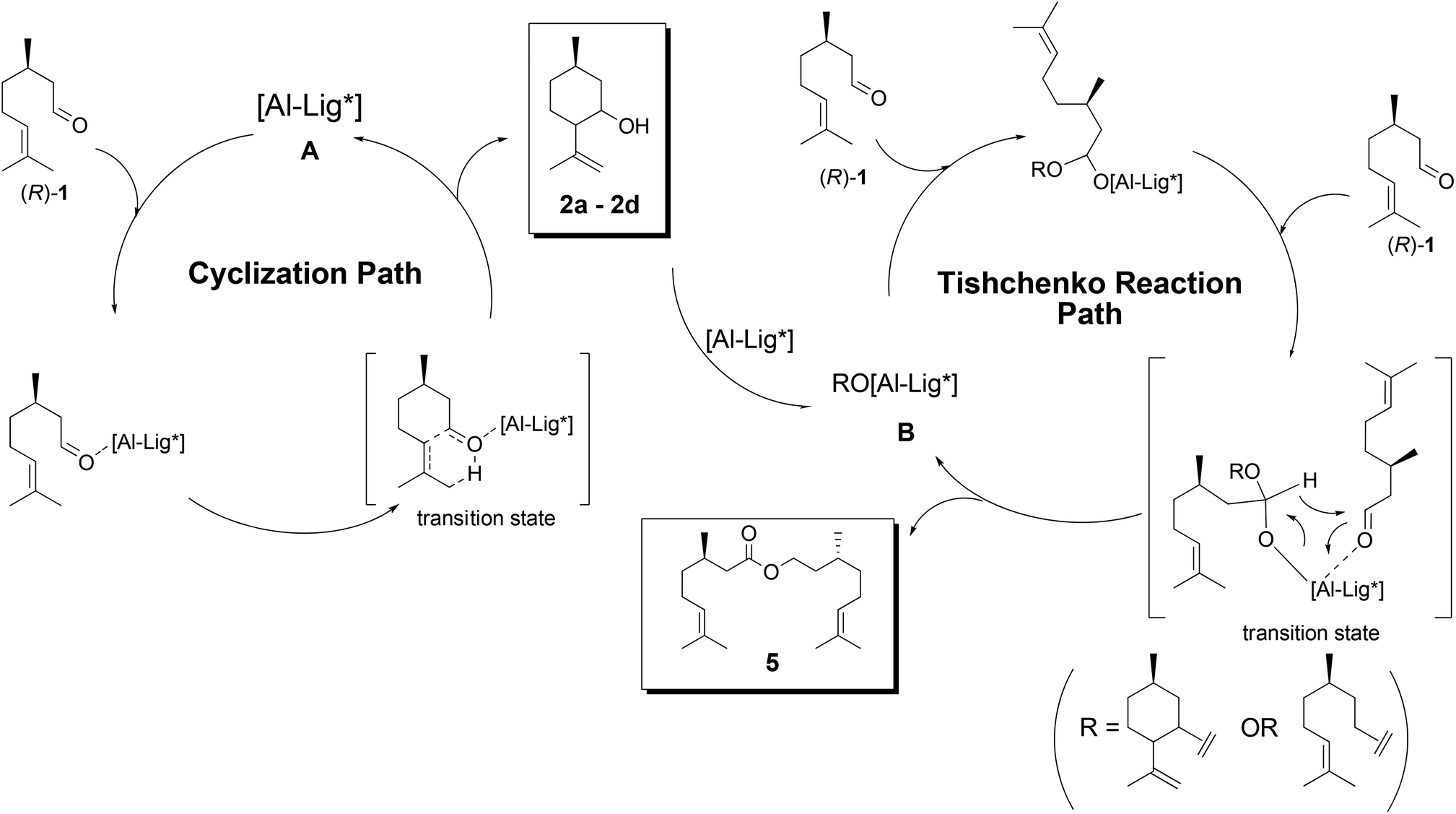 | ||
| Fig. 3 Tentative reaction mechanism of the ring-closing ene reaction by Al-catalysts.11 | ||
Diastereoselectivity with chiral recognition could be achieved using BINOL (4) or TADDOL (6). The molecular models of (R)-4a, (R)-4b, and (R)-6 complexes are shown in Fig. 4. These complexes have a metal center between two parallel aromatic rings. During the ring-closing ene reaction, the aromatic rings are in close proximity to citronellal in the transition state, and this narrow space results in excellent diastereo- and enantioselectivity of 2a. It is assumed that these aromatic rings are horizontally aligned with respect to each other, so that the edge of the aromatic rings can recognize the 3-methyl group of citronellal when the carbonyl group coordinates to the aluminum reaction site. (R)-1 can be fitted between the two parallel aromatic rings of (R)-4a-Al; therefore, optical resolution of citronellal occurs. The narrow space between the aromatic rings results in the formation of 2a with high enantioselectivity. When BINOL is substituted with bromine (4b), the edges of the BINOL ring expand horizontally to minimize chiral recognition from the rings, resulting in reduced chiral recognition.
