
Copper-catalyzed tandem trifluoromethylation/cyclization of internal alkynes†
Yun-Long
Ji
a,
Jin-Hong
Lin
*a,
Ji-Chang
Xiao
*a and
Yu-Cheng
Gu
b
aKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: jlin@sioc.ac.cn; jchxiao@sioc.ac.cn; Fax: (+86) 21-6416-6128; Tel: (+86) 21-5492-5380, (+86) 21-5492-5340
bSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire, RG42 6EY, UK
First published on 4th November 2014
Abstract
Copper-catalyzed tandem trifluoromethylation/cyclization of internal alkynes with Umemoto's reagent leads to 3-trifluoromethyl-1,2-dihydronaphthalene derivatives in moderate to good yields. The utility of this copper-catalyzed tandem reaction was demonstrated by oxidizing and reducing the trifluoromethylated product to give naphthalene and tetrahydronaphthalene, respectively, and the development of a short route to a trifluoromethylated analogue of Nafoxidine.
Introduction
The incorporation of a trifluoromethyl group into a drug candidate usually modifies its physical, chemical and biological properties through steric and electronic effects.1 Consequently, determined efforts have been directed towards the exploration of efficient and broadly applicable methods for trifluoromethylation.2 Transition metal-catalyzed or -mediated methods have proven to be highly attractive because of their high level of functional group tolerance and mild reaction conditions.2b,3 Copper4 and palladium5 complexes have been shown to be quite efficient for the trifluoromethylation of a wide variety of functional groups, leading to the construction of a new C–CF3 bond.Intensive studies have been devoted to the development of general approaches for the construction of the Cvinyl–CF3 bond via trifluoromethylation of olefins6 or alkynes.7 However, these methods suffer from the need to prefunctionalize olefinic substrates, or are limited to the trifluoromethylation of terminal alkynes. The direct trifluoromethylation of internal alkynes to construct a Cvinyl–CF3 bond remains a significant challenge. In continuation of our research interest in the chemistry of trifluoromethylation,8 we have now investigated the copper-catalyzed tandem direct trifluoromethylation/cyclization of internal alkynes to achieve the difunctionalization of alkynes.
Very recently, the use of copper-catalyzed direct trifluoromethylation of internal alkynes to construct Cvinyl–CF3 bonds has been reported by other groups.9 Liu and coworkers described the domino copper-catalyzed trifluoromethylation/Meyer–Schuster rearrangement of propargylic alcohols with Togni's reagent leading to α-trifluoromethyl enones as products (eqn (1), Scheme 1).9a Interestingly, trifluoromethylation of homopropargylic alcohols affords 3-trifluoromethyl-3-butenal derivatives via a quite different reaction route (eqn (2), Scheme 1).9b The construction of the Cvinyl–CF3 bond and difunctionalization of alkynes were achieved in the above two reactions. On the basis that cyclic structures are commonly found in natural products and drugs, studies on tandem trifluoromethylation/cyclization are also worthy