
Photoinduced HBr-catalyzed C–Si bond cleavage of benzylsilanes and their subsequent oxidation into benzoic acids with air as the terminal oxidant†
Jing
Sun‡
ab,
Yu
Wang‡
c,
Liqiong
Han
a,
Dawen
Xu
a,
Yiyong
Chen
a,
Xinhua
Peng
b and
Hao
Guo
*ad
aDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai, 200433, P. R. China. E-mail: Hao_Guo@fudan.edu.cn; Fax: +86-21-55664361; Tel: +86-21-55664361
bSchool of Chemical Engineering, Nanjing University of Science and Technology, Nanjing, 210094, P. R. China
cDepartment of Head and Neck Surgery, Fudan University Shanghai Cancer Center; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai, P. R. China
dKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, P. R. China
First published on 6th October 2014
Abstract
A photoinduced highly efficient C–Si bond cleavage reaction of benzylsilanes under the catalysis of HBr was developed. The in situ generated benzyl radical intermediates were aerobically oxidized into benzoic acids highly chemoselectively. In this transformation, HBr not only acted as the single electron transfer mediator for the initial C–Si bond cleavage, but also efficiently catalyzed the oxidation of benzaldehyde intermediates into benzoic acids.
The carbon–silicon bond is ubiquitous in organic chemicals.1 Its formation reactions have a rich history.2 Meanwhile, highly efficient and selective cleavage of C–Si bond and its subsequent functionalization have also received much attention.3 Besides the numerous achievements in thermal reactions, photochemical methods which provide an alternative way for C–Si bond cleavage and functionalization remain underdeveloped. Only a few research studies have been reported in this field.4–8 Compared to breaking of the C–Si bond, a more challenging issue in photodesilylation is to control its following transformation, especially for aerobic oxidation. For example, Otsuji and Mizuno4e reported a 9,10-dicyanoanthracene (DCA)-sensitized photooxidation of benzylsilanes, affording a mixture of aromatic aldehydes and carboxylic acids in poor chemoselectivities. Albini5a reported a TiO2-catalyzed photoreaction of benzylsilicons, which also yielded a mixture of alcohol, aldehyde and an acid as well as other byproducts. Although the selectivities in such cases are not satisfactory to be applied in synthetic organic chemistry, it provides an alternative but to be improved approach in this field. Two key steps are involved in this reaction: C–Si bond cleavage via a photoinduced single electron transfer process and oxidation of the in situ formed benzyl radicals. Thus, improvements of both C–Si bond cleavage efficiency and subsequent oxidation selectivity may lead to a new synthetically useful method for this transformation. Herein, we wish to report our recent observation on the photoinduced HBr-catalyzed C–Si bond cleavage of benzylsilanes and subsequent oxidation, affording only benzoic acid derivatives in good to excellent yields.
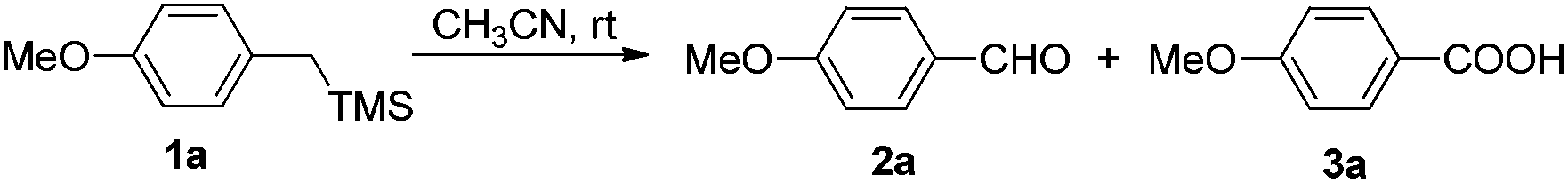
At the beginning of our study, (4-methoxybenzyl)trimethylsilane 1a was chosen as the model substrate to explore the feasibility of the reaction. Without a catalyst, the photoreaction of 1a in benzene irradiated by the Xe lamp at rt yielded a mixture of 4-methoxybenzaldehyde 2a and 4-methoxybenzoic acid 3a in a total yield of 63% (2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a = 38:25) (entry 1, Table 1). To improve the yield and selectivity, 20 mol% of HBr (aq., 40%) was applied, the total yield rose up to 80%; however, the chemoselectivity was still poor (entry 2, Table 1). Then, the solvent effect was studied carefully. When CH2Cl2, CH3OH, or DMSO was used as the solvent, 2a was formed as the major product (entries 3–5, Table 1). While for reactions in CH3NO2 or DMF, 3a was generated as the major product (entries 6 and 7, Table 1). In THF, the reaction gave the sole product of 3a but in a very low isolated yield (entry 8, Table 1). Fortunately, when the reaction was conducted in CH3CN, 3a was afforded as the only product in an excellent yield with an excellent selectivity (entry 9, Table 1). Then a Pyrex tube was used instead of a quartz tube, and a longer reaction time and a lower yield were observed (entry 10, Table 1). Finally, an Hg lamp (500 W) was used instead of an Xe lamp (300 W); however, 3a was formed as the minor product (entry 11, Table 1). Thus, we applied Condition A (20 mol% HBr (aq., 40%), CH3CN, an Xe lamp (300 W), quartz, air (1 atm), and rt) for the photooxidation of benzylsilanes.