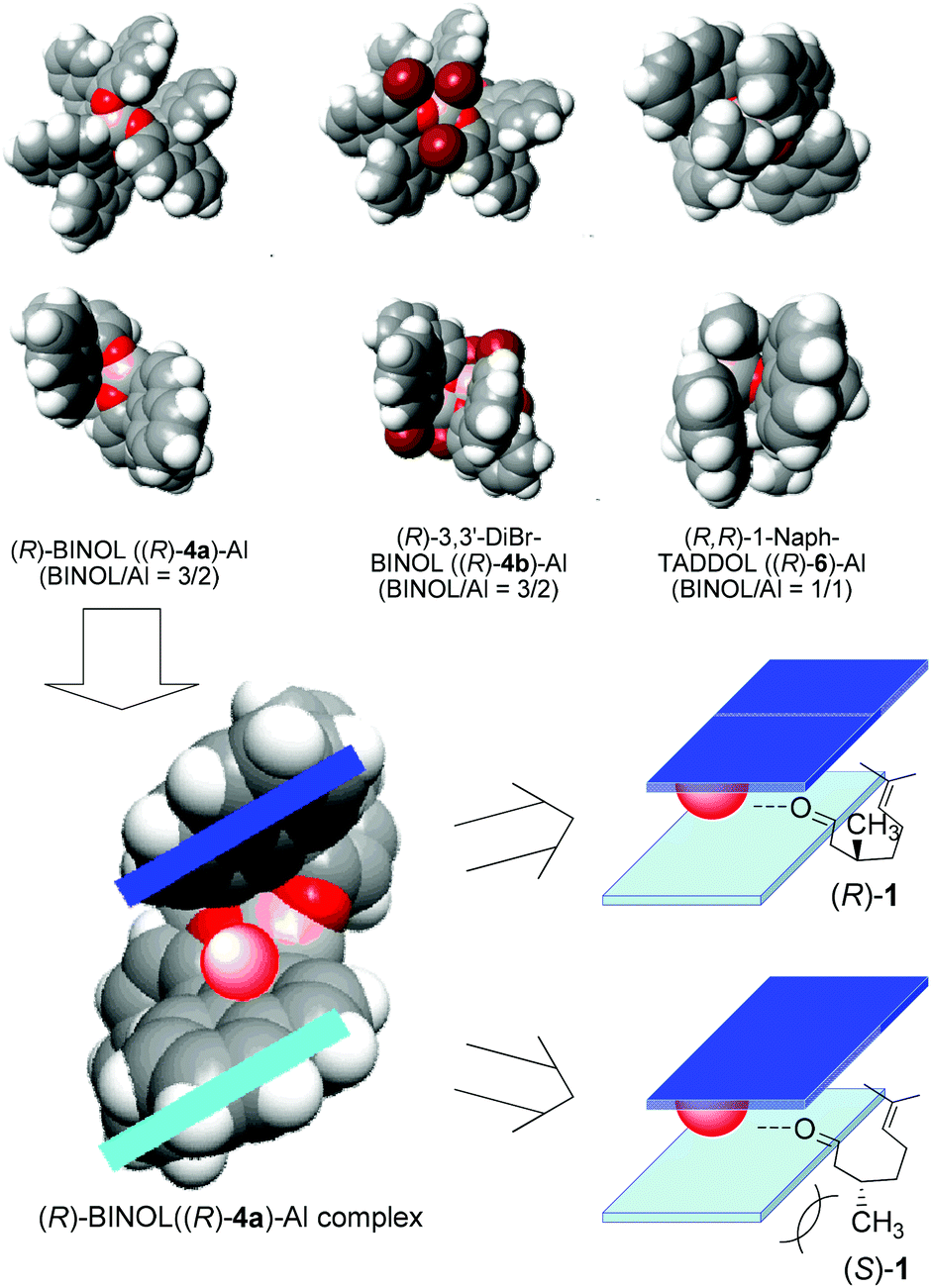 | ||
| Fig. 4 Expected 3D structures of the asymmetric Al-catalysts and their mechanism of chiral recognition. | ||
1-NaphTADDOL-Al also has aromatic rings; however, the 1-naphthyl substituents in TADDOL are more flexible than the BINOL rings. Therefore, 2a is formed with lower enantioselectivity.
We attempted to characterize these catalysts using NMR, but the NMR spectra for all the catalysts indicated complex mixtures. Based on this, we predict that the catalysts are associated.28 In addition, the remaining alkyl group on aluminum also affected the structure and performance of the catalysts.
In order to increase krel, we propose an expansion of the aromatic rings and the substituents on the other side of the aluminum metal center to keep the slit narrow. This would prevent the undesired enantiomer of citronellal from approaching the reaction site.
L-Menthol synthesis
We utilized this kinetic resolution for the synthesis of L-menthol (3). The synthesis path is shown in Schemes 3 and 4. Citral (10) was enantioselectively hydrogenated using a dual catalyst