of attention. The Hou group disclosed the trifluoromethylation of homopropargyl amines with Umemoto's reagent giving 4-trifluoromethyl-2,3-dihydro-pyrroliums (eqn (3)).9c During the preparation of this manuscript, Ding et al. reported the trifluoromethylation of propiolates resulting in trifluoromethylated coumarins as products (eqn (4)).9d Heterocycles were formed in these two processes (eqn (3) and (4)). Our efforts are aimed at the tandem trifluoromethylation/cyclization of internal alkynes to construct carbon rings (eqn (5)). Preliminary results are described herein.10
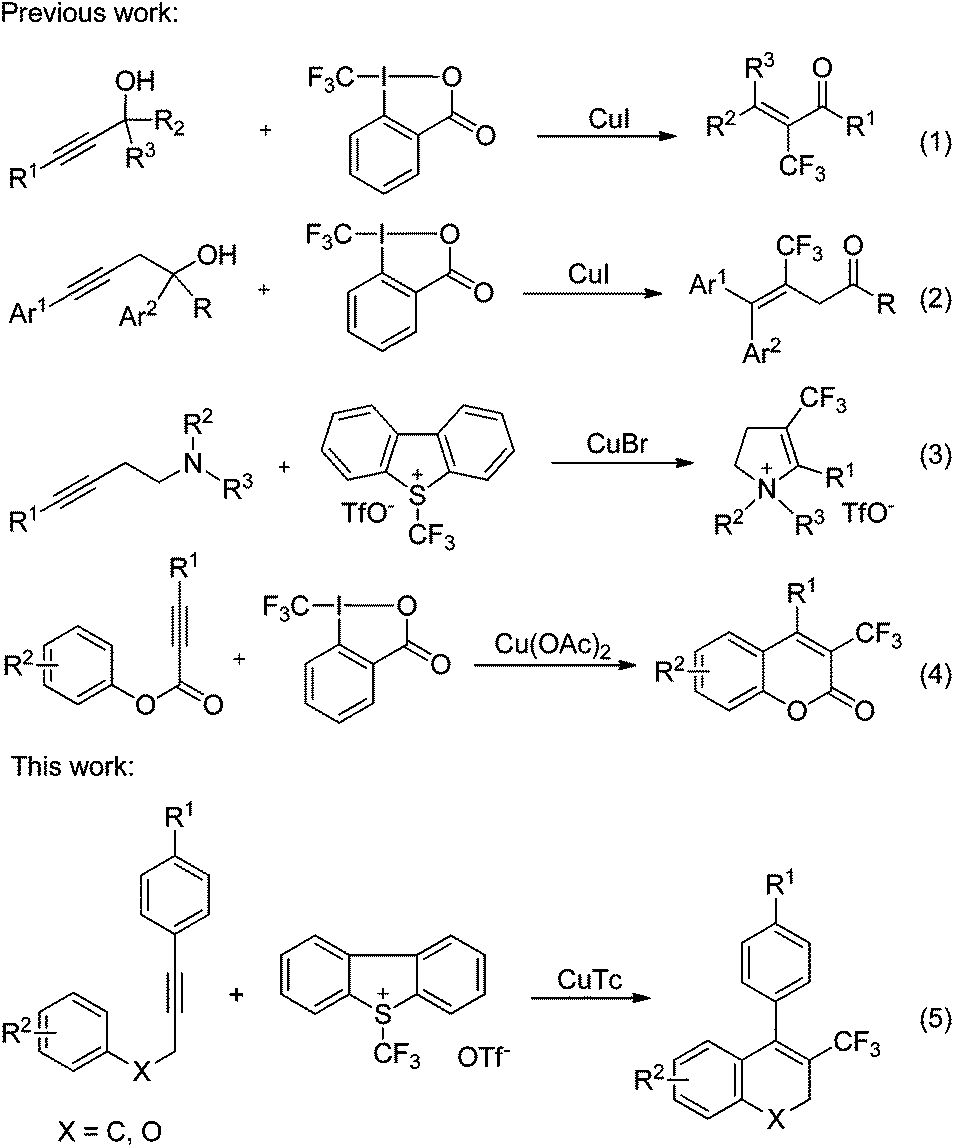 | ||
| Scheme 1 Trifluoromethylation of internal alkynes. | ||
Results and discussion
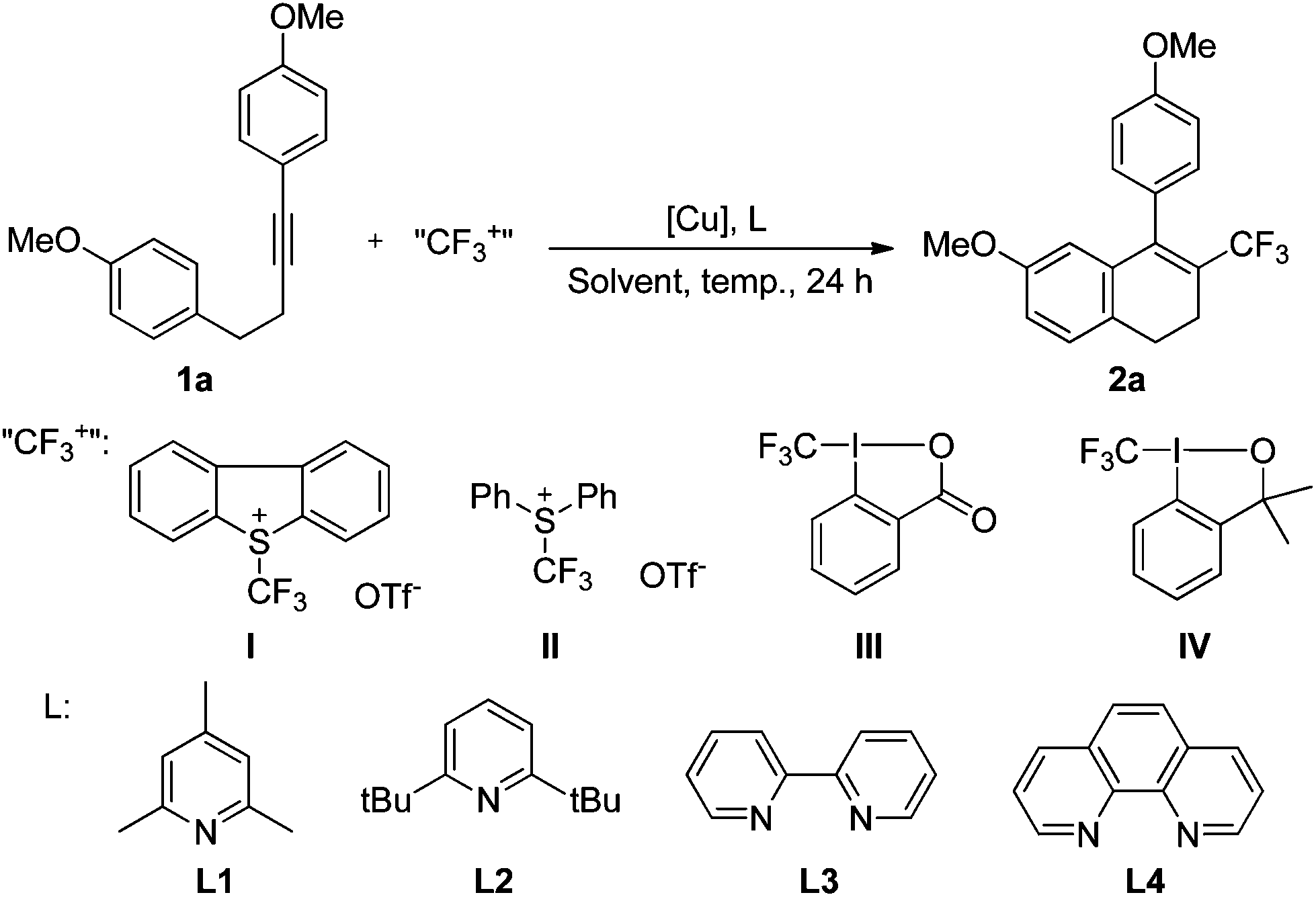
Our first attempt at trifluoromethylation of the internal alkyne 1a with Umemoto's reagent catalyzed by CuI in DCM was successful, leading to the desired product 2a, albeit in low yield (Table 1, entry 1). 2,4,6-Trimethyl pyridine (L1) was used both as the ligand and the base. An examination of other copper sources suggested that CuTc [copper(I)thiophene-2-carboxylate] was a suitable catalyst (entry 3), while Cu(0) and Cu(II) were not effective for this transformation (entries 2, 4, and 5). The reaction in 1,2-dichloroethane (DCE) gave essentially the same yields (entry 6 vs. entry 3). The use of other more polar solvents resulted in much lower yields or almost no reaction at all (entries 7–10). The use of the bulky ligand L2 or the bidentate ligand (L3 or L4) instead of L1 did not improve the yield (entries 11–13). Umemoto's reagent, in combination with the system CuTc/L3, was the only useful reagent for this conversion. Other trifluoromethylating reagents gave dramatically lower yields of the expected product (entry 14) or led to none of the expected product at all (entries 15 and 16). The loading of L3 had a significant effect on the reaction yield (entries 17–19 vs. entry 12), and the use of 0.8 equiv. of L3 improved the yield to 51% (entry 18). The yield was slightly improved when the reaction was carried out in DCE and the temperature was elevated to 80 °C (entry 20). A higher loading of Umemoto's reagent I increased the yield (entry 21), but the yield did not increase further with loadings beyond 1.5 equiv. of I (entry 22). Interestingly, in the presence of MeOH (0.1 mL), the reaction gave the expected product in good yield (entry 23 vs. entry 21). The desired transformation took place only to a very limited extent in the absence of a copper source (entry 24).|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | [Cu] | L | Solvent | Temp. (°C) | “CF3+” | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 1a (0.1 mmol), trifluoromethylating reagent (1 equiv.), copper source (20 mol%) and L (1.2 equiv.) in solvent (1 mL). b Determined by 19F NMR with the use of trifluoromethyl benzene as an internal standard. c 0.5 equiv. of L was used. d 2 equiv. of II was used. e 0.2 equiv. of L3 was used. f 0.8 equiv. of L3 was used. g 1 equiv. of L3 was used. h 1.5 equiv. of I was used. i 2 equiv. of I was used. j MeOH (0.1 mL) was added as an additive. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | CuI | L1 | DCM | 50 | I | 27 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | (MeCN)4CuPF6 | L1 | DCM | 50 | I | 5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | CuTc | L1 | DCM | 50 | I | 39 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Cu(OAc)2 | L1 | DCM | 50 | I | 4 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Cu | L1 | DCM | 50 | I | Complex | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | CuTc | L1 | DCE | 50 | I | 36 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | CuTc | L1 | THF | 50 | I | 8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | CuTc | L1 | CH3CN | 50 | I | 23 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | CuTc | L1 | MeOH | 50 | I | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | CuTc | L1 | DMF | 50 | I | 20 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | CuTc | L2 | DCM | 50 | I | 35 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12c | CuTc | L3 | DCM | 50 | I | 40 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13c | CuTc | L4 | DCM | 50 | I | 39 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14c,d | CuTc | L3 | DCM | 50 | II | 8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15c | CuTc | L3 | DCM | 50 | III | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16c | CuTc | L3 | DCM | 50 | IV | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17e | CuTc | L3 | DCM | 50 | I | 16 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18f | CuTc | L3 | DCM | 50 | I | 51 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 19g | CuTc | L3 | DCM | 50 | I | 31 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20f | CuTc | L3 | DCE | 80 | I | 54 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 21f,h | CuTc | L3 | DCE | 80 | I | 66 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 22f,i | CuTc | L3 | DCE | 80 | I | 64 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 23f,h,j | CuTc | L3 | DCE | 80 | I | 82 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 24f,h | — | L3 | DCE | 80 | I | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Since methanol had a positive effect on the conversion (Table 1, entry 23), we reasoned that other alcohols might also be favourable. To our surprise, under the standard conditions shown in entry 21 of Table 1, the addition of ethanol or isopropanol inhibited the desired transformation, and the reaction produced ethoxy- or isopropoxy-substituted side products, respectively (eqn (1) and (2), Scheme 2). The phenoxy-substituted substrate 1b could be converted to the desired product 2b in good yield under the standard conditions (eqn (3)). However, in the presence of methanol, compound 2b turned out to be a side product and the methoxy-substituted compound 2a was the major product (eqn (4)). These results indicated that the use of methanol as an additive was only favourable for the conversion of substrates in which the homopropargylic benzene ring was substituted by a 4-MeO group.