:3a = 38:25) (entry 1, Table 1). To improve the yield and selectivity, 20 mol% of HBr (aq., 40%) was applied, the total yield rose up to 80%; however, the chemoselectivity was still poor (entry 2, Table 1). Then, the solvent effect was studied carefully. When CH2Cl2, CH3OH, or DMSO was used as the solvent, 2a was formed as the major product (entries 3–5, Table 1). While for reactions in CH3NO2 or DMF, 3a was generated as the major product (entries 6 and 7, Table 1). In THF, the reaction gave the sole product of 3a but in a very low isolated yield (entry 8, Table 1). Fortunately, when the reaction was conducted in CH3CN, 3a was afforded as the only product in an excellent yield with an excellent selectivity (entry 9, Table 1). Then a Pyrex tube was used instead of a quartz tube, and a longer reaction time and a lower yield were observed (entry 10, Table 1). Finally, an Hg lamp (500 W) was used instead of an Xe lamp (300 W); however, 3a was formed as the minor product (entry 11, Table 1). Thus, we applied Condition A (20 mol% HBr (aq., 40%), CH3CN, an Xe lamp (300 W), quartz, air (1 atm), and rt) for the photooxidation of benzylsilanes.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time (h) | Isolated yield (%) | |
| 2a | 3a | |||
| a A solution of 1a (0.2 mmol) and 20 mol% HBr (aq., 40%) in the tested solvent (10 mL) in a quartz reaction tube was irradiated by a 300 W Xe lamp at rt in the open air. b The reaction was carried out without HBr. c A Pyrex reaction tube was used. d An Hg lamp (500 W) was used. | ||||
| 1b | Benzene | 12 | 38 | 25 |
| 2 | Benzene | 11 | 15 | 65 |
| 3 | CH2Cl2 | 4.5 | 37 | 26 |
| 4 | CH3OH | 10.5 | 15 | 7 |
| 5 | DMSO | 4 | 45 | 12 |
| 6 | CH3NO2 | 11 | 14 | 22 |
| 7 | DMF | 25 | 8 | 24 |
| 8 | THF | 24 | 0 | 17 |
| 9 | CH3CN | 3.5 | 0 | 91 |
| 10c | CH3CN | 11 | 0 | 86 |
| 11d | CH3CN | 21 | 66 | 16 |
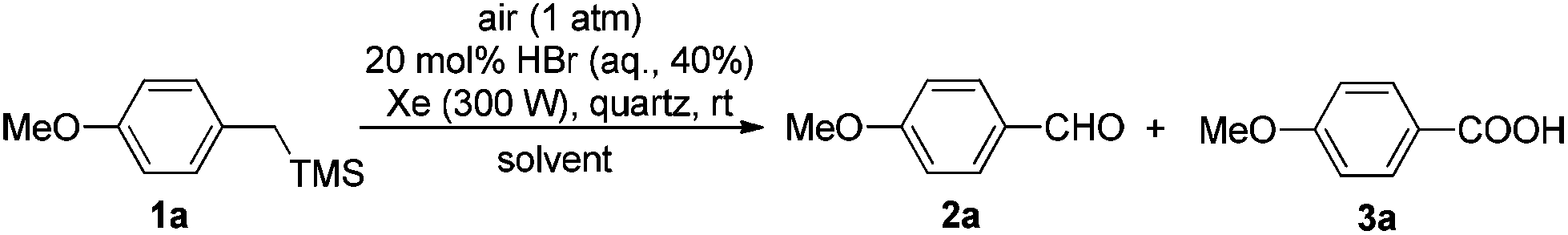