system.29 The new amine co-catalyst 9 was prepared from (R,R)-cis-hydroxyproline ester 7 for this synthesis (Scheme 3). This co-catalyst 9 afforded good enantioselectivity of (R)-1 and the preparation of the co-catalyst was inexpensive compared to other co-catalysts previously reported. We concluded that the co-catalyst is suitable for industrial use.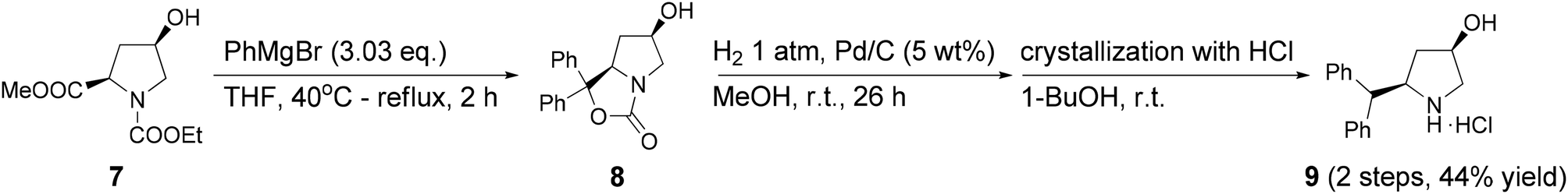 | ||
| Scheme 3 Preparation of a chiral amine co-catalyst. | ||
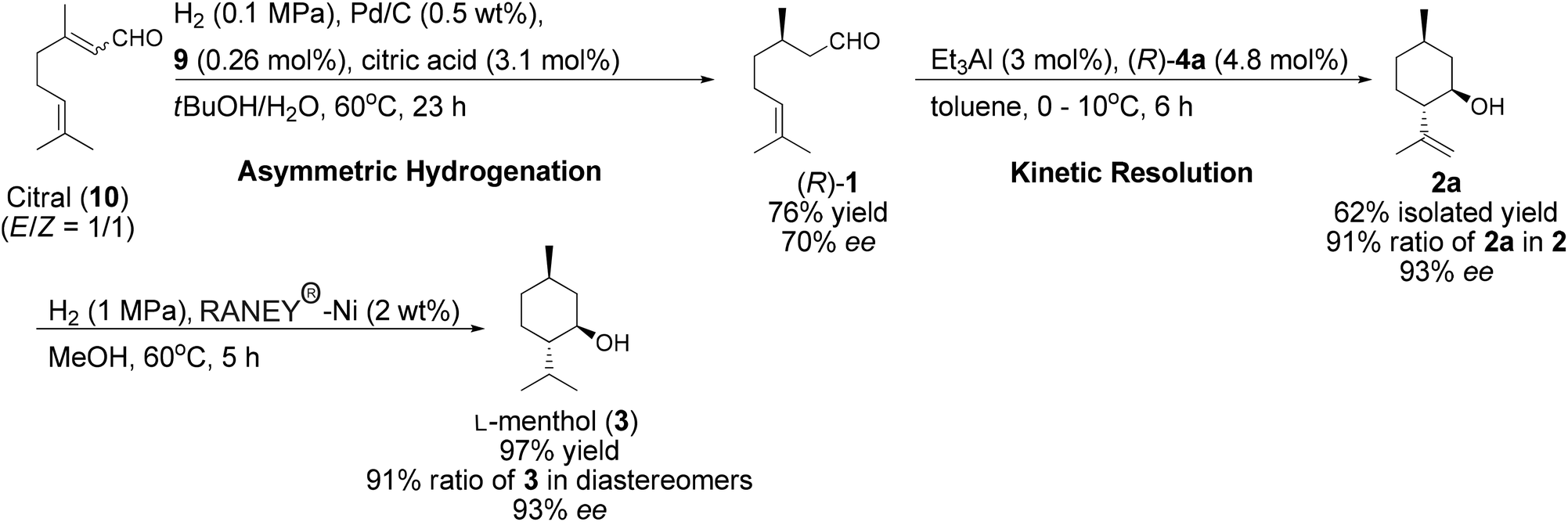 | ||
| Scheme 4 L-Menthol (3) synthesis with kinetic resolutions. | ||
The reaction afforded (R)-1 with 76% yield and 70% ee (Scheme 4). This kinetic resolution was used to amplify the enantioselectivity of 2a. The reaction was carried out in toluene and quenched at 78% conversion. This kinetic resolution afforded 2a in 62% yield. The ee of 2a was 93% and the ratio of 2a in 2 was 91%. Subsequently, 2a was hydrogenated with RANEY®-Ni and 3 was obtained in 97% yield. The diastereoselectivity and enantioselectivity of 3 were the same as those of 2a, indicating that chirality was maintained during this reaction.