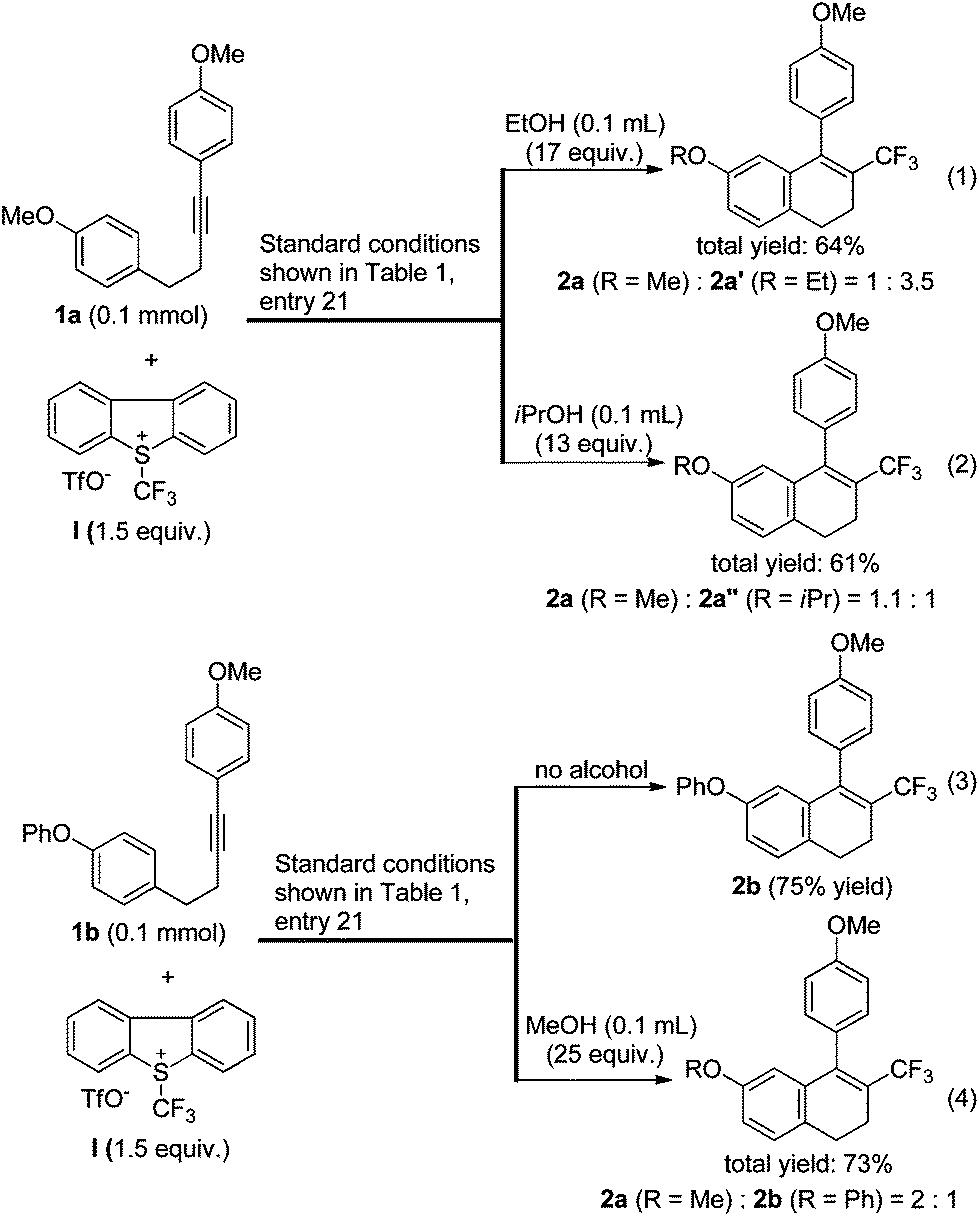 | ||
| Scheme 2 The use of alcohol as an additive. | ||
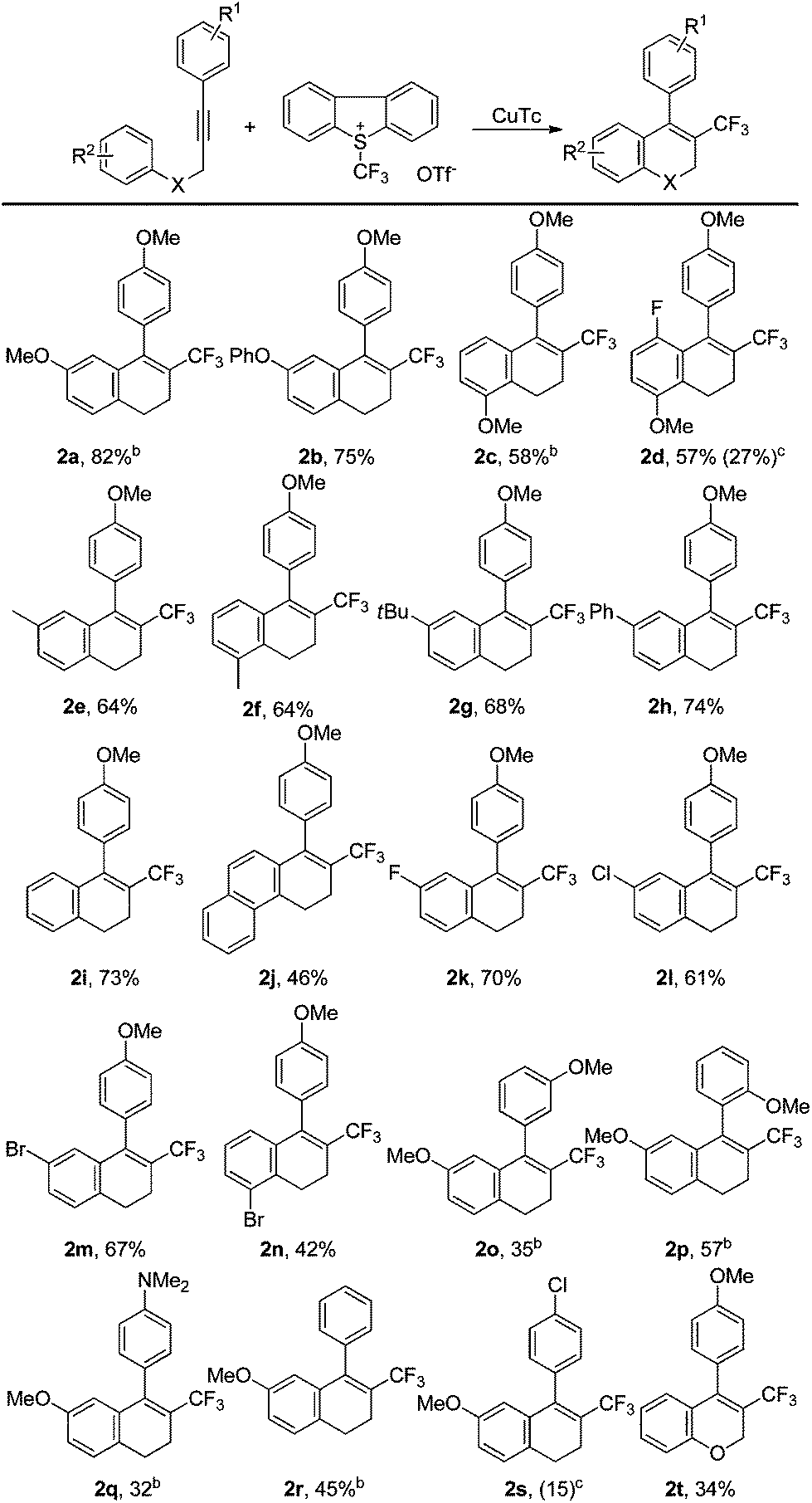
We then investigated the substrate scope of the tandem trifluoromethylation/cyclization under the optimized reaction conditions (Table 1, entry 21). As shown in Table 2, irrespective of whether the homopropargylic benzene ring is substituted by an electron-withdrawing group or an electron-donating group, the tandem reactions can proceed very well to give the desired products in moderate to good yields (2a–2n). For substrates substituted with a methoxy group on the homopropargylic benzene ring, superior results were obtained with the use of methanol as an additive (2a and 2c). In contrast, the presence of methanol in the reaction led to a lower yield for the conversion of 2d. Substrates substituted in the ortho-position of the homopropargylic benzene ring could also be converted smoothly to the corresponding products (2c, 2d, 2f and 2n). In the cases of substrates with a methoxy group on the meta- or ortho-position of the ethynyl benzene ring (2o–2p), or with the other electron-donating group on the ethynyl benzene ring (2q–2r), moderate yields of the expected products were obtained. But an electron-withdrawing group led to a dramatic decrease in the yield (2s). The reaction could also be applied to a propargylic ether, albeit affording the cyclized product in low yield (2t).
| a Reaction conditions: 1 (0.1 mmol), trifluoromethylating reagent (1.5 equiv.), CuTc (20 mol%) and L3 (0.8 equiv.) at 80 °C in DCE (1 mL). Isolated yields. b Methanol (0.1 mL) was used as an additive in the reaction. c The yield in parenthesis was determined by 19F NMR for the reaction with methanol (0.1 mL) as an additive. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The structure of the product 2h was determined by single crystal X-ray diffraction (Fig. 1).11 The structures of the other products were surmised by analogy.