With the optimized conditions in hand, we further investigated the scope of this reaction. A series of benzyltrimethylsilanes were subjected to the standard reaction conditions. The results are summarized in Table 2. It was observed that both electron-donating and -withdrawing groups could be installed to the phenyl ring of the benzyltrimethylsilanes affording the corresponding benzoic acids as the only product in good to excellent yields. When the methoxy group was introduced to the phenyl ring of the substrates, the relative position showed some influence. (3-Methoxybenzyl)-trimethylsilane 1b and (2-methoxybenzyl)trimethylsilane 1c gave much lower yields of the desired products than that of 1a (entries 1–3, Table 2), which might be due to a lower efficiency of the transformation of the aldehyde intermediate (vide infra) into benzoic acid, while for trifluoromethyl substituted reactants 1n–p, similar yields were generated (entries 14–16, Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | R | Time (h) | 3 | Isolated yield (%) |
| a A solution of 1 (0.2 mmol) and 20 mol% HBr (aq., 40%) in CH3CN (10 mL) in a quartz reaction tube was irradiated by a 300 W Xe lamp at rt in the open air. b No aldehydes were formed, determined for the crude reaction mixture by 400 MHz 1H NMR analysis. | |||||
| 1 | 1a | p-OMe | 3.5 | 3a | 91 |
| 2 | 1b | m-OMe | 7.5 | 3b | 46 |
| 3 | 1c | o-OMe | 4.5 | 3c | 50 |
| 4 | 1d | p-But | 3 | 3d | 66 |
| 5 | 1e | p-Ph | 24 | 3e | 83 |
| 6 | 1f | H | 5 | 3f | 74 |
| 7 | 1g | p-Cl | 5.5 | 3g | 70 |
| 8 | 1h | p-F | 9 | 3h | 82 |
| 9 | 1i | p-P(O)(Ph)2 | 17 | 3i | 62 |
| 10 | 1j | p-Ac | 10 | 3j | 81 |
| 11 | 1k | p-COOMe | 9 | 3k | 86 |
| 12 | 1l | p-COOEt | 8.5 | 3l | 90 |
| 13 | 1m | p-C(O)N(Pri)2 | 18.5 | 3m | 81 |
| 14 | 1n | p-CF3 | 3 | 3n | 80 |
| 15 | 1o | m-CF3 | 8 | 3o | 74 |
| 16 | 1p | o-CF3 | 6.5 | 3p | 77 |
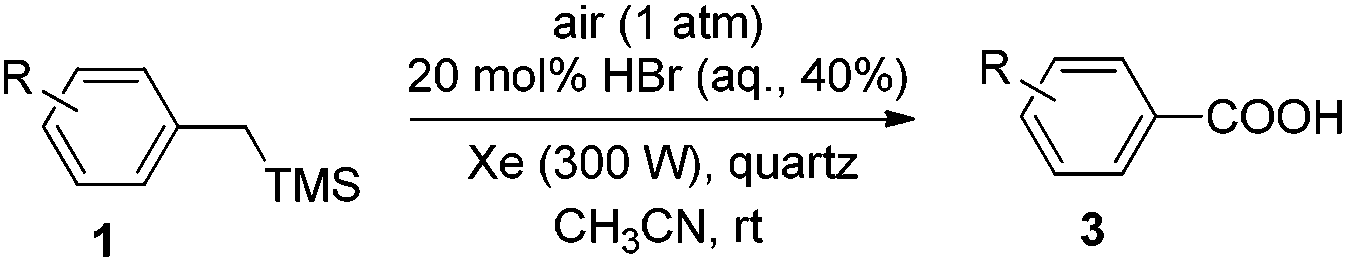
A scrutiny of the reactivity of different silyl groups was then conducted (Table 3). In all the tested cases, 3f was formed in good isolated yields as the only observed product, indicating that the substituents on the silicon atom showed no obvious influence on both the yield and selectivity.