Conclusions
Optically active BINOL and TADDOL ligands were reacted with an aluminum reagent to afford new catalysts for the ring-closing ene reaction. These catalysts successfully achieved the kinetic resolution of citronellal analogs. Racemic citronellal afforded optically active isopulegol with high diastereoselectivity. BINOL-Al catalysts showed the best performance in the kinetic resolution. The reaction mechanism is likely to involve chiral recognition of a citronellal enantiomer by the two parallel aromatic rings in the catalyst. The narrow space between the aromatic rings in the BINOL-Al and TADDOL-Al catalysts can recognize the methyl group in the 3-position of citronellal and these catalysts therefore afford high diastereoselectivity of isopulegol. L-Menthol was synthesized from citral with excellent diastereoselectivity and enantioselectivity using kinetic resolution.Experimental section
Gas chromatography (GC) was performed on a GC-2010AF system (Shimadzu) or GC-353B (GL science) and D-2500 (Hitachi) using DB-WAX (30 m × 0.32 mm × 0.5 μm), IC-1 (30 m × 0.25 mm × 0.25 μm), Chirasil-DEX-CB (25 m × 0.25 mm × 0.25 μm), Beta DEX™ 225 (30 m × 0.25 mm × 0.25 μm), and Beta DEX™ 325 (30 m × 0.25 mm × 0.25 μm) columns. Gas chromatography-mass spectrometry (GC-MS) was performed on a GC-QP2010 system (Shimadzu) using Rtx-1 (30 m × 0.25 mm × 0.25 μm) columns. 1H-NMR spectra were recorded on a Bruker 500 MHz spectrometer. Chloroform was used as the NMR solvent and chemical shifts are reported as δ values in parts per million relative to trimethylsilane (δ = 0). Optical rotations were determined using a JASCO P-1020 digital polarimeter (JASCO). Molecular orbital calculation was performed on SCIGRESS V2 powered by Fujitu. Graphic drawings were made using the Mathematica program.All other reagents were purchased from Sigma-Aldrich, Wako Pure Chemical Industries, Ltd, Nacalai Tesque, Inc., Takasago International Corporation, or Strem Chemicals Inc. They were used as received. All the compounds used were of commercial grade.
General procedure of (R)-citronellal ring-closing ene reaction using the BINOL (4a)-Al catalyst (Table 1)