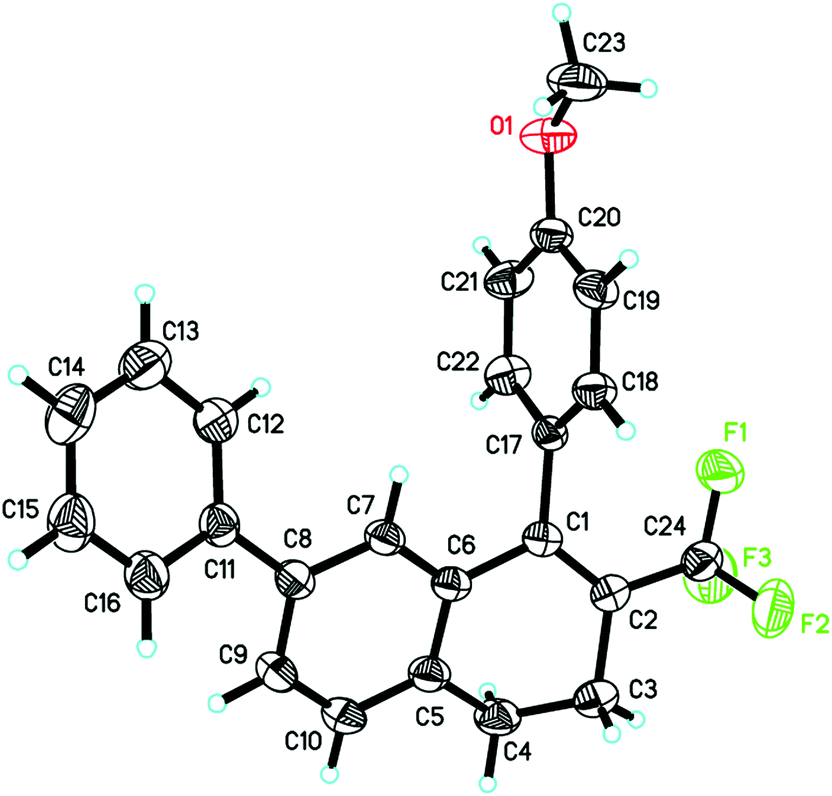 | ||
| Fig. 1 X-ray diffraction of 2h. | ||
The tandem trifluoromethylation/cyclization might not involve radical species, because the well-known radical scavenger, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), did not suppress the desired reaction (Scheme 3).
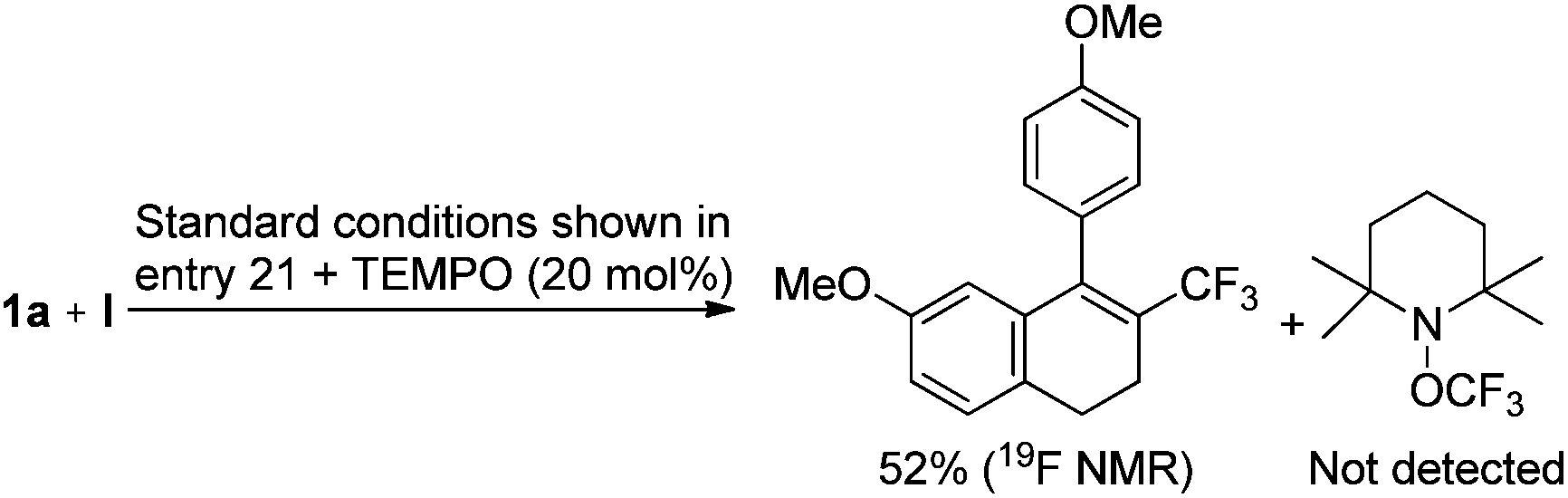 | ||
| Scheme 3 Mechanistic experiment involving a radical scavenger. | ||
On the basis of the above results, we propose that the reaction mechanism shown in Scheme 4 is plausible. The oxidation of Cu(I) by Umemoto's reagent produces CF3Cu(III), the electrophilic attack of which to substrate 1 generates intermediate A. Reductive elimination leads to intermediate B and regeneration of the catalyst Cu(I). The subsequent intramolecular cyclization gives intermediate C (path I), followed by aromatisation by loss of proton to furnish the final product 2. If the homopropargylic benzene ring is substituted by a 4-RO group, intermediate B might also undergo ipso Friedel–Crafts reaction to afford intermediate D (path II). The presence of methanol as an additive can stabilize intermediate D by converting this intermediate to E. The Wagner–Meerwein rearrangement of intermediate D then produces intermediate C, and facile aromatisation gives the final product 2. The second path explains the formation of the by-products 2a′ or 2a′′ with the use of other alcohols as additives, and also explains the conversion of substrate 1b to 2a in the presence of methanol (Scheme 2).