|
|
||||
|---|---|---|---|---|
| Entry | 1 | R | Time (h) | Isolated yield (%) |
| a A solution of 1 (0.2 mmol) and 20 mol% HBr (aq., 40%) in CH3CN (10 mL) in a quartz reaction tube was irradiated by a 300 W Xe lamp at rt in the open air. b No aldehydes were formed, determined for the crude reaction mixture by 400 MHz 1H NMR analysis. | ||||
| 1 | 1f | SiMe3 | 5 | 74 |
| 2 | 1q | SiEt3 | 3.5 | 83 |
| 3 | 1r | SiButMe2 | 3 | 70 |
| 4 | 1s | SiPhMe2 | 4.5 | 82 |
| 5 | 1t | SiPh2Me | 8 | 60 |
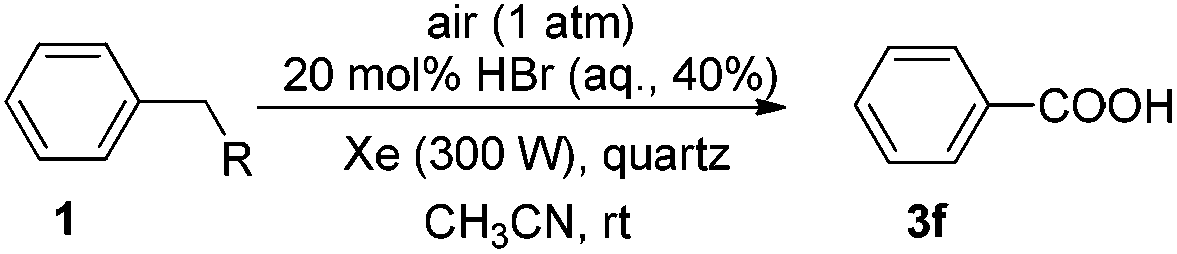
Then allylictrimethylsilanes 4 and aliphatictrimethylsilanes 5 were applied under Condition A. In both cases, only benzoic acid 3f was formed (Scheme 1).
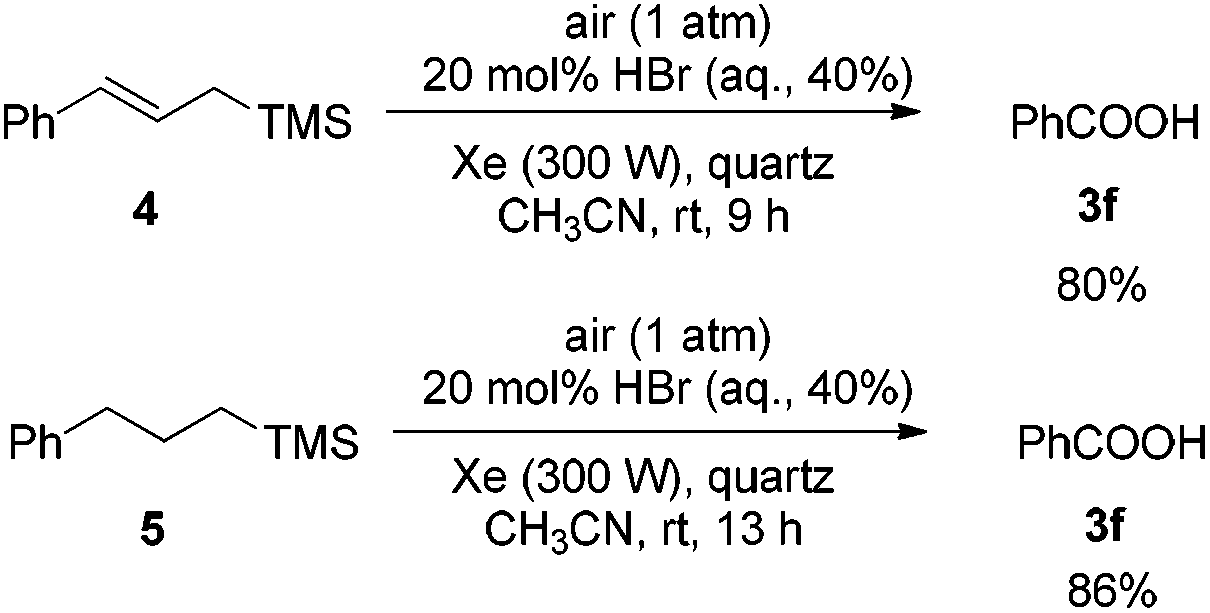 | ||
| Scheme 1 Photooxidation of 4 and 5 under Condition A. | ||
Notably, this reaction could easily proceed in a high yield on the gram scale (Scheme 2).