A mixture of BINOL (4a) (229 mg, 0.80 mmol, 8 mol%), a 1.0 mol L−1 toluene solution of triethylaluminum (0.50 mL, 0.50 mmol, 5 mol%), and the given solvent (4.5 mL) was added to a 50 mL Schlenk tube under a N2 atmosphere. After being stirred at r.t. for over 1 h, the solution was cooled to less than 10 °C. (R)-Citronellal (R)-1 (1.54 g, 10 mmol) was added dropwise slowly below 10 °C and stirred for 1 h. The reaction products were analyzed by GC. The authentic samples of (5R)-isopulegols 2a, 2b, 2c, and 2d were also prepared by silica-gel catalyzed cyclization.4,30General procedure of kinetic resolution of racemic citronellal ((±)-1) using ring-closing ene reaction by chiral Al-catalyst (Tables 2 and 3)
A mixture of chiral ligand 4 or 6 (0.80 mmol, 3.2 mol%), triethylaluminum in 1.0 mol L−1 toluene solution (0.50 mL, 0.50 mmol, 2 mol%), and the given solvent (11 mL) was added to a 50 mL Schlenk tube under a N2 atmosphere. After being stirred at r.t. for over 1 h, the solution was cooled to less than 10 °C. Racemic citronellal ((±)-1) (3.86 g, 25 mmol) was added dropwise slowly below 10 °C and stirred for a given amount of time. The solution samples were consecutively collected at regular intervals since the initiation of the dropwise addition and analyzed by GC.Preparation of (3R,5R)-5-benzhydrylpyrrolidin-3-ol·HCl (9) (Scheme 3)
Preparation of (R)-citronellal ((R)-1) with a dual catalyst system (Scheme 4)29
A mixture of citral (10) (E/Z = 50/50) (170 g, 1.12 mol), (3R,5R)-5-benzhydrylpyrrolidin-3-ol·HCl (9) (850 mg, 2.93 mmol, 0.262 mol%), citric acid (6.76 g, 35.2 mmol, 3.14 mol%), and 210 mg of 5%-Pd/C (0.5 wt%, Evonik Degussa type E 105 O/W 5% Pd, wetted with ca. 55% water) in t-BuOH–H2O (92:8 v/v) (170 mL) was stirred under a N2 atmosphere at 50 °C for 1 h. H2 gas was introduced into the mixture. After stirring for 18 h at 60 °C, the reaction mixture was filtered using Celite and the organic phase was analyzed by GC. The reaction was repeated three times and the reaction mixtures were gathered and evaporated (517 g). Distillation with a Claisen distillation apparatus afforded (R)-citronellal ((R)-1) as a colorless oil (bath 75 °C, bottom 68 °C, top 64 °C, 0.5 Torr) (obtained 387 g, 76% yield, 70% ee).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH3), 1.69 (br, 1H, CC–CH3), 1.94–2.12 (m, 3H), 2.23 (ddd, 1H, J = 16.0, 8.0, 3.0 Hz), 2.40 (ddd, 1H, J = 16.0, 5.5, 2.0 Hz), 5.06–5.11 (m, 1H, CHC), 9.75 (dd, 1H, J = 2.5, 2.0 Hz, CHO). 13C-NMR (125 MHz, CDCl3): 17.6 (CH3), 19.9 (CH3), 25.4 (CH2), 25.7 (CH3), 27.8 (CH), 36.9 (CH2), 51.0 (CH2), 124.0 (CH), 131.8 (C), 203.0 (CHO).