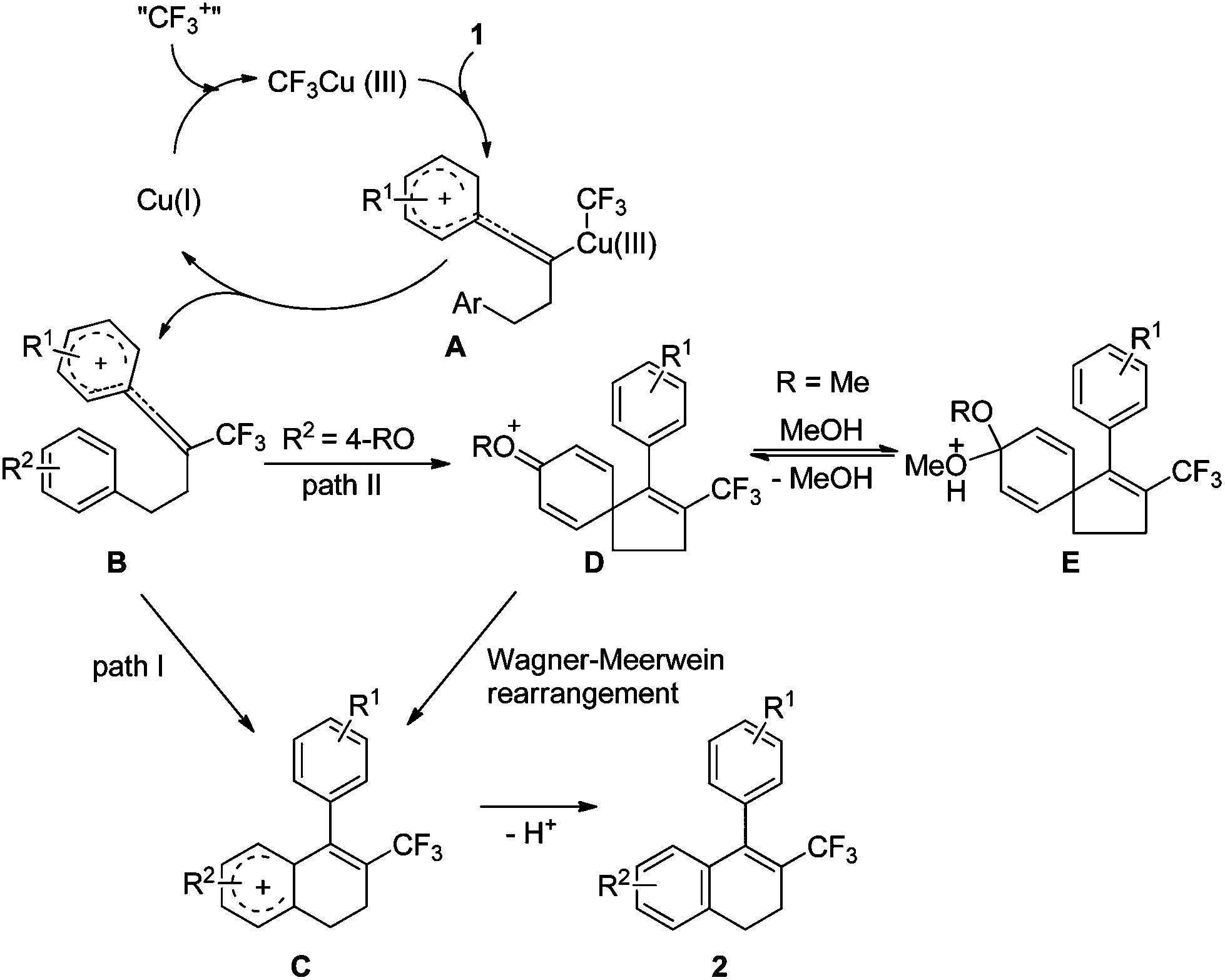 | ||
| Scheme 4 Proposed reaction mechanism. | ||
The utility of this copper-catalyzed tandem reaction was demonstrated by oxidizing and reducing trifluoromethylated product 2a to the naphthalene 3 and the tetrahydronaphthalene 4, respectively, and the development of a short route to a trifluoromethylated analogue of Nafoxidine, an anticancer agent (Scheme 5). Under the standard trifluoromethylating conditions, the substrate 1u was converted smoothly into a mixture of trifluoromethylated dihydronaphthalenes (52% yield) composed of para- and ortho-cyclized products (2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The treatment of the isolated para-cyclized dihydronaphthalene with pyrrolidine in ethanol afforded the Nafoxidine analogue 5 in 82% yield.
:1). The treatment of the isolated para-cyclized dihydronaphthalene with pyrrolidine in ethanol afforded the Nafoxidine analogue 5 in 82% yield.