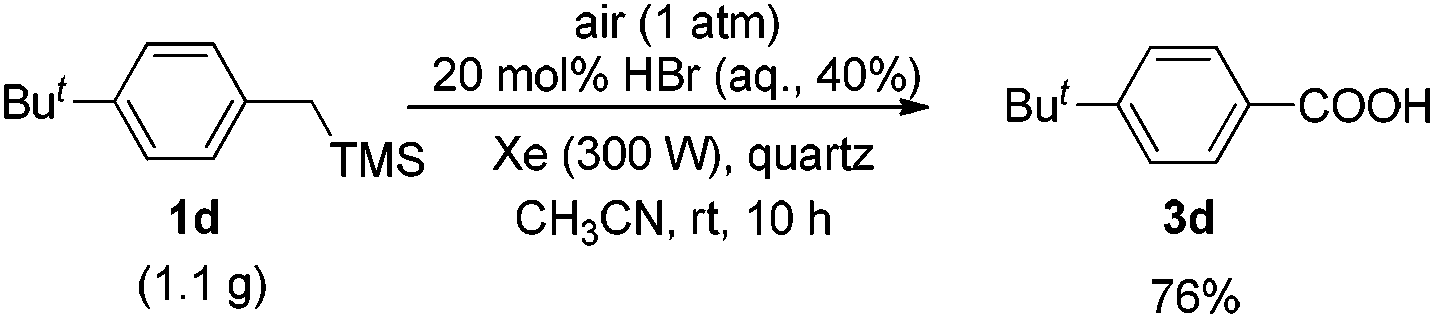 | ||
| Scheme 2 Gram-scale synthesis of 3d. | ||
To gain insight into the reaction mechanism, a series of control experiments were carried out (Table 4). The results showed that (i) under Condition A, this reaction proceeded smoothly to afford only 3a in an excellent yield (entry 1, Table 4); (ii) photoirradiation was required for the conversion (entry 2, Table 4); (iii) oxygen in air could be the terminal oxidant for the oxidation step (entry 3, Table 4); (iv) if the reaction under Condition A was quenched after 15 minutes, the formation of 2a was observed (entry 4, Table 4), indicating the intermediacy of 2a in the whole process; (v) if the photoirradiation was removed after 15 minutes, the reaction stopped sharply, since a similar recovered yield of 1a and isolated yields of 2a and 3a were observed (entry 5, Table 4); (vi) notably, without a catalyst, this photooxidation could occur to form a mixture of 2a and 3a in a poor chemoselectivity (entry 6, Table 4), which means that HBr not only improved the efficiency of the C–Si bond cleavage, but also catalyzed the transformation of benzaldehyde intermediates into benzoic acids.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 20 mol% HBr (aq., 40%) | Air (1 atm) | hv | Time (h) | Isolated yield (%) | ||
| 1a | 2a | 3a | |||||
| a A solution of 1a (0.2 mmol) in CH3CN (10 mL) in a quartz reaction tube was irradiated by a 300 W Xe lamp at rt. b The photoirradition was stopped after 15 minutes. c Reaction time under photoirradiation. d Total reaction time. | |||||||
| 1 | + | + | + | 3.5 | 0 | 0 | 91% |
| 2 | + | + | − | 3.5 | 87% | 0 | 0 |
| 3 | + | − | + | 3.5 | 0 | 0 | 0 |
| 4 | + | + | + | 0.25 | 18% | 45% | 17% |
| 5 | + | + | +b | 0.25c (3.5)d | 15% | 48% | 20% |
| 6 | − | + | + | 5 | 0 | 33% | 52% |
To further confirm the intermediacy of aldehyde, 2a was applied under Condition A. As expected, after full conversion of aldehyde, acid 3a was afforded in almost quantitative isolated yield (Scheme 3).