C–CH3), 1.69 (br, 1H, CC–CH3), 1.94–2.12 (m, 3H), 2.23 (ddd, 1H, J = 16.0, 8.0, 3.0 Hz), 2.40 (ddd, 1H, J = 16.0, 5.5, 2.0 Hz), 5.06–5.11 (m, 1H, CHC), 9.75 (dd, 1H, J = 2.5, 2.0 Hz, CHO). 13C-NMR (125 MHz, CDCl3): 17.6 (CH3), 19.9 (CH3), 25.4 (CH2), 25.7 (CH3), 27.8 (CH), 36.9 (CH2), 51.0 (CH2), 124.0 (CH), 131.8 (C), 203.0 (CHO).
Synthesis of L-menthol (3) from (R)-citronellal (70% ee of (R)-1) via kinetic resolution using BINOL-Al (Scheme 4)
A mixture of BINOL (R)-4a (742 mg, 2.59 mmol, 8 mol%), triethylaluminum in 1.0 mol L−1 toluene solution (1.6 mL, 1.62 mmol, 5 mol%), and toluene (14 mL) was added to a 100 mL Schlenk tube under a N2 atmosphere. After being stirred at r.t. for over 1 h, the solution was cooled to less than 10 °C. (R)-Citronellal (70% ee of (R)-1) (5.00 g, 32.4 mmol) was added dropwise slowly below 10 °C and stirred for 1.5 h. The reaction mixture was poured into toluene–dil. HCl after quenching. The oil layer was washed with brine and dried on MgSO4. After filtration and evaporation, the residue was obtained as a colorless oil. After purification by column chromatography (heptane–AcOEt = 6/1), 3.11 g of (5R)-n-isopulegol (2a) (62% yield, 93% ee, 91% ratio of 2a in 2) was obtained.(5R)-n-Isopulegol (2a) (500 mg, 3.24 mmol) was hydrogenated with RANEY®-Ni (10 mg, 2 vol%) in the presence of H2 (1 MPa) and MeOH (3 mL) at 60 °C for 5 h. After filtration and evaporation, 493 mg of L-menthol (3) (97% yield, 93% ee, 91% ratio of 3 in diastereoisomers) was obtained.
Compound characterization
CH2), 1.84 (d, 1H, J = 2.0 Hz), 1.85–1.91 (m, 1H), 2.02–2.07 (m, 1H), 3.43–3.49 (m, 1H, CH–OH), 4.78–4.95 (m, 2H, CCH2). 13C-NMR (125 MHz, CDCl3): 19.2 (CH3), 22.2 (CH3), 29.6 (CH2), 31.5 (CH), 34.3 (CH2), 42.7 (CH2), 54.2 (CH), 70.4 (CH), 112.8 (CH2), 146.7 (C).