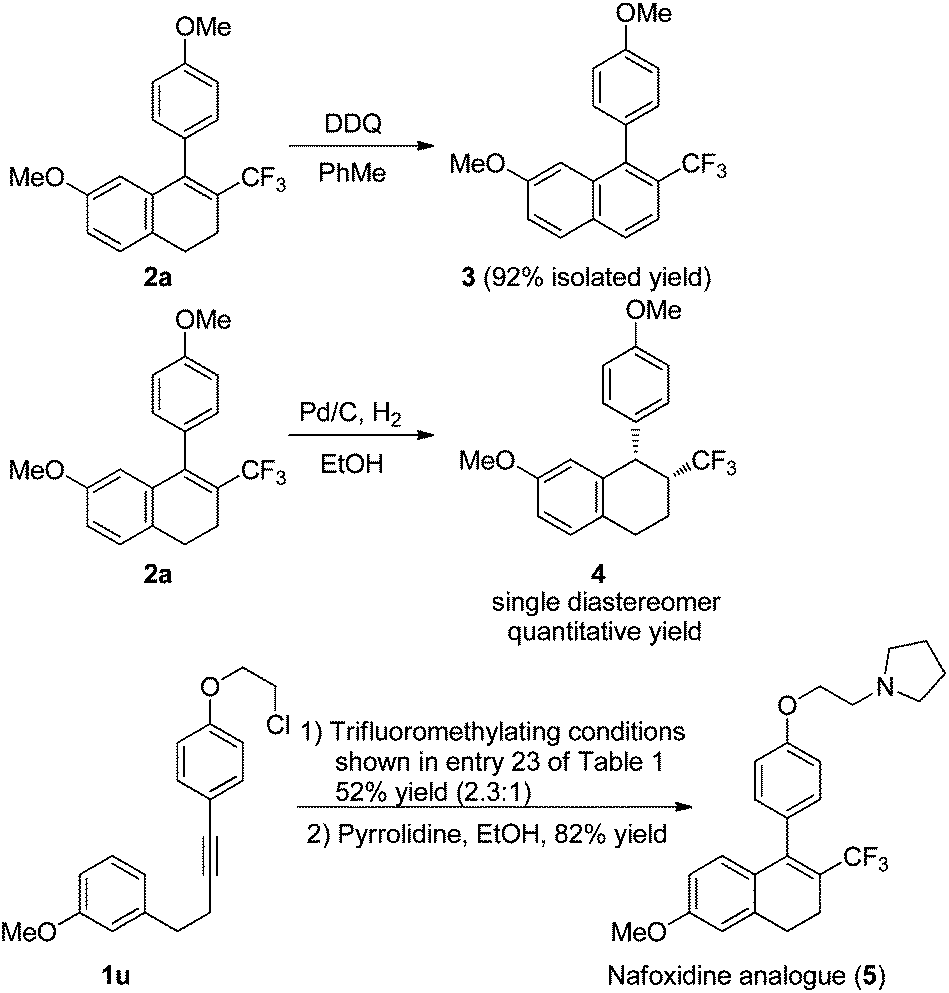 | ||
| Scheme 5 The utilities of the tandem reaction. | ||
Conclusions
In conclusion, we have described the copper-catalyzed tandem trifluoromethylation/cyclization of internal alkynes with Umemoto's reagent leading to 3-trifluoromethyl-1,2-dihydronaphthalene derivatives in moderate to good yields. The new method can be applied to the synthesis of a trifluoromethylated analogue of Nafoxidine, demonstrating the utility of this tandem trifluoromethylation/cyclization. This tandem transformation represents a convenient method for the difunctionalization-type trifluoromethylation of internal alkynes.Acknowledgements
We thank the National Natural Science Foundation (21032006, 21172240, 21421002, 21472222), the National Basic Research Program of China (2015CB931900), the Science and Technology Commission of Shanghai Municipality (14ZR1448800), the Chinese Academy of Sciences and the Syngenta PhD Fellowship Awarded to Y.-L. Ji. We thank Dr John Clough of Syngenta at Jealott's Hill International Research Centre for proof reading the manuscript.Notes and references
-
(a)
K. Uneyama, Organofluorine Chemistry, Blackwell Publishing, Oxford, 2006 Search PubMed
; (b) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed
- For recent reviews, please see:
(a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed
- For recent reviews, please see:
(a) H. Liu, Z. Gu and X. Jiang, Adv. Synth. Catal., 2013, 355, 617–626 CrossRef CAS
-
(a) F. Wang, D. Wang, X. Mu, P. Chen and G. Liu, J. Am. Chem. Soc., 2014, 136, 10202–10205 CrossRef CAS PubMed
-
(a) E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679–1681 CrossRef CAS PubMed
-
(a) E. Cho and S. Buchwald, Org. Lett., 2011, 13, 6552–6555 CrossRef CAS PubMed
-
(a) P. G. Janson, I. Ghoneim, N. O. Ilchenko and K. J. Szabo, Org. Lett., 2012, 14, 2882–2885 CrossRef CAS PubMed
-
(a) C.-P. Zhang, Z.-L. Wang, Q.-Y. Chen, C.-T. Zhang, Y.-C. Gu and J.-C. Xiao, Angew. Chem., Int. Ed., 2011, 50, 1896–1900 CrossRef CAS PubMed
-
(a) Y. P. Xiong, M. Y. Wu, X. Y. Zhang, C. L. Ma, L. Huang, L. J. Zhao, B. Tan and X. Y. Liu, Org. Lett., 2014, 16, 1000–1003 CrossRef CAS PubMed
- When the manuscript was complete, the group of Fu also reported the copper-catalyzed tandem trifluoromethylarylation/cyclization of internal alkynes: J. Xu, Y.-L. Wang, T.-J. Gong, B. Xiao and Y. Fu, Chem. Commun., 2014, 50, 12915–12918 RSC
- The structure of the product 2h was determined by single crystal X-ray diffraction (see ESI†). Summary of data CCDC 1018298.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1018298. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00240g |
| This journal is © the Partner Organisations 2014 |