 | ||
| Scheme 3 The conversion of 2a to 3a under Condition A. | ||
Next, the following test reactions were carried out. A 0.1 M solution of HBr in CH3CN was irradiated by a 300 W Xe lamp at rt in the open air. After 30 minutes, it was tested with starch iodide paper which turned blue quickly, revealing the in situ formation of Br2. Then 10 mol% of Br2 was applied instead of HBr (aq., 40%). After reacting for 3.5 hours under Condition A, 26% of 2a together with 65% of 3a were afforded (Scheme 4), which showed that Br2 could also catalyze this photooxidation but with a lower efficiency.
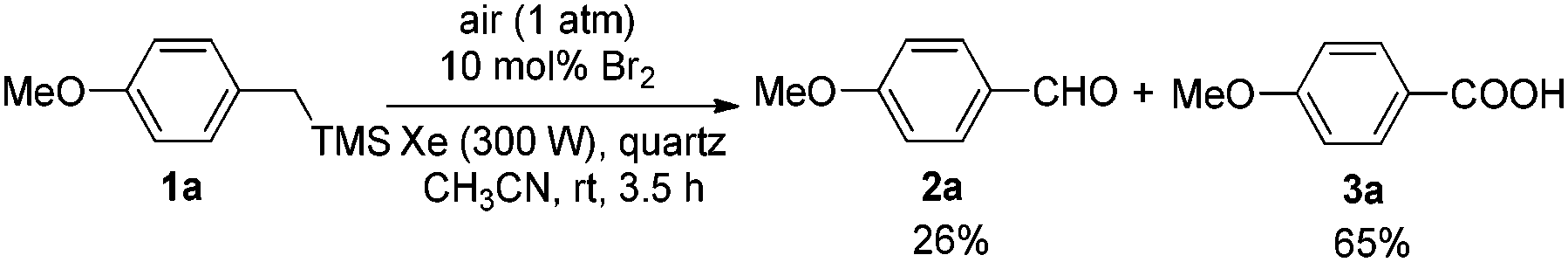 | ||
| Scheme 4 Br2-catalyzed photooxidation of 1a. | ||
Based on the above data and literature precedents,9–13 a possible mechanism was proposed as shown in Scheme 5. Bromine was formed by aerobic photooxidation of hydrogen bromide (eqn (1))9 and further converted into the bromine radical under continuous photoirradiation (eqn (2)).6a,9,10 It is known that the bromine radical is a good oxidant (Eo = 2.0 V vs. NHE in H2O)6b,11 for the oxidation of benzylsilane derivatives.6a,b Thus, in the next step, single electron transfer (SET) from 1 to the bromine radical resulted in a pair of radical ions ArCH2SiR3⊕•6 and Br⊖ (eqn (3)).6a,6b Here, HBr was a key factor in improving the oxidizing ability of the whole system, since the stronger acidity of the reaction mixture would lead to a higher E° value of the Br•/Br⊖ couple, due to better solvation for Br⊖via hydrogen bonds.6b This might be the reason why Br2 could catalyze this photooxidation but with a lower efficiency than that of HBr (Scheme 4). The radical cation 6 underwent fast desilylation probably via a nucleophile-assisted cleavage of the C–Si