C–CH3), 1.68 (s, 6H, CC–CH3), 1.90–2.04 (m, 6H), 2.10 (dd, 1H, J = 14.6, 8.3 Hz), 2.30 (dd, 1H, J = 14.6, 8.3 Hz), 4.05–4.18 (m, 2H, COO–CH2), 5.05–5.15 (m, 2H, CCH). 13C-NMR (125 MHz, CDCl3): 17.6 (CH3), 19.4 (CH3), 19.6 (CH3), 25.4 (CH2), 25.4 (CH2), 25.7 (CH3), 29.5 (CH), 30.1 (CH), 30.9 (CH), 35.5 (CH2), 36.8 (CH2), 37.0 (CH2), 41.9 (CH2), 62.7 (CH2), 124.3 (CH), 124.6 (CH), 131.3 (C), 131.5 (C), 173.4 (CH), 206.9 (CO).
Notes and references
- K. Mikami and M. Shimizu, Chem. Rev., 1992, 92, 1021 CrossRef CAS; E. Arundale and L. A. Mikeska, Chem. Rev., 1952, 51, 505 CrossRef; D. R. Adams and S. P. Bhatnagar, Synthesis, 1977, 661 CrossRef; B. B. Snider, Compr. Org. Synth., 1991, 2, 527 Search PubMed.
- O. Corminboeuf, L. E. Overman and L. D. Pennington, J. Am. Chem. Soc., 2003, 125, 6650 CrossRef CAS PubMed.
- D. J. Kopecky and S. D. Rychnovsky, J. Am. Chem. Soc., 2001, 123, 8420 CrossRef CAS; H. H. Jung, J. R. Seiders and P. E. Floreancig, Angew. Chem., Int. Ed., 2007, 46, 8464 CrossRef PubMed.
- H. B. Glass, US2117414, 1936 Search PubMed.
- Y. Nakatani and K. Kawashima, Synthesis, 1978, 147 CrossRef CAS; P. N. Davey and C. Tse (P. N. Davey, Quest International B. V., C. Tse), PCTWO2000/069777, 2000 Search PubMed.
- T. K. Sarkar and S. K. Namdy, Tetrahedron Lett., 1996, 37, 5195 CrossRef CAS.
- S. Sakane, K. Maruoka and H. Yamamoto, Tetrahedron Lett., 1985, 46, 5535 CrossRef CAS; S. Sakane, K. Maruoka and H. Yamamoto, Tetrahedron, 1986, 42, 2203 CrossRef.
- H. Du, J. Long, J. Hu, X. Li and K. Ding, Org. Lett., 2002, 24, 4349 CrossRef PubMed.
- K. S. Jeong, Y. B. Go, S. M. Shin, S. J. Lee, J. Kim, O. M. Yaghi and N. Jeong, Chem. Sci., 2011, 2, 877 RSC.
- K. Mikami, E. Sawa and M. Terada, Tetrahedron: Asymmetry, 1991, 2, 1403 CrossRef CAS; K. Mikami, M. Terada, E. Sawa and T. Nakai, Tetrahedron Lett., 1991, 32, 6571 CrossRef.
- B. B. Sinder, M. Karras, R. T. Price and D. J. Rodini, J. Org. Chem., 1982, 47, 4538 CrossRef CAS; B. B. Sinder, D. J. Rodini, M. Karras, T. C. Kirk, E. A. Deutsch, R. Cordova and R. T. Prince, Tetrahedron, 1981, 37, 3927 CrossRef; M. Karras and B. B. Sinder, J. Am. Chem. Soc., 1980, 102, 7951 CrossRef.
- Y. Hori, T. Iwata and Y. Okeda, (Takasago International Corporation) US20020133046, 2002 Search PubMed.
- M. Nobls, Lyss (SYMRISE GmbH & Co. KG) PCTWO2007/039342, 2007 Search PubMed.
- K. Ebel, C. Jakel, N. Kashani-Shirazi and M. Rauls, (Basf Se) PCTEP2006/065322, 2006 Search PubMed.
- Y. Kikukawa, S. Yamaguchi, Y. Nakagawa, K. Uehara, S. Uchida, K. Yamaguchi and N. Mizuno, J. Am. Chem. Soc., 2008, 130, 15872 CrossRef CAS PubMed.