bond by the solvent of acetonitrile4d,e,12 to form benzyl radical 7 (eqn (4)).4d,e,5a,12,13 Considering that this photooxidation could occur without a catalyst under continuous photoirradiation (entry 6, Table 4), the transformation from 1 into 7 might also be promoted by light. It is known that 7 could be aerobically oxidized into aldehyde 2 easily (eqn (5)).4d,e,10 Finally, 2 was further oxidized into 3 under Condition A (Scheme 3) (eqn (6)).14
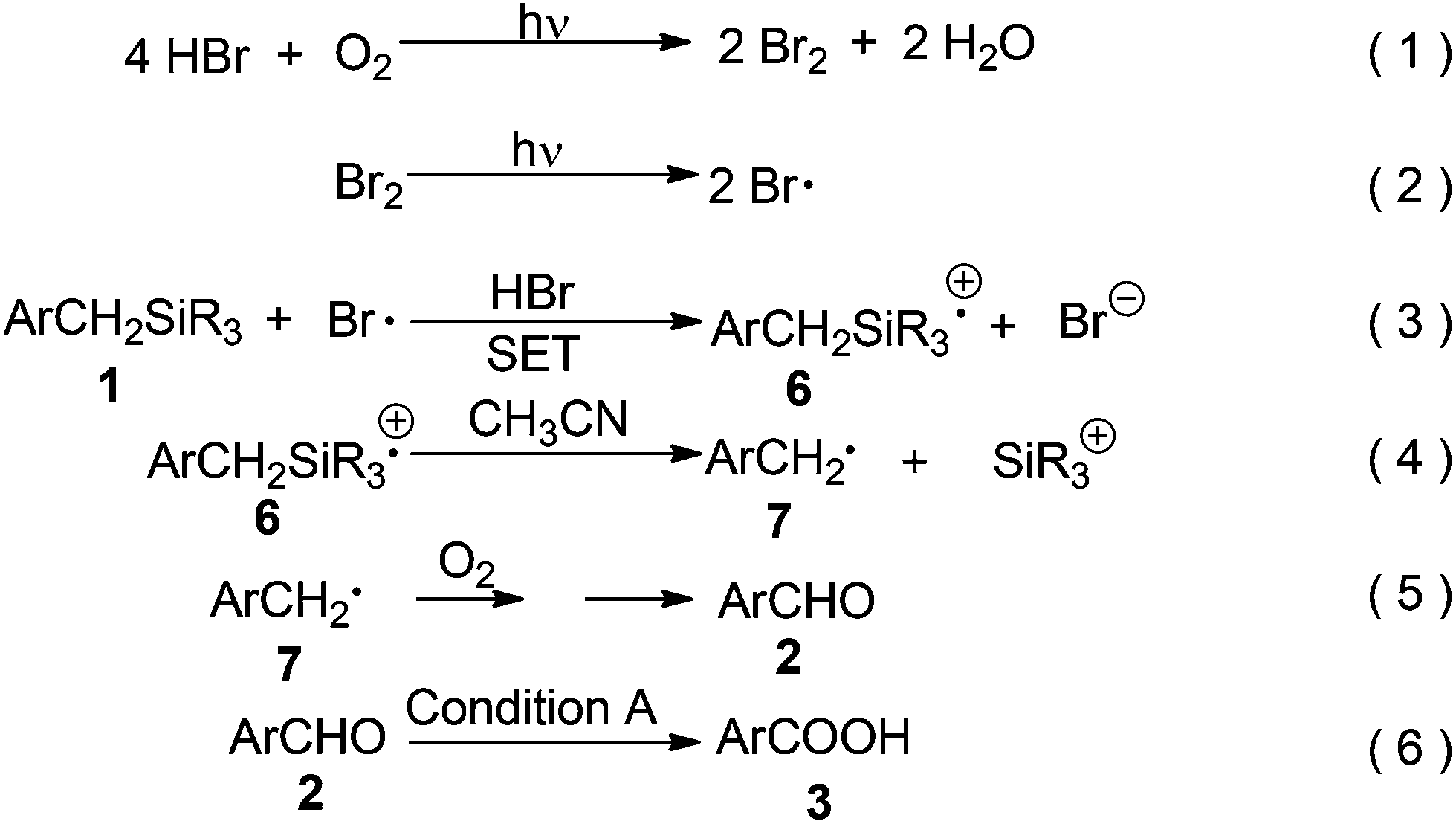 | ||
| Scheme 5 Plausible mechanism. | ||
Conclusions
In summary, we have developed a photoinduced HBr-catalyzed highly efficient C–Si bond cleavage reaction of benzylsilanes. The resulting benzyl radical intermediates were highly chemoselectively oxidized into benzoic acids using air as the terminal oxidant. A possible reaction pathway was proposed based on a series of control experiments. Notably, HBr not only acts as the single electron transfer mediator for the initial C–Si bond cleavage, but also efficiently catalyzes the transformation of benzaldehyde intermediates into benzoic acids. Further investigations in this field are in progress in our laboratory.Acknowledgements
We greatly acknowledge financial support from Shanghai Rising-Star Program (14QA1400500), National Basic Research Program of China (973 Program, 2012CB720300), National Nature Science Foundation of China (21102016 and 21274023), and Shanghai Scientific and Technological Innovation Project (13520711500).Notes and references
- For reviews, see: (a) H. J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8450 Search PubMed; (b) J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef CAS PubMed; (c) S. Farkas, CNS Drug Rev., 2006, 12, 218 CrossRef CAS PubMed.
- For reviews, see: (a) J. K. Puri, R. Singh and V. K. Chahal, Chem. Soc. Rev., 2011, 40, 1791 RSC; (b) L. W. Xu, L. Li, G. Q. Lai and J. X. Jiang, Chem. Soc. Rev., 2011, 40, 1777 RSC; (c) P. C. Bulman Page, S. S. Klair and S. Rosenthal, Chem. Soc. Rev., 1990, 19, 147 RSC.
- For reviews, see: (a) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845 RSC; (b) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC; (c) A. Hosomi, Acc. Chem. Res., 1988, 21, 200 CrossRef CAS.
- (a) T. Hayamizu, H. Maeda and K. Mizuno, J. Org. Chem., 2004, 69, 4997 CrossRef CAS PubMed; (b) T. Hayamizu, H. Maeda, M. Ikeda and K. Mizuno, Tetrahedron Lett., 2001, 42, 2361 CrossRef CAS; (c) T. Hayamizu, M. Ikeda, H. Maeda and K. Mizuno, Org. Lett., 2001, 3, 1277 CrossRef CAS PubMed; (d) T. Tamai, K. Mizuno, I. Hashida and Y. Otsuji, Bull. Chem. Soc. Jpn., 1993, 66, 3747 CrossRef CAS; (e) T. Tamai, K. Mizuno, I. Hashida and Y. Otsuji, Chem. Lett., 1992, 21, 781 CrossRef; (f) K. Mizuno, M. Ikeda and Y. Otsuji, Chem. Lett., 1988, 1507 CrossRef CAS; (g) E. Hasegawa, M. A. Brumfield, P. S. Mariano and U. C. Yoon, J. Org. Chem., 1988, 53, 5435 CrossRef CAS.
- (a) L. Cermenati, M. Fagnoni and A. Albini, Can. J. Chem., 2003, 81, 560 CrossRef CAS; (b) L. Cermenati and A. Albini, J. Adv. Oxid. Technol., 2002, 5, 58 CAS.
- (a) E. Baciocchi, M. Crescenzi and T. D. Giacco, J. Chem. Soc., Perkin Trans. 1, 1991, 3377 RSC; (b) E. Baciocchi and M. Crescenzi, Angew. Chem., 1990, 102, 667 ( Angew. Chem., Int. Ed. Engl. , 1990 , 29 , 658 ) CrossRef.
- S. Montanaro, D. Ravelli, D. Merli, M. Fagnoni and A. Albini, Org. Lett., 2012, 14, 4218 CrossRef CAS PubMed.
- Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2012, 48, 6966 RSC.
- S. Hirashima and A. Itoh, Chem. Pharm. Bull., 2007, 55, 156 CrossRef CAS.
- T. Sugai and A. Itoh, Tetrahedron Lett., 2007, 48, 2931 CrossRef CAS PubMed.
- L. Eberson, Adv. Phys. Org. Chem., 1982, 18, 1 ff CrossRef.
- J. P. Dinnocenzo, S. Farid, J. L. Goodman, I. R. Gould, W. P. Todd and S. L. Mattes, J. Am. Chem. Soc., 1989, 111, 8973 CrossRef CAS.
- L. Cermenati, M. Mella and A. Albini, Tetrahedron, 1998, 54, 2575 CrossRef CAS.
- For other bromide-catalyzed aerobic photooxidation of benzaldehydes into benzoic acids, see: S. Hirashima and A. Itoh, Chem. Pharm. Bull., 2006, 54, 1457 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental methods, experimental procedures, and the characterization data of compounds. See DOI: 10.1039/c4qo00229f |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2014 |