- M. Tokunaga reported the details of a variety of kinetic resolution examples by a variety of methods; M. Tokunaga, J. Kiyosu, Y. Obora and Y. Tsuji, J. Am. Chem. Soc., 2006, 128, 4481 CrossRef CAS PubMed.
- M. Sakamoto, A. Unosawa, S. Kobaru, A. Saito, T. Mino and T. Fujita, Angew. Chem., Int. Ed., 2005, 44, 5523 CrossRef CAS PubMed; M. Sakamoto, U. A. Nosawa, S. Kobaru, K. Fujita, T. Mino and T. Fujita, Chem. Commun., 2007, 3586 RSC; M. Sakamoto, M. Kato, Y. Aida, K. Fujita, T. Mino and T. Fujita, J. Am. Chem. Soc., 2008, 130, 1132 CrossRef PubMed; M. Sakamoto, K. Fujita, F. Yagishita, A. Unosawa, T. Mino and T. Fujita, Chem. Commun., 2011, 47, 4267 RSC.
- M. I. Klauck, S. G. Patel and S. L. Wiskur, J. Org. Chem., 2012, 77, 3570 CrossRef CAS PubMed.
- H. Aoyama, M. Tokunaga, J. Kiyosu, T. Iwasawa, Y. Obora and Y. Tsuji, J. Am. Chem. Soc., 2005, 127, 10474 CrossRef CAS PubMed.
- T. O. Luukas, C. Girard, D. R. Fenwick and H. B. Kagan, J. Am. Chem. Soc., 1999, 121, 9299 CrossRef CAS.
- E. Vedejs and M. Jure, Angew. Chem., Int. Ed., 2005, 44, 39741 CrossRef PubMed.
- J. B. Alexander, D. S. La, D. R. Cefalo, A. H. Hoveyda and R. R. Schrock, J. Am. Chem. Soc., 1998, 120, 4041 CrossRef CAS; D. S. La, J. B. Alexander, D. R. Cefalo, D. D. Graf, A. H. Hoveyda and R. R. Schrock, J. Am. Chem. Soc., 1998, 120, 9720 CrossRef.
- P. R. Staff, J. Org. Chem., 1973, 38, 1433 CrossRef CAS; K. J. Ralston and A. N. Hulme, Synthesis, 2012, 2310 Search PubMed.
- Y. Hon, Y. Wong, C Chang and C. Hsieh, Tetrahedron, 2007, 63, 11325 CrossRef CAS PubMed.
- T. Arai, H. Sasai, K. Aoe, K. Okamura, T. Date and M. Shibasaki, Angew. Chem., Int. Ed. Engl., 1996, 35, 104 CrossRef CAS; T. Arai, M. Bougauchi, H. Sasai and M. Shibasaki, J. Org. Chem., 1996, 61, 2926 CrossRef.
- J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5 CrossRef CAS.
- J. W. Faller and M. Tokunaga, Tetrahedron Lett., 1993, 34, 7359 CrossRef CAS.
- I. J. Worrall, J. Chem. Educ., 1969, 46, 510 CrossRef CAS; C. R. Graves, H. Zhou, C. L. Stern and S. T. Nguyen, J. Org. Chem., 2007, 72, 9121 CrossRef PubMed.
- H. Maeda, S. Yamada, H. Itoh and Y. Hori, Chem. Commun., 2012, 48, 1772 RSC.
- G. Indo, Goseikoryo, 2005, 81 Search PubMed; J. S. Yadav, E. V. Bhasker and P. Srihari, Tetrahedron, 2010, 66, 1997 CrossRef CAS PubMed.
- D. J. Bailey, D. O'Hagan and M. Tavasli, Tetrahedron: Asymmetry, 1997, 8, 149 CrossRef CAS; Y. Li, X. Liu, Y. Yang and G. Zhao, J. Org. Soc., 2007, 72, 288 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analysis data for products; 1H, 13C-NMR spectra and GC chart of compounds (R)-1, 2a, 3 and 5. See DOI: 10.1039/c4qo00222a |
| This journal is © the Partner Organisations 2014 |